

Abstract
Signals from extracellular matrix (ECM) to growth factor receptors regulate glomerular epithelial cell (GEC) proliferation. Epidermal growth factor (EGF), basic fibroblast growth factor, hepatocyte growth factor (HGF), or thrombin stimulated proliferation of GECs when the cells were adherent to collagen matrices, but not plastic substratum. Furthermore, EGF, HGF, or thrombin activated p42 mitogen-activated protein (MAP) kinase in collagen-adherent GECs, whereas activation was weak in GECs on plastic. To further examine the interaction of ECM with the Ras-MAP kinase cascade, GECs were stably transfected with a constitutively active Ras mutant (V12Ras). Low or moderate levels of V12Ras expression did not affect basal MAP kinase activity but, unlike parental GECs, in clones that express V12Ras, EGF was able to induce proliferation and activate MAP kinase when these cells were adherent to plastic. In parental and V12Ras-transfected GECs, MAP kinase activation was inhibited by cytochalasin D. Thus, adhesion of GECs to ECM facilitates proliferation and MAP kinase activation by mitogens acting via tyrosine kinase or non-tyrosine kinase receptors. Activation of pathway(s) downstream of V12Ras supplants signals from ECM that enable proliferation. These signals may involve the actin cytoskeleton.
Adhesion of cells to extracellular matrix (ECM) can modulate proliferative responses of cells to polypeptide growth factors and promote cell differentiation. 1-3 We and others have studied intracellular signaling mechanisms that are activated by adhesion of cells to ECM, as well as interactions of ECM with growth factors. 3-11 Many growth factors stimulate cell proliferation through binding to cell surface receptors that possess intrinsic tyrosine kinase activity. 12,13 Growth factors that are mitogenic for epithelial cells include epidermal growth factor (EGF), transforming growth factor-α, and heparin-binding EGF, which are structurally and functionally related polypeptides that bind to the EGF receptor (EGF-R) 14,15 as well as hepatocyte growth factor (HGF) and basic fibroblast growth factor (bFGF), which bind to Met and the FGF-Rs, respectively. 16,17 It is believed that the initial events involve binding of growth factor to a receptor tyrosine kinase and receptor oligomerization. 12,13 This results in transmembrane activation of the cytoplasmic tyrosine kinase, receptor autophosphorylation, and phosphorylation of substrate proteins. 12,13 The signal is then transmitted to nuclear or cytoplasmic effectors through a series of serine/threonine protein kinases, collectively known as the mitogen-activated protein (MAP) kinase pathway. 18,19 Briefly, receptor tyrosine kinases usually activate p21Ras (Ras) via Grb-2/Sos. Ras induces translocation of Raf-1 to the plasma membrane, where Raf-1 is activated by an undefined kinase. Raf-1 activates MEK (MAP or extracellular signal-regulated kinase (ERK) kinase), which then activates p42 (ERK2) and/or p44 (ERK1) MAP kinases via dual phosphorylation on threonine and tyrosine. The ERKs have multiple potential actions, which include the triggering of gene expression required for cell proliferation.
Visceral and parietal glomerular epithelial cells (GECs) are intrinsic components of the kidney glomerulus, and both cell types are in contact with ECM. 20,21 Turnover of GECs is normally low, and it has been suggested that visceral GECs do not proliferate. 20,22 However, proliferation of parietal and possibly visceral GECs and expansion of the ECM may occur in immune glomerular injury and may lead to impaired glomerular function and/or permselectivity. 21,23,24 For example, urine samples from children with Henoch-Schönlein purpura nephritis (a nephritis often associated with glomerular proliferation) contain a factor that resembles transforming growth factor-α, suggesting that the presence of this factor in the glomerulus may be stimulating epithelial proliferation. 25 In previous studies, we have demonstrated that adhesion to ECM triggers signals that can regulate proliferation of cultured rat GECs in a positive or negative fashion. β1-Integrin-mediated turnover of inositol phospholipids was associated with a reduction in GEC proliferation. 4,5 ECM also facilitated proliferation and enhanced EGF-dependent activation of EGF-R. 6,8 Specifically, EGF stimulated EGF-R autophosphorylation, the activity and tyrosine phosphorylation of ERK2, and proliferation in GECs adherent to collagen matrices but not to plastic substratum. Furthermore, an inhibitor of MEK, PD98059, blocked EGF-induced ERK2 activity and proliferation of collagen-adherent GECs. 6,8 The differences in EGF-R activation between substrata could not be accounted for by differences in ligand binding, EGF-R protein content, or EGF-R degradation and appeared to be due to regulation of EGF-R kinase activity and/or trafficking by factors extrinsic to the receptor. 6
The aims of the present study were to determine whether the modulation of receptor tyrosine kinases and MAP kinase activation by ECM occur with diverse GEC mitogens, and to define the role of the Ras in the regulation of GEC proliferation by ECM. We demonstrate that by analogy to EGF, HGF or thrombin induced proliferation and stimulated ERK2 activity significantly in collagen-adherent GECs but not in GECs on plastic. Second, stable expression of a constitutively active Ras mutant (V12Ras) allowed GECs to proliferate on plastic, indicating that sustained activation of pathway(s) downstream of Ras enabled proliferation that was independent of ECM.
Materials and Methods
Materials
Tissue culture media, Transfinity CaPO4 transfection system, and G418 (geneticin) were obtained from Life Technologies (Burlington, Ontario, Canada). Pepsin-solubilized bovine dermal collagen (Vitrogen) was from Collagen Corp. (Palo Alto, CA). NuSerum, EGF, HGF, bFGF, and collagen IV were purchased from Collaborative Research (Bedford, MA). Myelin basic protein and thrombin were obtained from Sigma Chemical Co. (St. Louis, MO). Anti-phosphotyrosine monoclonal antibody, PY20, was from Transduction Laboratories (Lexington, KY). Rabbit anti-ERK2, rabbit anti-FGF-R2, and rabbit anti-Met antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit anti-EGF-R antibody, RK2, was described previously. 6,8 Anti-phospho-ERK antibody was purchased from New England Biolabs (Mississauga, Ontario, Canada). [γ-32P]ATP (3000Ci/mmol) was from New England Nuclear (Boston, MA). Electrophoresis and immunoblotting reagents were from Biorad Laboratories (Mississauga, Ontario, Canada). Plasmid HO6T1, which contains the constitutively active Ras gene, 26 was kindly provided by Dr. Morag Park (McGill University, Montreal, Canada).
Extracellular Matrix and GEC Culture
Type I collagen gel matrices were prepared by combining RPMI-1640 (10X) medium, 7.5% NaHCO3, pepsin-solubilized bovine dermal collagen (∼3 mg/ml in 0.012 N HCl), and 0.1 N NaOH in proportions of 10:4:80:10 at 4°C. The mixture was then poured into tissue culture dishes (∼0.06 ml/cm2) and allowed to gel at 37°C, as described previously. 6 Collagen IV solution was applied to culture wells at 0.02 mg/cm 2 and was allowed to air dry at 22°C.
Primary cultures of rat GECs were established from explants of rat glomeruli, as described previously. 4-8,27 Studies were done with cells between passages 25 and 70. According to established criteria, the cells demonstrated polygonal shape and cobblestone appearance at confluency, cytotoxic susceptibility to low doses of aminonucleoside of puromycin, presence of junctional complexes by electron microscopy, 27 and positive immunofluorescence staining for a variety of GEC antigens. 28 Presently, it is not possible to determine specifically whether GECs in culture originate from visceral or parietal epithelium. Under standard conditions, GECs were cultured on collagen matrices in K1 medium, which consisted of Dulbecco’s modified Eagle’s medium/Ham F10 (1:1), containing 5% NuSerum and hormone supplements. 6,27 To remove GECs from collagen substrata, collagen gels with adherent cells were scraped from culture dishes into a test tube and were incubated with collagenase and trypsin-EDTA to produce a cell suspension. 6,27 For passaging of cultures, GECs were replated onto collagen gels; for experiments, GECs were replated onto collagen gels or plastic substrata.
Measurement of GEC Proliferation
Cell number was determined by visual counting. For the proliferation experiments, GECs were cultured in serum-poor medium (Dulbecco’s modified Eagle’s medium/Ham F10, 1:1, with 0.5% fetal calf serum), with or without growth factor (bFGF was added together with heparin, 10 U/ml). Cells adherent to collagen gels (35-mm plates) were placed into single-cell suspension with collagenase and trypsin-EDTA, as described above. Cells on plastic substratum or collagen IV were placed into suspension by incubation with trypsin-EDTA. Suspended cells were then counted in a hemacytometer. 6
A soft agar assay was used to evaluate anchorage-independent proliferation. Tissue culture plates (60 mm) were coated with 4 ml of 0.6% agar in K1 medium. The bottom layer was allowed to solidify and was then overlaid with 2 ml of 0.3% agar in K1 medium containing 10,000 GECs. After the top layer solidified, the GECs were cultured at 37°C. The number of GEC colonies was determined after 14 days of culture by counting 10 randomly selected, low-power microscopic fields.
Assay of ERK2 Activity and Tyrosine Phosphorylation
For ERK2 assays, GECs were plated onto substrata and were incubated in serum-poor medium for 18 hours before addition of growth factor. Preliminary studies were performed in collagen-adherent GECs to determine the incubation time required to maximally stimulate ERK2 activity. After incubation with growth factor, GECs were scraped from culture dishes and were solubilized in buffer containing 0.5% Triton X-100, 50 mmol/L β-glycerophosphate, 2 mmol/L MgCl2, 1 mmol/L dithiothreitol, 20 μmol/L leupeptin, 20 μmol/L pepstatin, 0.2 mmol/L phenylmethylsulfonyl fluoride, 1 mmol/L Na3VO4, and 1 mmol/L EGTA, pH 7.2 (4°C). 8 Proteins (50 to 100 μg) were immunoprecipitated with rabbit anti-ERK2 antibody (1 hour at 4°C) or nonimmune IgG (background control), followed by absorption with agarose-coupled protein A (1 hour at 4°C). The immunoprecipitates were then assayed for ERK2 activity by monitoring phosphorylation of myelin basic protein. 8 In addition to the immunoprecipitates, the assay contained 20 mmol/L MOPS, pH 7.2, 25 mmol/L β-glycerophosphate, 5 mmol/L EGTA, 1 mmol/L Na3VO4, 1 mmol/L dithiothreitol, 0.5 mg/ml bovine brain myelin basic protein, 7.5 mmol/L MgCl2, and 50 μmol/L [γ-32P]ATP (10 μCi). After 10 minutes of incubation at 30°C, the mixture was spotted onto phosphocellulose paper. The paper was washed with 0.75% phosphoric acid and acetone, and bound radioactivity was quantitated in a β-scintillation counter. Background radioactivity was subtracted from total radioactivity of each sample, and results are expressed as fold increase as compared with control. To assess ERK2 tyrosine phosphorylation, GECs were scraped from culture dishes. Lysates (200 to 400 μg of protein) were immunoprecipitated with anti-ERK2 antibody or nonimmune IgG in controls. Immunoprecipitates were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). After transfer to nitrocellulose paper, the immunoprecipitates were immunoblotted with anti-phosphotyrosine antibody, as described previously. 6,8 In some experiments, activation of ERK2 was monitored by immunoblotting with anti-phospho-ERK antibody, ie, antibody that reacts with ERK phosphotyrosine204. Tyrosine204 phosphorylation was quantitated by densitometry, as described previously. 6
GEC Transfection
Plasmid HO6T1 contains a constitutively active Ha-Ras gene (Gly12→Val 12 mutation) and neomycin-resistance gene. 26 GECs adherent to collagen were transfected with plasmid HO6T1 (2 μg of DNA per 100-mm plate), using the CaPO4 technique, as described previously. 7 GECs were then cultured in K1 medium containing 0.5 mg/ml G418. GEC clones resistant to G418 were isolated and replated onto plastic substratum. Clones that proliferated on plastic in K1 medium were selected, passaged, and assessed for expression of Ras protein by immunoblotting.
Statistics
Data are presented as means ± SEM. One-way analysis of variance (ANOVA) was used to determine significant differences among groups. Where significant differences were found, individual comparisons were made between groups using the t statistic and adjusting the critical value according to the Bonferroni method.
Results
Collagen I Enables Growth-Factor-Induced Proliferation
The first series of experiments assessed whether ECM regulated proliferation in the presence of growth factors that are known to act via receptor tyrosine kinases. As earlier studies demonstrated that EGF-induced proliferation in GECs on collagen I increases steadily over 3 days after plating, 6,8 the 3-day time point was chosen to study the effects of growth factors. EGF, HGF, and bFGF significantly stimulated proliferation of GECs adherent to collagen but not plastic (Figure 1 ▶ , upper panel). HGF and bFGF, although used at high doses, were not as potent as EGF. Addition of growth factors to GECs on plastic did not increase the number of cells significantly, as compared with serum-poor medium alone, and collagen did not independently stimulate proliferation in the absence of growth factors 6 (data not shown). Studies on the expression of receptor tyrosine kinases demonstrated that there were no differences in the protein content of EGF-R, Met (the receptor for HGF), or FGF-R2 between GECs on collagen and plastic (Figure 2) ▶ . Thus, differences in GEC proliferation between substrata could not be accounted for by differences in receptor expression. It should be noted that in GECs, EGF-R was detectable by immunoblotting, but to detect Met and FGF-R2, samples required immunoprecipitation with receptor-specific antibodies before immunoblotting. Thus, Met and FGF-R2 were probably expressed at levels lower than EGF-R, explaining why the effects of EGF on proliferation may have been more potent than those of HGF or bFGF. In other experiments, we demonstrated that in GECs, expression of FGF-R1, FGF-R3, and FGF-R4 was trivial (results not shown). Therefore, the effect of bFGF on proliferation is most likely mediated through FGF-R2.
Figure 1.
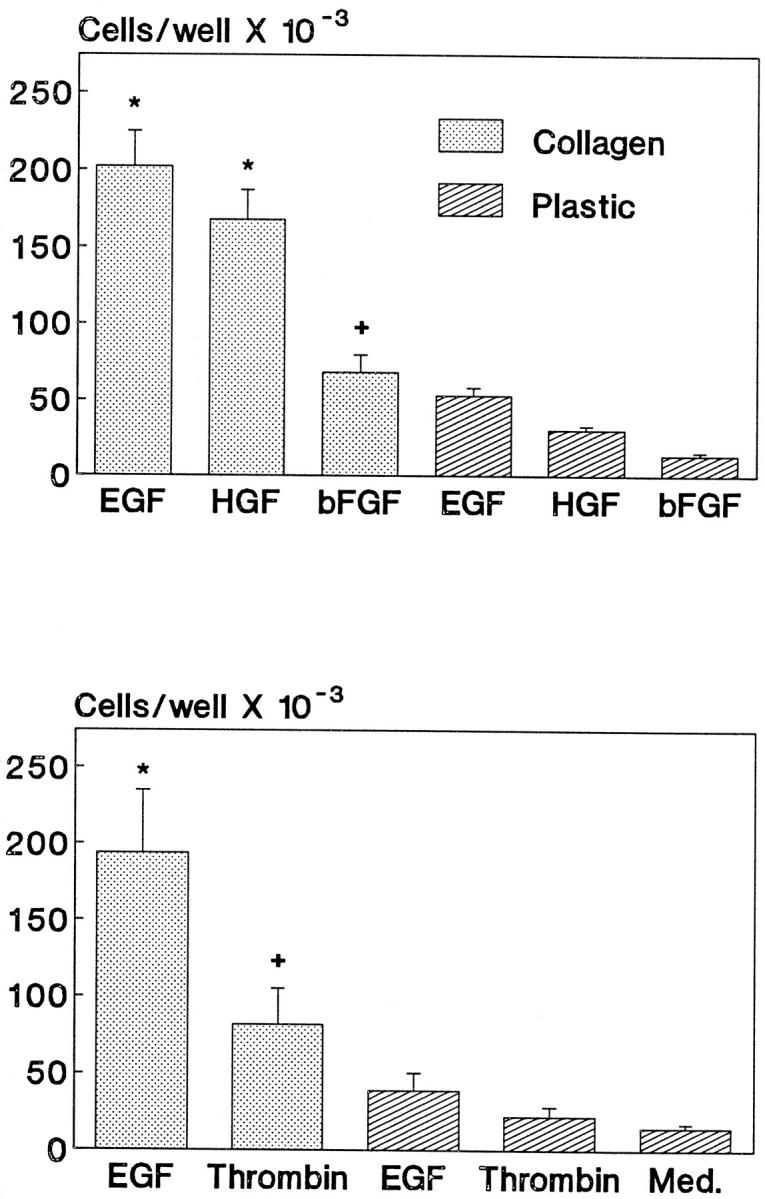
ECM facilitates growth-factor-induced proliferation. GECs were plated into culture wells that were coated with collagen or were uncoated (plastic) in serum-poor medium that contained EGF (100 ng/ml), HGF (50 ng/ml), bFGF (50 ng/ml), thrombin (5 U/ml), or medium alone (Med). Cell number was determined 3 days after plating. All growth factors stimulated proliferation only when GECs were adherent to collagen. Significant differences were present among groups (upper panel: P < 0.0001 ANOVA; *P < 0.0001, +P < 0.01 collagen versus plastic; lower panel: P < 0.0001 ANOVA; *P < 0.0001, +P < 0.05 collagen versus plastic). Each bar represents mean ± SEM of 8 to 12 wells.
Figure 2.

ECM does not affect expression of receptor tyrosine kinases. GECs on collagen (C) or plastic (P) were solubilized in detergent-containing buffer. For EGF-R, samples (125 μg of protein) were subjected to SDS-PAGE and immunoblotting with anti-EGF-R receptor antibody. The arrow points to the 170-kd EGF-R. For Met and FGF-R2, samples (1.5 mg of protein) were immunoprecipitated with receptor-specific antibodies before SDS-PAGE and immunoblotting with receptor-specific antibodies. In the Met blot, the arrow points to the 145-kd Met. The upper band (∼190 kd) probably represents the unprocessed Met precursor, and the lower band may be a degradation product. In the FGF-R2 blot, the arrow points to the 150-kd (3 Ig domain) FGF-R2. The lower band at ∼125 kd probably represents the 2 Ig domain FGF-R2. 17
To determine whether the effect of collagen I on GEC proliferation was restricted to receptor tyrosine kinases, we also studied the mitogenic response to thrombin, which acts through a seven-transmembrane domain receptor coupled to G-proteins. 29 Similar to EGF, thrombin stimulated proliferation in GECs adherent to collagen but not plastic (Figure 1 ▶ , lower panel). Thus, ECM regulates induction of GEC proliferation by multiple growth factors, acting via tyrosine kinase and non-tyrosine kinase receptors.
ERK2 Activity Is Stimulated by Growth Factors in GECs Adherent to Collagen I
In keeping with previous results, 8 EGF stimulated ERK2 activity effectively only in collagen-adherent GECs (Figure 3) ▶ . By analogy, HGF and thrombin stimulated ERK2 activity significantly in GECs on collagen, but not plastic, whereas bFGF induced an upward trend only in collagen-adherent cells (Figure 3) ▶ . Therefore, similar to proliferation, ECM regulated ERK2 activation by growth factors acting via tyrosine kinase and non-tyrosine kinase receptors, and the potency of stimulating ERK2 activity paralleled that of proliferation. It should also be noted that before stimulation of GECs with growth factors (ie, at ∼18 hours after plating), there were no significant differences in basal ERK2 activity between collagen and plastic substrata (data not shown).
Figure 3.

ERK2 activity is stimulated by growth factors in GECs adherent to ECM. GECs were plated onto collagen or plastic and were cultured in serum-poor medium for 18 hours. GECs were then incubated with or without EGF (100 ng/ml, 1 hour), HGF (50 ng/ml, 10 minutes), bFGF (50 ng/ml, 10 minutes), or thrombin (5 U/ml, 1 hour) at 37°C. Cell lysates were immunoprecipitated with anti-ERK2 antibody, and the immunoprecipitates were assayed for ERK2 activity by monitoring phosphorylation of myelin basic protein. Values are expressed as fold increase, growth-factor-stimulated versus unstimulated. Significant stimulation of ERK2 activity was evident in GECs adherent to collagen in the presence of EGF (P < 0.004, nine experiments), HGF (P < 0.006, five experiments), and thrombin (P < 0.025, four experiments). bFGF induced an upward trend in GECs on collagen (five experiments).
The MEK inhibitor PD98059 was used to determine whether activation of the Ras-ERK2 pathway in collagen-adherent GECs was required for proliferation. PD98059 (50 μmol/L) blocked GEC proliferation induced by EGF (95 ± 5% inhibition), HGF (84 ± 11% inhibition), bFGF (90 ± 10% inhibition), and thrombin (87 ± 6% inhibition; three experiments, P < 0.0001 for each growth factor). This concentration of PD98059 inhibits EGF-stimulated ERK2 activity in GECs by 80%. 8
Stable Expression of V12Ras in GECs
We postulated that expression of a constitutively active Ras gene, which would result in sustained activation of Ras and its downstream pathways, 6 may supplant the effect of ECM and lead to proliferation of GECs on plastic. GECs adherent to collagen were stably transfected with V12Ras (a constitutively active Ha-Ras) and neomycin-resistance genes (see Methods). Clones resistant to G418 were then replated onto plastic, and 22 clones that proliferated on plastic were isolated, passaged, and assessed for expression of Ras protein by immunoblotting. Among these 22 transfected GEC clones, all stably expressed V12Ras protein, 2 at high levels, 11 at intermediate levels, and 9 at low levels. Four clones that express high (R514), intermediate (R25, R34), or low levels of V12Ras (R311) were chosen for additional studies (Figure 4) ▶ . It should be noted that expression of endogenous Ras in parental (untransfected) GECs is not readily detectable by immunoblotting.
Figure 4.

Overexpression of V12Ras. GECs adherent to collagen were transfected with a constitutively active Ha-Ras gene (V12Ras). Clones that proliferated on plastic were selected and assessed for expression of Ras protein by SDS-PAGE and immunoblotting (25 μg of protein per lane). Expression of endogenous Ras in parental (untransfected) GECs is not readily detectable by immunoblotting. Four clones express high (R514), intermediate (R25, R34), or low levels of V12Ras (R311).
By light microscopy, the morphology of V12Ras-transfected GEC clones did not appear to be different from that of parental cells, described previously; 27 ie, the transfected GECs featured a polygonal shape and cobblestone appearance at confluency. In addition, the ultrastructural features of clone R514 (which stably expresses a high level of V12Ras) were evaluated using electron microscopy. Differentiated features of epithelial cells, including apical microvilli and junctional complexes, were preserved in clone R514 GEC (Figure 5) ▶ . This ultrastructural appearance is similar to that of parental GECs adherent to collagen. 27 Although, the other V12Ras-transfected GEC clones were not studied by electron microscopy, it is unlikely that lower levels of V12Ras expression would have induced morphological alterations or dedifferentiation.
Figure 5.
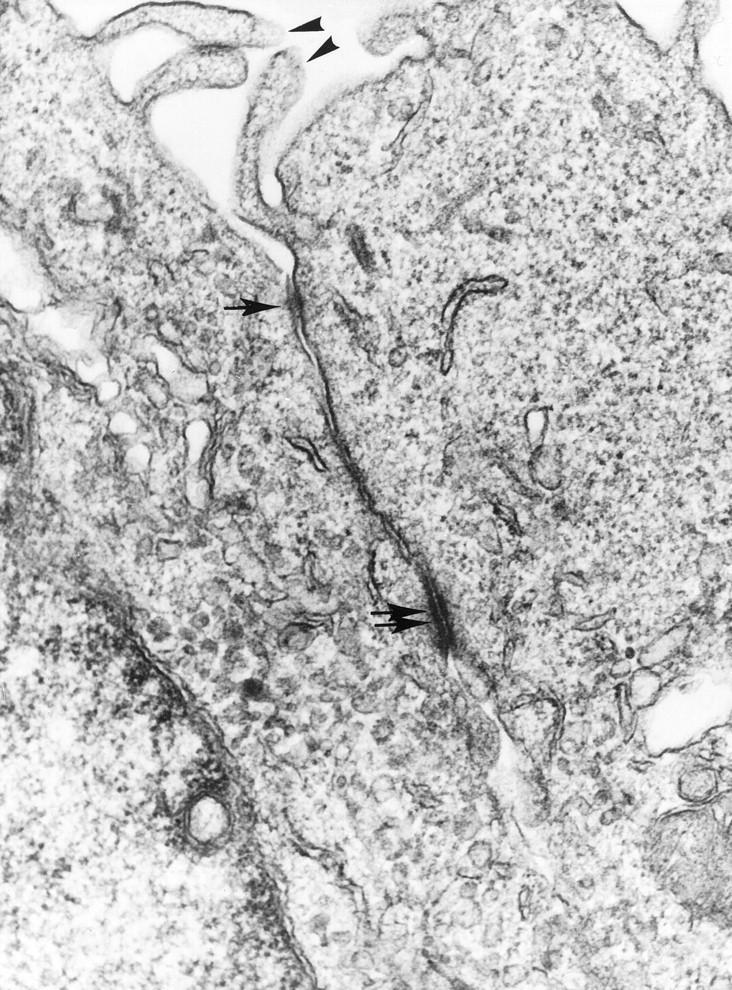
Ultrastructure of GECs that stably express V12Ras (clone R514). The electron micrograph shows portions of two GECs with apical microvilli (arrowheads) and a junctional complex consisting of a tight junction (arrow) and desmosome (two arrows). Magnification, ×50,000.
The V12Ras-transfected GEC clones were also tested for anchorage dependence of proliferation, by using a soft agar assay. Clone R514 (which expresses a high level of V12Ras) was able to form colonies in soft agar (Table 1) ▶ . Although the colonies were relatively few in number, nevertheless, this clone appeared to have undergone transformation. The three other V12Ras-transfected clones, as well as parental GECs, did not form colonies in agar (Table 1) ▶ .
Table 1.
Anchorage Dependence of Proliferation in V12Ras-Transfected GECs
| Number of colonies | Number of wells | |
|---|---|---|
| Parental GECs | 0 ± 0 | 6 |
| Ras-transfected GECs | ||
| R514 | 4.0 ± 0.7* | 6 |
| R25 | 0 ± 0 | 3 |
| R34 | 0 ± 0 | 3 |
| R311 | 0 ± 0 | 3 |
GECs were plated in soft agar in K1 medium (10,000 cells/60-mm well). The number of colonies was determined in 10 randomly selected, low-power microscopic fields after 14 days of culture. Significant differences were present among groups (P < 0.0001, ANOVA).
*Only clone R514, which stably expresses a high level of V12Ras, proliferated in an anchorage-independent manner (P < 0.0001 versus other groups).
Expression of V12Ras Enables Growth-Factor-Dependent Proliferation of GECs on Plastic
Proliferation of V12Ras-transfected GEC clones on plastic was studied 3 days after plating, in the presence or absence of EGF (the mitogen that appeared to be most potent). In the absence of EGF, clones R311, R34, and R25 (low to moderate expression of V12Ras) were not able to proliferate, as cell number was not significantly different from that of parental GECs on plastic (Figure 6 ▶ , upper panel). In contrast, these V12Ras-transfected GEC clones proliferated on plastic in the presence of EGF, at rates similar to parental GECs on collagen, whereas the parental GECs did not proliferate on plastic (Figure 6 ▶ , lower panel). By analogy to EGF, when clone R25 GECs were cultured for 3 days on plastic in the presence of HGF or thrombin, cell number increased 2.43 ± 0.50-fold and 1.80 ± 0.07-fold, respectively (P < 0.05, three wells per group). Thus, low to moderate expression of V12Ras supplants the requirement for ECM but not growth factor. Clone R514, which featured a high level of V12Ras expression and anchorage-independent proliferation in the presence of growth factors, was able to proliferate on plastic in an EGF-independent manner (Figure 6 ▶ , upper panel). The increase in cell number was, however, smaller than the increase in parental GECs cultured on collagen in the presence of EGF.
Figure 6.
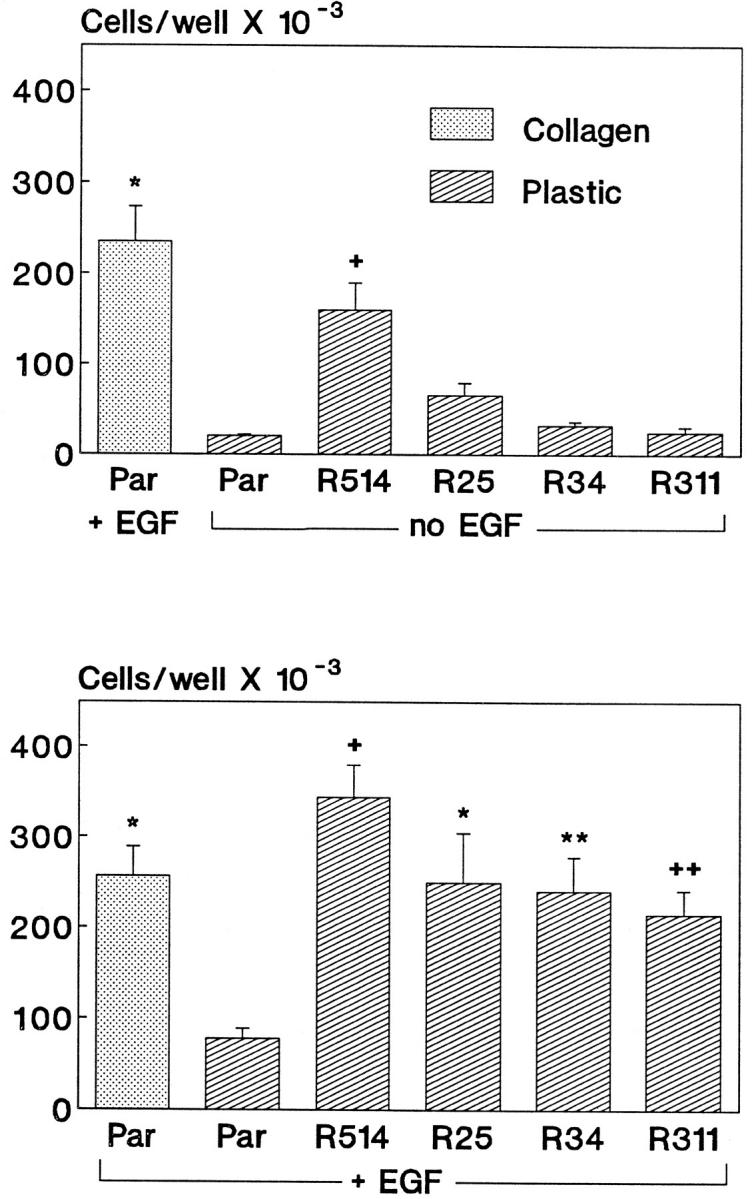
V12Ras supplants the effect of ECM on proliferation. Parental GECs (Par) or GEC clones that express V12Ras were plated into culture wells that were coated with collagen or were uncoated (plastic) in serum-poor medium, with or without EGF (100 ng/ml). Cell number was determined 3 days after plating. Unlike parental GECs, V12Ras-transfected GEC clones were able to proliferate on plastic in the presence of EGF (lower panel). Except for clone R514, proliferation of V12Ras-transfected GECs did not occur in the absence of EGF (upper panel). Significant differences were present among groups (upper panel: P < 0.0001 ANOVA; *P < 0.0001 versus parental GECs on plastic and P = 0.05 versus R514, +P < 0.005 versus parental GECs on plastic, mean ± SEM of four wells; lower panel: P < 0.0001 ANOVA; +P < 0.0001, *P < 0.003, **P < 0.006, ++P < 0.025 versus parental GECs on plastic, mean ± SEM of eight wells).
Growth Factors Activate ERK2 in Plastic-Adherent GECs That Express V12Ras
As proliferation of parental GECs on collagen was dependent on the activation of the ERK2 pathway, we assessed whether expression of V12Ras resulted in changes in basal or stimulated ERK2 activity. In the absence of EGF, basal ERK2 activity in clones R25 and R34 (which express moderate levels of V12Ras) was not significantly different from parental GECs (Table 2) ▶ . As stated above, basal ERK2 activity in parental GECs was not affected significantly by adhesion to collagen I; consequently, basal ERK2 activity in the V12Ras-transfected GECs on plastic was also similar to unstimulated parental GECs on collagen. Addition of EGF stimulated ERK2 activity significantly in both clones R25 and R34 (Table 2) ▶ . Thus, moderate expression of V12Ras enabled EGF to stimulate ERK2 activity in the absence of ECM.
Table 2.
ERK2 Activity in V12Ras-Transfected GECs
| ERK2 activity (fold increase) | |||
|---|---|---|---|
| R25 | R34 | R514 | |
| No EGF | 1.17 ± 0.33 | 1.25 ± 0.25 | 2.11 ± 0.57* |
| + EGF | 1.84 ± 0.13† | 2.24 ± 0.56‡ | 2.59 ± 0.84 |
Parental and V12Ras-transfected GECs (clones R25, R34, and R514) were plated onto plastic and were cultured in serum-poor medium for 18 hours. R25, R34, and R514 GECs were then incubated with or without EGF (100 ng/ml) for 1 hour at 37°C. Parental GECs were incubated without EGF. ERK2 activity was determined as in Figure 3 ▶ . Values are expressed as fold increase, V12Ras-transfected versus parental (five to six experiments per clone). EGF stimulated ERK2 activity significantly in R25 and R34 GECs. Basal ERK2 activity was increased significantly only in R514 GECs.
*P < 0.045 versus parental.
†P < 0.001 versus no EGF.
‡P < 0.05 versus no EGF.
The pattern of ERK2 activity was different in GEC clone 514 (high level of V12Ras expression). In the absence of EGF, clone R514 GECs adherent to plastic demonstrated an approximately twofold increase in ERK2 activity, as compared with parental GECs on plastic (Table 2) ▶ . In the presence of EGF, there was a further increase in ERK2 activity in R514 GECs, which was not statistically significant (Table 2) ▶ .
Results obtained in assays of ERK2 activity were confirmed by monitoring tyrosine phosphorylation of ERK2. In keeping with changes in ERK2 activity (Figure 3) ▶ , EGF induced tyrosine phosphorylation of ERK2 in parental GECs adherent to collagen, but there was little change in cells on plastic (Figure 7A) ▶ . Furthermore, in contrast to parental GECs on plastic, ERK2 was endogenously tyrosine phosphorylated in V12Ras-transfected GECs adherent to plastic (clone R514; Figure 7B ▶ ), consistent with changes in ERK2 activity (Table 2) ▶ . The basal level of ERK2 tyrosine phosphorylation in V12Ras-transfected GECs was comparable to the level of phosphorylation in EGF-stimulated GECs adherent to collagen (Figure 7B) ▶ . Based on three experiments, there were no significant differences in ERK2 protein between transfected and parental GECs, although the amount of ERK2 protein may be slightly greater in the V12Ras-transfected GECs shown in Figure 7C ▶ .
Figure 7.
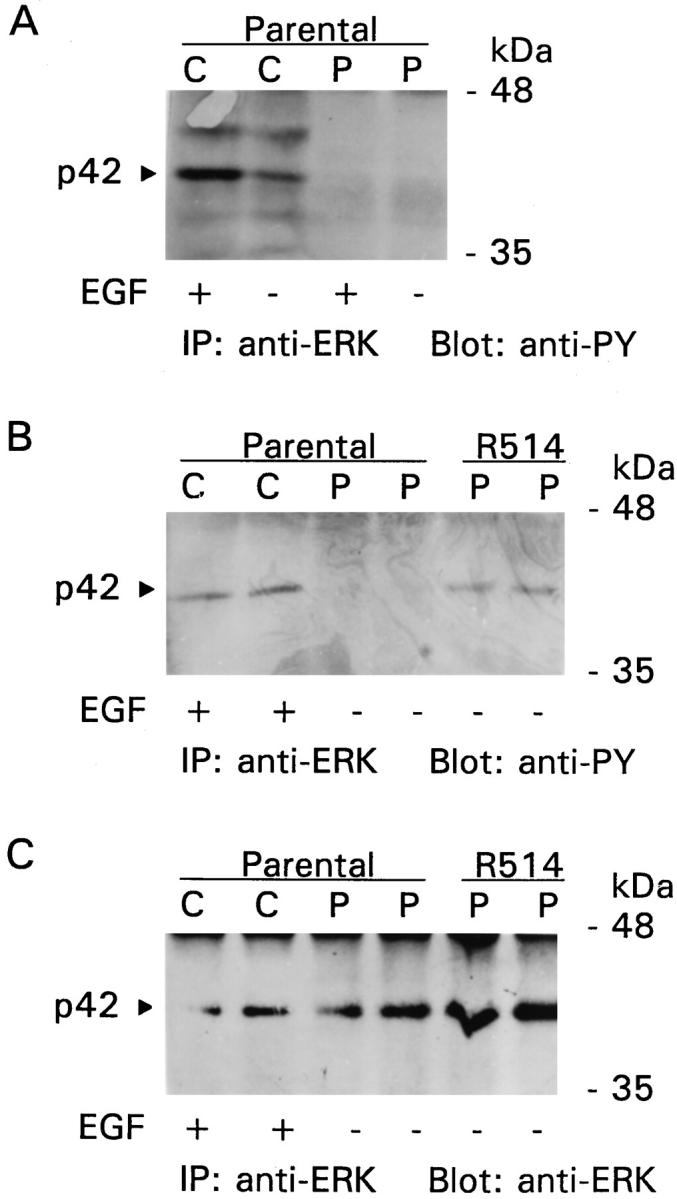
Effects of EGF, ECM, and V12Ras on ERK2 tyrosine phosphorylation. A: EGF stimulates ERK2 tyrosine phosphorylation in parental GECs adherent to collagen. GECs were plated onto collagen (C) or plastic (P) and were cultured in serum-poor medium for 18 hours. GECs were then incubated with (+) or without (−) EGF (100 ng/ml) for 1 hour at 37°C. Cell lysates were immunoprecipitated (IP) with anti-ERK antibody, and the immunoprecipitates were subjected to SDS-PAGE and immunoblotting with anti-phosphotyrosine (PY) antibody. B and C: ERK2 is tyrosine phosphorylated in V12Ras-transfected GECs (clone R514). GECs were cultured in serum-poor medium for 18 hours. Parental GECs on collagen were incubated with or without EGF (as in A). V12Ras-transfected GECs were incubated without EGF. Cell lysates (in duplicate) were immunoprecipitated and immunoblotted as indicated.
Proliferation of V12Ras-transfected GECs (clone R514) on plastic was blocked with 50 μmol/L PD98059, confirming the functional importance of the ERK2 pathway. Cell counts in two experiments carried out 3 days after plating were as follows: serum-poor medium (no EGF), 103,750; serum-poor medium (no EGF) plus PD98059, 49,375; EGF, 165,000; EGF plus PD98059, 78,125 (averages of two experiments). These results are analogous to collagen-adherent parental GECs, cultured in the presence of EGF.
In the next series of experiments, we compared ERK2 activation in V12Ras-transfected GEC clones after addition of growth factors that had stimulated ERK2 activity in parental GECs on collagen. We used GECs adherent to plastic, which express low (clone R311) or moderate levels of V12Ras (clones R34 and R25), as studies described above suggested that basal ERK2 activity was not increased significantly in these clones. Both EGF and HGF activated ERK2 significantly, whereas thrombin induced an upward trend in each of the clones (Table 3) ▶ . Basal ERK2 activity was not significantly different from parental GECs (Table 3) ▶ , confirming results in Table 2 ▶ . Thus, the pattern of ERK2 activation in V12Ras-transfected GECs on plastic is similar to that of parental GECs on collagen (Figure 3) ▶ , except that the effect of HGF was relatively more potent in some transfected cells (although not significantly greater than the effect of EGF), and the effect of thrombin appeared to be relatively more potent in parental cells.
Table 3.
Effects of Growth Factors on ERK2 Activation in V12Ras-Transfected GECs
| ERK2 tyrosine204 phosphorylation (fold increase) | |||
|---|---|---|---|
| R25 | R34 | R311 | |
| Basal | 0.81 ± 0.19 | 1.54 ± 0.38 | 1.43 ± 0.66 |
| EGF | 4.29 ± 0.88* | 6.01 ± 1.44† | 4.59 ± 0.37† |
| HGF | 3.10 ± 0.28‡ | 8.30 ± 3.07§ | 9.43 ± 2.41§ |
| Thrombin | 1.82 ± 0.19 | 1.86 ± 0.14 | 1.68 ± 0.16 |
Parental and V12Ras-transfected GECs (clones R25, R34, and R311) were plated onto plastic and were cultured in serum-poor medium for 18 hours. R25, R34, and R311 GECs were then incubated with or without growth factors (see legend to Figure 3 ▶ ). Parental GECs were incubated without growth factors. Lysates were subjected to SDS-PAGE and immunoblotting with anti-phospho-ERK antibody. Tyrosine204 phosphorylation, which reflects ERK2 activation, was quantitated by densitometry. Values are expressed as fold increase, V12Ras-transfected versus parental cells (three to five experiments per clone). Significant differences were present among groups (ANOVA: R25 P < 0.005, R34 P < 0.04, R311 P < 0.01). EGF and HGF activated ERK2 significantly, whereas thrombin induced an upward trend in each clone. Basal ERK2 activity was not increased significantly as compared with parental GECs.
*P < 0.001 versus control.
†P < 0.04 versus control.
‡P < 0.01 versus control.
§P < 0.005 versus control.
Cytochalasin D Inhibits ERK2 Activation
Moderate expression of V12Ras enabled EGF to stimulate ERK2 activation and proliferation in the absence of ECM. To determine whether the signal from ECM or V12Ras that facilitates ERK2 activation may have involved the cytoskeleton, we monitored the activation of ERK2 after incubation of GECs with cytochalasin D, a compound that disrupts the actin cytoskeleton. In the presence of cytochalasin D, EGF-induced ERK2 activation was inhibited significantly in parental GECs adherent to collagen and was abolished completely in V12Ras-transfected GECs on plastic (clones R25 and R34; Table 4 ▶ ).
Table 4.
Effect of Cytochalasin D on ERK2 Activation in Parental and V12Ras-Transfected GECs
| ERK2 tyrosine204 phosphorylation (fold increase) | |||
|---|---|---|---|
| Parental | R25 | R34 | |
| EGF | 2.54 ± 0.37 | 3.79 ± 0.82 | 2.62 ± 0.67 |
| Cytochalasin D+ EGF | 1.59 ± 0.23* | 1.00 ± 0.00† | 1.00 ± 0.00* |
Parental GECs on collagen and V12Ras-transfected GECs on plastic (clones R25 and R34) were cultured in serum-poor medium for 18 hours. GECs were incubated with or without cytochalasin D (20 μmol/L) for 15 minutes at 37°C and then with or without EGF (100 ng/ml) for 1 hour at 37°C. Lysates were subjected to SDS-PAGE and immunoblotting with anti-phospho-ERK antibody. Tyrosine204 phosphorylation, which reflects ERK2 activation, was quantitated by densitometry. Values are expressed as fold increase as compared with incubation without EGF (four to six experiments). Cytochalasin D inhibited EGF-stimulated ERK2 activity significantly.
*P < 0.03 versus EGF.
†P < 0.01 versus EGF.
EGF Stimulates Proliferation and ERK2 Activation in GECs on Collagen IV
These experiments were carried out to determine whether collagen IV facilitated GEC proliferation and ERK2 activation. Parental GECs were plated onto collagen IV or plastic substrata and were cultured in the presence of EGF (see legend to Figure 1 ▶ ). After 3 days, cell number increased 4.94 ± 0.36-fold on collagen IV, as compared with plastic (P < 0.001, five wells). In GECs adherent to collagen IV, EGF stimulated ERK2 activation 2.65 ± 0.61-fold (as determined by immunoblotting with antibody to ERK tyrosine204; P < 0.04, four wells). Thus, effects of collagen IV paralleled those of collagen I.
Discussion
This study demonstrates that ECM facilitates GEC proliferation and ERK2 activation. In keeping with earlier studies, 6,8 in GECs adherent to collagen, EGF induced a marked increase in cell number, whereas in GECs on plastic, there was no significant change (Figure 1) ▶ . Activation of growth factor receptor tyrosine kinases typically leads to the activation of the Ras-ERK2 cascade. 12,13 Stimulation of ERK2 activity by EGF was significantly greater in collagen-adherent GECs, as compared with plastic substratum (Figure 3) ▶ . The present study extends our previous observations by demonstrating that bFGF and HGF also stimulated proliferation significantly only in collagen-adherent GECs. In addition, HGF activated ERK2 in GECs on collagen, and a similar trend was observed with bFGF (Figure 3) ▶ . Furthermore, inhibition of MEK blocked GEC proliferation, indicating that the Ras-ERK2 pathway is functionally important. The effects of thrombin on GEC proliferation and ERK2 activation (Figures 1 and 3) ▶ ▶ were similar to those of EGF and HGF. Together, these results indicate that the regulatory effect of ECM in GECs involves multiple tyrosine kinase and non-tyrosine kinase receptors, and thus, it appears to be directed at Ras or downstream kinases of the ERK2 cascade, although our earlier studies have indicated that ECM may also regulate receptor activation. 9,10 By analogy to GECs, a recent study has shown that EGF and platelet-derived growth factor can activate ERK1 and ERK2 more efficiently in NIH 3T3 cells adherent to fibronectin, as compared with cells in suspension. 30 In contrast to other cells, 9,10 we did not detect direct ECM-induced activation of ERK2 in GECs, but our measurements were carried out several hours after plating of cells, and it is possible that ECM had activated the Ras-ERK2 pathway transiently or at very early time points after plating of GECs or that there was chronic, low-grade activation.
The principal novel observation in this study is that GECs, which had been stably transfected to express low or moderate levels of V12Ras (clones R25, R34, R311) proliferated when adherent to plastic in an EGF-dependent manner (Figure 6) ▶ . Thus, the signal(s) provided by V12Ras was sufficient to supplant the signal(s) from ECM, but not growth factor. In these cells, EGF (and other growth factors) were also able to activate ERK2; however, the moderate expression of V12Ras did not independently enhance basal ERK2 activity (Tables 2 and 3) ▶ ▶ . These results suggest that EGF activated ERK2 through endogenous Ras, whereas V12Ras was not activating the ERK2 pathway directly, but was activating different downstream effectors that secondarily facilitated the EGF-dependent activation of endogenous Ras and ERK2. The effectors of V12Ras may include phosphatidylinositol-3-kinase, Rho family GTPases, and others, which affect cytoskeletal remodeling. 31,32 The latter are supported by the studies showing that cytochalasin D abolished EGF-mediated activation of ERK2 in GECs (Table 4) ▶ . By analogy, cytochalasin D inhibited activation of the cell cycle in fibroblasts, and the authors concluded that growth factors and cytoskeletal integrity jointly contribute to cell cycle activation. 33 Thus, in the plastic-adherent GECs that express moderate levels of V12Ras, EGF was able to stimulate ERK2 activity similarly to parental GECs on collagen, suggesting that ECM and V12Ras may be activating analogous downstream pathways involving the actin cytoskeleton. Organization of the cytoskeleton may enable growth-factor-induced activation of endogenous Ras and ERK2 and cell proliferation. Additional studies will be required to define the relevant targets of ECM and V12Ras more precisely. In contrast, a high level of V12Ras expression (clone 514) led to the loss of anchorage dependence as well as growth-factor-independent proliferation. High levels of V12Ras expression also resulted in collagen- and growth-factor-independent increases in ERK2 activity and tyrosine phosphorylation, ie, basal ERK2 activation (Table 2 ▶ ; Figure 7 ▶ ). This basal activation may have been due to direct action of V12Ras on the ERK2 cascade (which did not occur with lower V12Ras expression), or possibly, high levels of V12Ras may have induced production of growth factors, which then acted in an autocrine fashion to activate endogenous Ras and ERK2 and stimulate proliferation. 19
The Ras-ERK2 pathway is critical for GEC proliferation, but it is important to note that this pathway is generally not sufficient. In earlier studies, we showed that brief incubation with phorbol myristate acetate (PMA, 250 ng/ml) could stimulate ERK2 activity in GECs on plastic as well as collagen (presumably via activation of protein kinase C). PMA, however, was not able to induce proliferation of GECs. 8 These results imply that although the ERK2 pathway is necessary for proliferation, there is also a requirement for growth factors to activate other pathways. Alternatively, growth factors and PMA may be activating ERK2 in distinct subcellular compartments or with different kinetics, thereby differentially regulating downstream specificity. 19
The magnitude of growth-factor-induced increases in ERK2 activity in GECs was relatively small as compared with certain other cell types, although such responses generally tend to be small in epithelial cells, 31 eg, renal inner medullary collecting tubule cells. 34 This may be related to low levels of expression of growth factor receptors in GECs; eg, GECs contain only ∼3 × 10 4 EGF-Rs/cell. 6 Alternatively, GECs may express only small amounts of proteins of the Ras-ERK2 cascade. For example, endogenous levels of Ras appeared to be very low (Figure 4) ▶ , and even very high overexpression of V12Ras (clone R514) did not stimulate ERK2 activity markedly (Table 2) ▶ . GECs might also contain abundant negative regulators of the Ras-ERK2 cascade, such as phosphatases. 35 Nonetheless, the relatively small amount of ERK2 activation in GECs is biologically significant, as functional inhibition of this pathway abolishes proliferation.
Collagen I was used as the ECM in most experiments in the present study. Adhesion of GECs to collagen I in vivo is generally limited to pathological conditions (eg, glomerular inflammation), whereas in the normal glomerulus, GECs are adherent to collagen IV. 21 In cultured GECs, collagens I and IV exert similar effects on proliferation, whereas proliferation is not evident on laminin. 4 In the present study, we confirmed that collagen IV can support GEC proliferation, and we showed that EGF can also activate ERK2 in collagen-IV-adherent GECs. However, it was not possible perform all of the experiments using collagen IV, because it is not practical to produce collagen IV in amounts sufficient for these experiments. One can speculate on how the effects of ECM on growth factor receptor signaling might regulate GEC proliferation in vivo. There appears to be minimal turnover of GECs, and there is a low concentration of epithelial growth factors in normal glomeruli. Proliferation of GECs may occur in pathological states, including experimental membranous nephropathy in rats (Heymann nephritis). 21,24 After initial sublethal injury of GECs by the complement membrane attack complex, GECs express proliferating cell nuclear antigen, in the absence of inflammatory cell infiltrate. 24 At present, the endogenous growth factor(s) responsible for this proliferative response have not been defined. Nevertheless, exogenously administered bFGF can increase the number of mitoses in the injured GECs in experimental membranous nephropathy. 36 Furthermore, rat glomeruli express various forms of FGFs and FGFRs. 37 In other types of glomerulonephritis, glomeruli may become infiltrated with inflammatory cells, eg, macrophages or platelets, which are sources of epithelial growth factors, including transforming growth factor-α, EGF, and bFGF. 38,39 Production of HGF in intrinsic glomerular cells (eg, mesangial cells) might increase. 40 Enhanced procoagulant activity in glomerulonephritis can lead to an increase in thrombin. 41 Finally, accumulation of basement membrane and interstitial collagens is often evident in glomerulopathies. As a result, the concentration of factors that can potentially modulate growth factor receptor activation may increase and may lead to enhanced GEC proliferation. The present study may provide additional insights into the regulation of GEC proliferation in glomerular injury.
Acknowledgments
We thank Dr. M. Park for providing plasmid HO6T1.
Footnotes
Address reprint requests to Dr. Andrey V. Cybulsky, Division of Nephrology, Royal Victoria Hospital, 687 Pine Avenue West, Montreal, Quebec, Canada H3A 1A1. E-mail: cybulsky@pathology.lan.mcgill.ca.
Supported by research grants from the Medical Research Council of Canada and the Kidney Foundation of Canada. A.V. Cybulsky holds a scholarship from the Fonds de la Recherche en Santé du Québec. T. Takano holds a fellowship from the Medical Research Council of Canada.
References
- 1.Kleinman HK, Klebe RJ, Martin GR: Role of collagenous matrices in the adhesion and growth of cells. J Cell Biol 1981, 88:473-485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ingber DE, Folkman J: How does extracellular matrix control capillary morphogenesis? Cell 1989, 58:803-805 [DOI] [PubMed] [Google Scholar]
- 3.Ingber DE: The riddle of morphogenesis: a question of solution chemistry or molecular cell engineering? Cell 1993, 75:1249-1252 [DOI] [PubMed] [Google Scholar]
- 4.Cybulsky AV, Bonventre JV, Quigg RJ, Wolfe LS, Salant DJ: Extracellular matrix regulates proliferation and phospholipid turnover in glomerular epithelial cells. Am J Physiol 1990, 259:F326-F337 [DOI] [PubMed] [Google Scholar]
- 5.Cybulsky AV, Carbonetto S, Cyr MD, McTavish AJ, Huang Q: Extracellular matrix-stimulated phospholipase activation is mediated by β1 integrin. Am J Physiol 1993, 264:C323-C332 [DOI] [PubMed] [Google Scholar]
- 6.Cybulsky AV, McTavish AJ, Cyr MD: Extracellular matrix modulates epidermal growth factor receptor activation in rat glomerular epithelial cells. J Clin Invest 1994, 94:68-78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cybulsky AV, McTavish AJ, Papillon J: Extracellular matrix stimulates production and breakdown of inositol phospholipids. Am J Physiol 1996, 271:F579-F587 [DOI] [PubMed] [Google Scholar]
- 8.Cybulsky AV, McTavish AJ: Extracellular matrix is required for MAP kinase activation and proliferation of rat glomerular epithelial cells. Biochem Biophys Res Commun 1997, 231:160-166 [DOI] [PubMed] [Google Scholar]
- 9.Juliano RL, Haskill S: Signal transduction from the extracellular matrix. J Cell Biol 1993, 120:577-585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clark EA, Brugge JS: Integrins and signal transduction pathways: the road taken. Science 1995, 268:233-239 [DOI] [PubMed] [Google Scholar]
- 11.Ruoslahti E: Proteoglycans in cell regulation. J Biol Chem 1989, 264:13369-13372 [PubMed] [Google Scholar]
- 12.Schlessinger J, Ullrich A: Growth factor signaling by receptor tyrosine kinases. Neuron 1992, 9:383-391 [DOI] [PubMed] [Google Scholar]
- 13.Malarkey K, Belham CM, Paul A, Graham A, McLees A, Scott PH, Plevin R: The regulation of tyrosine kinase signalling pathways by growth factor and G-protein-coupled receptors. Biochem J 1995, 309:361-375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carpenter G: Receptors for epidermal growth factor and other polypeptide mitogens. Annu Rev Biochem 1987, 56:881-914 [DOI] [PubMed] [Google Scholar]
- 15.Massagué J, Pandiella A: Membrane-anchored growth factors. Annu Rev Biochem 1993, 62:515-541 [DOI] [PubMed] [Google Scholar]
- 16.Cantley LG, Cantley LC: Signal transduction by the hepatocyte growth factor receptor, c-met: activation of the phosphatidylinositol 3-kinase. J Am Soc Nephrol 1995, 5:1872-1881 [DOI] [PubMed] [Google Scholar]
- 17.Johnson DE, Williams LT: Structural and functional diversity in the FGF receptor multigene family. Adv Cancer Res 1993, 60:1-41 [DOI] [PubMed] [Google Scholar]
- 18.Seger R, Krebs EG: The MAPK signaling cascade. FASEB J 1995, 9:726-735 [PubMed] [Google Scholar]
- 19.Marshall CJ: Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell 1995, 80:179-185 [DOI] [PubMed] [Google Scholar]
- 20.Salant DJ: The structural biology of glomerular epithelial cells in proteinuric diseases. Curr Opin Nephrol Hypertens 1994, 3:569-574 [DOI] [PubMed] [Google Scholar]
- 21.Adler S: Glomerular epithelial cells. Neilson EG Couser WG eds. Immunologic Renal Diseases. 1997, :pp 655-667 Lippincott-Raven, Philadelphia [Google Scholar]
- 22.Kriz W: Progressive renal failure: inability of podocytes to replicate and the consequences for development of glomerulosclerosis. Nephrol Dial Transplant 1996, 11:1738-1742 [PubMed] [Google Scholar]
- 23.Morita T, Susuki Y, Churg J: Structure and development of the glomerular crescent. Am J Pathol 1973, 72:349-368 [PMC free article] [PubMed] [Google Scholar]
- 24.Floege J, Johnson RJ, Alpers CE, Fatemi-Nainie S, Richardson CA, Gordon K, Couser WG: Visceral glomerular epithelial cells can proliferate in vivo and synthesize platelet-derived growth factor B-chain. Am J Pathol 1993, 142:637-650 [PMC free article] [PubMed] [Google Scholar]
- 25.Goodyer P, Fata J, Goodyer CG: Excretion of epidermal growth factor-like material in acute Henoch-Schonlein purpura nephritis. Pediatr Nephrol 1990, 4:101-104 [DOI] [PubMed] [Google Scholar]
- 26.Spandidos DA, Wilkie NM: Malignant transformation of early passage rodent cells by a single mutated human oncogene. Nature 1984, 310:469-475 [DOI] [PubMed] [Google Scholar]
- 27.Quigg RJ, Cybulsky AV, Jacobs JB, Salant DJ: Anti-Fx1A produces complement-dependent cytotoxicity of glomerular epithelial cells. Kidney Int 1988, 34:43-52 [DOI] [PubMed] [Google Scholar]
- 28.Coers W, Reivinen J, Miettinen A, Huitema S, Vos JT, Salant DJ, Weening JJ: Characterization of a rat glomerular visceral epithelial cell line. Exp Nephrol 1996, 4:184-192 [PubMed] [Google Scholar]
- 29.Vu TK, Hung DT, Wheaton VI, Coughlin SR: Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 1991, 64:1057-1068 [DOI] [PubMed] [Google Scholar]
- 30.Lin TH, Chen Q, Howe A, Juliano RL: Cell anchorage permits efficient signal transduction between Ras and its downstream kinases. J Biol Chem 1997, 272:8849-8852 [PubMed] [Google Scholar]
- 31.Marshall CJ: Ras effectors. Curr Opin Cell Biol 1996, 8:197-204 [DOI] [PubMed] [Google Scholar]
- 32.Joneson T, White MA, Wigler MH, Bar-Sagi D: Stimulation of membrane ruffling and MAP kinase activation by distinct effectors of Ras. Science 1996, 271:810-812 [DOI] [PubMed] [Google Scholar]
- 33.Böhmer RM, Scharf E, Assoian RK: Cytoskeletal integrity is required throughout the mitogen stimulation phase of the cell cycle and mediates the anchorage-dependent expression of cyclin D1. Mol Biol Cell 1996, 7:101-111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heasley LE, Senkfor SI, Winitz S, Strasheim A, Teitelbaum I, Berl T: Hormonal regulation of MAP kinase in cultured rat inner medullary collecting tubule cells. Am J Physiol 1994, 267:F366-F373 [DOI] [PubMed] [Google Scholar]
- 35.Nebreda AR: Inactivation of MAP kinases. Trends Biochem Sci 1994, 19:1-2 [DOI] [PubMed] [Google Scholar]
- 36.Floege J, Kriz W, Schulze M, Susani M, Kerjaschki D, Mooney A, Couser WG, Koch KM: Basic fibroblast growth factor augments podocyte injury and induces glomerulosclerosis in rats with experimental membranous nephropathy. J Clin Invest 1995, 96:2809-2819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ford MD, Cauchi J, Greferath U, Bertram JF: Expression of fibroblast growth factors and their receptors in rat glomeruli. Kidney Int 1997, 51:1729-1738 [DOI] [PubMed] [Google Scholar]
- 38.Madtes DK, Raines EW, Sakariassen KS, Assoian RK, Sporn MB, Bell GI, Ross R: Induction of transforming growth factor-α in activated human alveolar macrophages. Cell 1988, 53:285-293 [DOI] [PubMed] [Google Scholar]
- 39.Rifkin DB, Moscatelli D: Recent developments in the cell biology of basic fibroblast growth factor. J Cell Biol 1989, 109:1-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harris RC, Burns KD, Alattar M, Homma T, Nakamura T: Hepatocyte growth factor stimulates phosphoinositide hydrolysis and mitogenesis in cultured renal epithelial cells. Life Sci 1993, 52:1091-1100 [DOI] [PubMed] [Google Scholar]
- 41.Brentjens JR: Glomerular procoagulant activity and glomerulonephritis. Lab Invest 1987, 57:107-111 [PubMed] [Google Scholar]