

Abstract
A multiplex system of Western blotting is presented in which most of the current muscular dystrophy proteins can be analyzed simultaneously on one pair of blots. This represents a significant improvement in efficiency and cost for this type of analysis. The final diagnosis is more quickly achieved in patients where several possible diagnoses are indicated after clinical appraisal, and those with unusual presentations may be quickly resolved. The method uses a biphasic polyacrylamide gel system, which enables the corresponding blot to be probed simultaneously with a cocktail of monoclonal antibodies. The gel is optimized so that large proteins of more than 200 kd (eg, dystrophin, dysferlin, and myosin heavy chain) can be analyzed in the top part, while smaller proteins under 150 kd (eg, calpain 3, the 80-kd fragment of laminin α2 chain, all of the sarcoglycans, and caveolin 3) are separated in the lower phase. This basic system could be used for different combinations of antibodies as new muscular dystrophy proteins are identified and require examination. In addition, analysis of the laminin α2 chain of merosin showed that this protein was expressed as a doublet or triplet set of bands in many patients with active muscle pathology. This may indicate the existence of an embryonic isoform, which is re-expressed in regenerating fibers.
Great progress has been made in the last 10 years toward the differential diagnosis of the recessive muscular dystrophies. Previously, a diagnosis based solely on clinical criteria was feasible for severe X-linked Duchenne dystrophy (DMD), which had a very characteristic profile of muscle involvement and clinical progression. However, differentiation of the other recessive dystrophies was more problematical because the clinical symptoms were more heterogeneous, and the age of onset and rate of progression were more variable. If muscle weakness was present from a very early stage, the possible diagnoses might include a form of congenital muscular dystrophy (CMD) or even a severe childhood autosomal recessive muscular dystrophy (SCARMD). For later onset, the choices included Becker muscular dystrophy (BMD) or, if nothing else seemed appropriate, patients might be given a tentative diagnosis of limb-girdle muscular dystrophy (LGMD), which encompassed a group of diseases. Even the diagnosis of dystrophy would need to be separated from other potentially confusing neuromuscular disorders such as forms of spinal muscular atrophy and metabolic myopathies. The advent of dystrophin analysis revealed that misdiagnoses had occurred, and the lack of clear clinical definitions, particularly among the limb-girdle dystrophies, 1 led to a gene/protein-based system of nomenclature (Table 1) ▶ .
Table 1.
Muscular Dystrophy Genes and Proteins
| Muscular Dystrophy (MD) | Abbreviation or alternative names | Gene Location | Recessive (r) or dominant (d) | Protein | Other names for protein | Approximate molecular mass (kd) |
|---|---|---|---|---|---|---|
| Duchenne/Becker MD | DMD/BMD, Dystrophinopathy | Xp21 | r | Dystrophin | 400 | |
| Emery-Dreifuss MD | EDMD, EMD | Xq28 | r | Emerin | 34 | |
| Limb-Girdle MD type 1A | LGMD1A | 5q | d | ? | ||
| Limb-Girdle MD type 1B | LGMD1B | 1q11-21 | d | ? | ||
| Limb-Girdle MD type 1C | LGMD1C | 3p25 | d | Caveolin 3 | 18 | |
| Limb-Girdle MD type 2A | LGMD2A | 15q15 | r | Calpain 3 | p94 | 94 |
| Limb-Girdle MD type 2B/Miyoshi Myopathy | LGMD2B/MM | 2p13 | r | Dysferlin | 230 | |
| αSarcoglycanopathy | LGMD2D, SCARMD2 | 17q21 | r | α-Sarcoglycan | Adhalin, 50DAG, A2 | 50 |
| βSarcoglycanopathy | LGMD2E | 4q12 | r | β-Sarcoglycan | A3b | 43 |
| γSarcoglycanopathy | LGMD2C, SCARMD1 | 13q12 | r | γ-Sarcoglycan | 35DAG | 35 |
| δSarcoglycanopathy | LGMD2F | 5q33 | r | δ-Sarcoglycan | 35 | |
| Merosin-negative congenital MD | Merosinopathy | 6q22 | r | Laminin α2 chain | Merosin, laminin-2 | 400 (80/300) |
| Fukuyama congenital MD | FCMD | 9q31 | r | Fukutin | 53 | |
| Integrin-negative congenital myopathy (or MD) | 12q13 | r | Integrin α7 | 130 | ||
| Myotonic dystrophy | DM | 19q13 | d | Myotonin protein kinase | DMPK | 55/64/71/80 |
| Facioscapulohumeral MD | FSHD, FSH | 4q35 | d | ? | ||
| Oculopharyngeal MD | OPMD | 14q11 | d | Poly(A) binding protein 2 | 35 | |
| Epidermolysis bullosa simplex with MD | EB-MD, EBS-MD | 8q24 | r | Plectin | 500 |
Note that the laminin α2 chain of merosin fragments into two polypeptides of 80 and 300 kd and that the size of myotonin protein kinase varies in different reports.
Unlike the dystrophin gene where the vast majority of mutations are large multiexonic deletions, most of the mutations reported in the genes for the sarcoglycans, calpain 3, and α2 laminin are more subtle and difficult to detect. It would therefore be helpful if protein analysis could be used to indicate where to start the search for gene mutations. 2 Analysis of muscular dystrophy proteins obviously requires a muscle biopsy to be taken, but these are commonly performed as part of the routine diagnostic procedures undertaken for new patients presenting with muscle problems.
At Newcastle we now undertake immunocytochemical and immunoblotting analysis of biopsies from all new muscular dystrophy patients using a panel of monoclonal antibodies. Our experiences with BMD demonstrated just how broad the clinical phenotype could be, and as the other recessive dystrophies are still being defined at the clinical level, we feel that it is appropriate to test all biopsies with all of the antibodies that are currently available. This sort of approach allows for unexpected diagnoses in patients with atypical clinical presentation 3,4 and can sometimes lead to the definition of clinical subgroups with characteristic abnormalities of protein expression, but where the primary gene defect is still unknown. 5,6
Labeling sections with a panel of antibodies is straightforward (so long as the antibodies recognize native protein in sections), but it would not be cost-effective to run a blot for every antibody. We have therefore evolved a biphasic gel system, which enables the corresponding blot to be probed with several monoclonal antibodies simultaneously. The gel is optimized so that large proteins of more than 200 kd (eg, dystrophin, dysferlin, and myosin heavy chain) can be analyzed in the top part, while smaller proteins under 150 kd (eg, calpain 3, the 80-kd fragment of laminin α2 chain/merosin, all of the sarcoglycans, and caveolin 3) are separated in the lower phase. This basic system could be used for different combinations of antibodies as new muscular dystrophy proteins are identified and require analysis.
Materials and Methods
Patients
Muscle biopsies (usually from vastus lateralis) were taken from patients as part of the routine diagnostic procedure. Muscle samples from normal control subjects were also obtained, with consent, from legs amputated at the knee (gastrocnemius, soleus, etc) or abdominal operations (rectus abdominis). Diagnosis at the molecular level was primarily undertaken in the Department of Human Genetics (now the Human Molecular Genetics Unit in the School of Biochemistry and Genetics) of the University of Newcastle in Newcastle on Tyne.
Antibodies
The following mouse monoclonal antibodies were used on blots: Dy8/6C5 (dystrophin carboxy terminus), Dy4/6D3 (dystrophin rod domain), Ad1/20A6 (α-sarcoglycan), βSarc1/5B1 (β-sarcoglycan), 35DAG/21B5 (γ-sarcoglycan), γSarc3/12C1 (δ-sarcoglycan), 43DAG/8D5 (α-dystroglycan), Calp3d/2C4 (calpain 3 exon 1), and Calp3c/12A2 (calpain 3 exon 8). 7 These antibodies are also available commercially from Novocastra Laboratories, Newcastle on Tyne, UK. An antibody to the laminin α2 chain of merosin was purchased from Chemicon International, Harrow, UK (MAb 1922, which recognizes the 80-kd fragment), and the Transduction Laboratories antibody C38320 to caveolin 3 was purchased from Affiniti Research Products, Mamhead, UK. A monoclonal antibody to dysferlin (NCL-hamlet) is currently being characterized in our laboratory, and this was also tested in this multiplex system.
Polyacrylamide Gel Electrophoresis and Western Blotting
BioRad Protean II equipment was used to cast two 16-cm gels 1.5 mm thick. The resolving gel was poured in two phases: the first (bottom) phase was 7 cm of a plain 7% polyacrylamide gel (30% acrylamide plus 0.8% bisacrylamide cross-linker) in 0.375 mol/L Tris/HCl buffer, pH 8.8, containing 0.2% sodium dodecyl sulfate (SDS). As soon as the bottom part of the gel was poured, the rest of the gel was pumped on above it so that the two phases set together. The upper gradient phase was 4.5 cm of a 5.5% to 4% polyacrylamide gradient in the 0.375 mol/L Tris/HCl buffer containing 0.2% SDS. There was a gradient of glycerol throughout to help stabilize the phases: the 7% gel contained 10% glycerol, the 5.5% gel contained 7.5% glycerol, and the 4% gel contained 3% glycerol. After the resolving gel was set, the 2-cm-deep stacking gel consisting of 3% acrylamide in 0.125 mol/L Tris/HCl, pH 6.8, containing 3% glycerol and 0.2% SDS, was poured above the resolving gel and allowed to polymerize around the comb that was inserted to form the sample lanes (10 × 1 cm). 8
The tissue samples were prepared while the gels were setting. Frozen muscle samples (∼20 to 30 mg) were quickly weighed and homogenized twice for 15 seconds each in an Ultra Turrax homogenizer with 19 vol of treatment buffer containing 0.125 mol/L Tris/HCl buffer, pH 6.4, 10% glycerol, 4% SDS, 4 mol/L urea, 10% mercaptoethanol, and 0.001% bromophenol blue (final pH of treatment buffer was 6.8). Additional protease inhibitors are not required as all of the proteins present in the sample are denatured by the SDS and urea. Autolysis does not appear to be a problem, as judged by the satisfactory results obtained with calpain 3 analysis in this system. 7 The samples were placed in boiling water for 3 minutes and centrifuged at 9500 × g for 3 minutes before 30-μl aliquots of the supernatants (equivalent to ∼200 μg of non-collagen protein in control samples 9 ) were applied to each lane. The gels were run at 21 mA overnight in a tank buffer containing 1.44% glycine, 0.3% Tris, and 0.2% SDS, using a thermocirculator set at 10°C.
The next day the gels were blotted with a limiting current of 1 A for 7 hours on to Schleicher and Schuell (Keene, NH) 0.45-μm nitrocellulose using a transfer buffer that contained 0.6% Tris, 2.88% glycine, 0.01% SDS, and 20% methanol. 10 The BioRad (Richmond, CA) blotting tank contained plate electrodes and a supercooling coil. The equipment was attached to a thermocirculator set at −13.5°C. After blotting, the gels were fixed in 20% trichloroacetic acid, stained in 0.115% Coomassie brilliant blue R250 in 25% ethanol/10% acetic acid, and destained in 10% ethanol/10% acetic acid. The density of the myosin heavy chain band on the dried, post-blotted gel was used to indicate how much muscle protein (as opposed to fat and fibrous connective tissue) had been loaded in each sample lane. 11
Multiplex Immunolabeling of Blots
Unreacted binding sites on the blots were blocked by incubation in 5% low-fat dried milk powder (supermarket) in a pH 8 buffer containing 10 mmol/L Tris/HCl, 0.15 mol/L NaCl, and 0.05% Tween 20 (TBST) on a rocking table for 1 hour at room temperature. This milk solution was retained. The primary mouse monoclonal antibodies were added together in a cocktail. The antibodies may be used in any combination, so long as the size of all of the potentially immunoreactive bands is taken into consideration. Some proteins, such as dystrophin or calpain 3, produce more than one band, and the laminin α2 chain of merosin is detected as 80-kd or 300-kd fragments according to the antibody used. It is also obviously not possible to analyze proteins of the same size on the same blot. The dilutions of antibodies used are shown in Table 2 ▶ , but these suggestions are for guidance only; higher dilutions may well give good results, depending on the sensitivity of the visualization system used. It is possible to retain, freeze, and reuse the antibody cocktail, but this is not a recommended procedure, as some antibody activity is lost at each freeze/thaw cycle.
Table 2.
Examples of Monoclonal Antibody Combinations That Could Be Used On Pairs of Multiplex Blots
| Protein | Antibody name | Commercial designation | Dilution used | Volume used in 10 ml* |
|---|---|---|---|---|
| Multiplex A | ||||
| Dystrophin C-term | Dy8/6C5 | NCL-DYS2 | 1 /50 | 200 μl |
| Calpain 3 exon 1 | Calp3d/2C4 | NCL-CALP-2C4 | 1 /10 | 1 ml |
| α-Sarcoglycan | Ad1/20A6 | NCL-a-SARC | 1 /10 | 1 ml |
| β-Dystroglycan | 43DAG/8D5 | NCL-b-DG | 1 /200 | 50 μl |
| γ-Sarcoglycan | 35DAG/21B5 | NCL-g-SARC | 1 /2 | 5 ml |
| Caveolin 3 | C38320 | 1 /1000 | 10 μl | |
| Multiplex B | ||||
| Dystrophin rod | Dy4/6D3 | NCL-DYS1 | 1 /50 | 200 μl |
| Calpain 3 exon 8 | Calp3c/12A2 | NCL-CALP-12A2 | 1 /10 | 1 ml |
| Laminin α2 | MAB 1922 | 1 /1000 | 10 μl | |
| β-Sarcoglycan | βSarc1/5B1 | NCL-b-SARC | 1 /2 | 5 ml |
| δ-Sarcoglycan | δSarc3/12C1 | NCL-d-SARC | 1 /3 | 3.3 ml |
*Ten milliliters is the volume required to just cover one blot after it is trimmed. The volume of antibody solution is made up to 10 ml with the TBST washing buffer, but this spare volume could be used to incorporate additional antibodies; eg, 1/300 NCL-hamlet (33 μl) could be used to label dysferlin in multiplex A.
After incubation for 1 hour at room temperature, the blots were washed in TBST for 15 minutes, in the retained milk solution for 15 minutes, and in TBST for 15 minutes. After the final wash, peroxidase-conjugated anti-mouse secondary antibody (Dako P260) was diluted 1/300 in TBST and added to the blot for 1 hour. After three 15-minute washes with TBST, the immunoreactive bands on the blot were visualized by exposure to freshly prepared 0.05% diaminobenzidine in phosphate buffer, pH 7.2, to which 0.1% hydrogen peroxide had been added just before use. When dry, the blots were stuck onto thick filter paper and stored in envelopes in the dark. Blots stored under such conditions have shown no deterioration in band/background labeling intensity in the 10 years since we started diagnostic blotting work. The blots can be scanned and the images stored on optical disks for future densitometric analysis 7 or for illustration.
Results
Figure 1 ▶ illustrates strips from a blot of human skeletal muscle labeled with the multiplex antibody combinations shown in Table 2 ▶ . This demonstrates what can be achieved, but in practice various simpler combinations of antibodies may be more useful. Figure 2 ▶ shows a blot labeled for the carboxy terminus of dystrophin, the laminin α2 chain of merosin, α-sarcoglycan, and α-dystroglycan. Samples from normal control subjects, shown in the lanes labeled C, demonstrate the clear separation of the bands representing the proteins of diagnostic importance. Lane 1 shows a patient with partial merosin deficiency, and lane 2 shows a sample from a CMD patient where the merosin labeling is absent. Lane 3 shows no labeling for α-sarcoglycan, and this patient was later confirmed as having a mutation in this gene. In contrast, the patient in lane 4 shows a partial reduction in α-sarcoglycan labeling and was found to have a mutation in the γ-sarcoglycan gene. The patient in lane 5 has Duchenne muscular dystrophy, and although labeling for dystrophin is absent, labeling for all of the other proteins is reduced too. The myosin heavy chain labeling in the bottom panel confirms that this reduction is not simply due to insufficient sample loading.
Figure 1.

Strips from a Western blot of human skeletal muscle labeled with antibodies to the proteins indicated. The antibody combinations and dilutions are detailed in Table 2 ▶ . The left-hand strip is labeled with multiplex A, and the right-hand strip is labeled with multiplex B. Many dystrophin metabolites are labeled, 9 as are various calpain 3 bands. 7 The unidentified band at 120 kd is an additional singlet/doublet labeled by antibody 35DAG/21B5 to γ-sarcoglycan. 14
Figure 2.

Blot labeled with antibodies to the carboxy terminus of dystrophin (400 kd), the laminin α2 chain of merosin (80 kd), α-sarcoglycan (50 kd), and β-dystroglycan (43 kd). Lane C, normal control muscle samples; lanes 1 and 2, samples from patients with congenital muscular dystrophy; lanes 3 and 4, samples from patients with sarcoglycanopathies; lane 5, sample from a patient with Duchenne muscular dystrophy; lane 6, sample from a patient with polymyositis. The bottom panel shows the corresponding myosin heavy chain bands on the post-blotted gel, stained with Coomassie blue.
The sample in lane 6 of Figure 2 ▶ is from a patient with polymyositis and demonstrates a doublet of bands for the laminin α2 chain of merosin. As we have used the multiplex approach to blotting, we have seen many examples of multiple laminin α2 chain bands in patients with muscle diseases where there is a lot of fiber degeneration and regeneration going on. Figure 3 ▶ illustrates some of the variation observed, from overexpression in one LGMD patient (band 7) to very weak bands in a DMD patient (band 13). The most intensely labeled components of the doublet and triplet bands are often lower than the band in the normal control samples (band 3, for example). These bands are not seen in normal control muscle but do occur in fetal tissue (results not shown). Thus, this may be evidence for an embryonic isoform of laminin α2 chain, with re-expression in regenerating fibers.
Figure 3.

Close-up of the 80-kd laminin α2 chain/merosin band (Chemicon MAb 1922) in samples on three blots. Bands labeled 1, 5, 6, 9, 11, and 14 are from normal control muscle samples. Samples 2, 3, and 4 are from patients with inflammatory muscle disease (polymyositis and dermatomyositis); 7, 8, 10, 12, and 14 are from patients with forms of limb-girdle muscular dystrophy; 13 is from a patient with Duchenne muscular dystrophy, and 15 is from a patient with faciosculohumeral muscular dystrophy.
The differential diagnosis of adult patients with undefined ?LGMD is difficult, and Figure 4 ▶ illustrates the results obtained with a group of six patients who presented with muscle weakness in a limb-girdle distribution and high creatine kinase levels. For this analysis, one multiplex contained antibodies to the carboxy terminus of dystrophin, exon 1 of calpain 3, α-sarcoglycan, and β-dystroglycan (Figure 4A) ▶ , whereas the second multiplex contained antibodies to the rod domain of dystrophin, exon 8 of calpain 3, the α2 chain of laminin (merosin), β-dystroglycan, and γ-sarcoglycan (Figure 4B) ▶ . Labeling for β-dystroglycan is useful as a control for the diagnostic proteins and was therefore included in both. The patients in lanes 1 and 6 remained unresolved, with band patterns that were indistinguishable from those in normal control subjects. Normal labeling was also observed on sections labeled with antibodies to all of the proteins currently known to show primary or secondary abnormalities in muscular dystrophy patients. The patient in lane 2 had no detectable laminin α2 chain on blots (see panel A at 80 kd) and had a pattern of muscle involvement that matched the small group of patients previously described with this secondary abnormality of protein expression. 6 The patient in lane 3 had dystrophin bands of abnormal size (see panels A and B at 400 kd), which are characteristic of patients with Becker MD. The patient in lane 4 was unresolved by this combination of antibodies, but the addition of the dysferlin antibody to the multiplex in panel A revealed that this patient had LGMD2B (manuscript in preparation). Finally, the patient in lane 5 was found to have abnormal expression of calpain 3 (see panel A at 94 kd and 30 kd and panel B at 94 kd and 60 kd), and the diagnosis of LGMD2A was later confirmed by the finding of a mutation in the CAPN3 gene.
Figure 4.
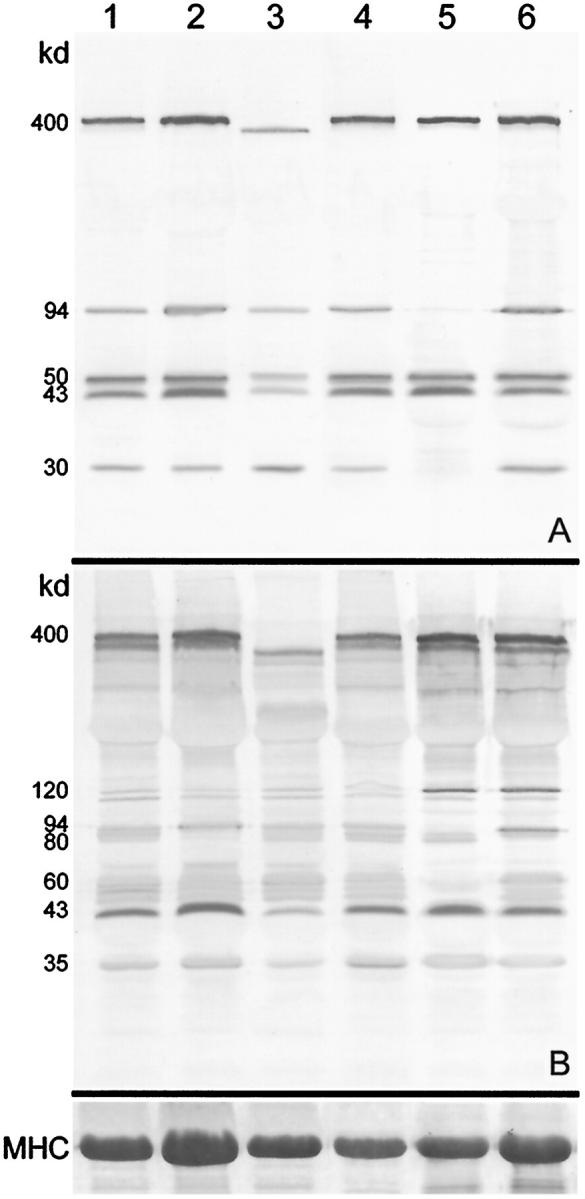
Immunoanalysis of muscle homogenates from six patients presenting with possible limb-girdle muscular dystrophy. A: Blot labeled with antibodies to the carboxy terminus of dystrophin (400 kd), exon 1 of calpain 3 (94 and 30 kd), α-sarcoglycan (50 kd), and β-dystroglycan (43 kd). B: Blot labeled with antibodies to the rod domain of dystrophin (400 kd), exon 8 of calpain 3 (94 and ∼60 kd), the laminin α2 chain of merosin (80 kd), β-dystroglycan (43 kd), and γ-sarcoglycan (35 kd). The 120-kd band (present as a doublet in these patients) is labeled by the antibody to γ-sarcoglycan. The corresponding myosin heavy chain bands on the post-blotted gel, stained with Coomassie blue, are shown at the bottom.
The occurrence of additional bands is always eye-catching, but there is not always an underlying genetic cause. Figure 5 ▶ illustrates an additional band that we have observed in several cases, all but one being in a sarcoglycanopathy. An odd band at ∼30 kd was detected in the multiplex system, but subsequent analysis with the individual antibodies alone showed that this band was labeled by antibody 43DAG/8D5 to β-dystroglycan. Careful examination of our records showed that, in every case, the sample had been sent from outside Newcastle and that there had been some delay in transit. It appears that this band is therefore a degradation product of β-dystroglycan, and it seems likely that instability of the dystrophin-glycoprotein complex due to sarcoglycan deficiency may predispose β-dystroglycan to break down in this manner. A mutation in the γ-sarcoglycan gene has been confirmed in the patient shown in Figure 5 ▶ .
Figure 5.

Lanes from a Western blot of skeletal muscle showing muscle from two normal control subjects (C) and a patient (P) with γ-sarcoglycanopathy (limb-girdle muscular dystrophy type 2C). In the patient the labeling for dystrophin is normal, the laminin α2 chain of merosin is represented by a triplet with the middle band being most intensely labeled, α-sarcoglycan labeling is reduced, and β-dystroglycan is partially broken down to a 30-kd fragment.
Discussion
A multiplex system of Western blotting is presented in which many of the current muscular dystrophy proteins could be analyzed simultaneously on one pair of blots. This represents a significant improvement in efficiency and cost for this type of analysis. Final diagnosis is more quickly achieved in patients where several possible diagnoses are indicated after clinical appraisal (eg, ?LGMD), and those with unusual presentations may be quickly resolved. In addition, the approach of labeling all biopsies with all antibodies has enabled us to identify a clinical subgroup of LGMD patients who are characterized by having abnormal expression of the laminin α2 chain of merosin on blots. 6
We found that our modified methods of electrophoresis and blotting were better for analyzing the proteins in skeletal muscle samples than the classical methods of Laemmli 12 and Towbin. 13 Running the resolving gel slowly overnight and increasing the SDS in the polyacrylamide gel matrix to 0.2% improved the resolution of the high molecular mass proteins. Incorporating glycerol in to the stacking gel also seemed to improve the resolution of the high molecular mass bands and reduced the flare from the myosin heavy chain band (which is effectively grossly overloaded in proportion to the other muscle proteins present). The addition of 4 mol/L urea to a treatment buffer already containing 4% SDS aided the dissociation and solubilization of large protein complexes in skeletal muscle, particularly those of myosin and laminin. The addition of 0.01% SDS to the blotting buffer encouraged large proteins out from the gel matrix (but did not appear to inhibit binding to the nitrocellulose, as judged by the detection of dystrophin in DMD patients 11 ), whereas the 20% methanol helped to fix the proteins to the nitrocellulose sheets.
During the development of this method we have identified two common sources of disappointing results. First, if there is a change in sample composition between adjacent lanes, one lane may appear skinny and distorted. Thus, if a sample has a low myosin content so that all of the protein bands are faint, it is not feasible to simply double the volume of sample applied. This results in an imbalance in the SDS and urea content between adjacent lanes. To analyze a sample like this, the best approach is to homogenize an additional sample with a 1:10 rather than 1:20 volume of treatment buffer. Alternatively, the sample volume may be doubled so long as an extra volume of treatment buffer is added in all of the other sample wells. Too large a volume will result in poor band resolution, however. The other common problem arises from using muscle samples that have been set in mountant for sectioning. Contamination with mountant results in band smearing, and this should be avoided by slicing as much mountant as possible off while the tissue is frozen. As a last resort the muscle sample may have to be thawed and the mountant quickly wiped off on a paper tissue before homogenization.
Footnotes
Address reprint requests to Dr. Louise V.B. Anderson, Neurobiology Department, University Medical School, Framlington Place, Newcastle upon Tyne NE2 4HH, UK. E-mail: L.V.B.Anderson@ncl.ac.uk.
Supported by grants from the Muscular Dystrophy Group of Great Britain and the Association Française contre les Myopathies.
References
- 1.Bushby KMD, Beckmann JS: The limb-girdle muscular dystrophies - proposal for a new nomenclature (30th and 31st ENMC Workshops Report). Neuromuscul Disord 1995, 5:337-343 [DOI] [PubMed] [Google Scholar]
- 2.Anderson LVB: Optimised protein diagnosis in the autosomal recessive limb-girdle muscular dystrophies. Neuromuscul Disord 1996, 6:443-446 [DOI] [PubMed] [Google Scholar]
- 3.Tan E, Topaloglu H, Sewry C, Zorlu Y, Naom I, Erdem S, D’Alessandro M, Muntoni F, Dubowitz V: Late onset muscular dystrophy with cerebral white matter changes due to partial merosin deficiency. Neuromuscul Disord 1997, 7:85-89 [DOI] [PubMed] [Google Scholar]
- 4.Muntoni F, Lichtarowicz-Krynska EJ, Sewry CA, Manilal S, Recan D, Llense S, Taylor J, Morris GE, Dubowitz V: Early presentation of X-linked Emery-Dreifuss muscular dystrophy resembling limb-girdle muscular dystrophy. Neuromuscul Disord 1998, 8:72-76 [DOI] [PubMed] [Google Scholar]
- 5.Taylor J, Muntoni F, Robb S, Dubowitz V, Sewry C: Early onset autosomal dominant myopathy with rigidity of the spine: a possible role for laminin β1? Neuromuscul Disord 1997, 7:211-216 [DOI] [PubMed] [Google Scholar]
- 6.Bushby KMD, Anderson LVB, Pollitt C, Naom I, Muntoni F, Bindoff L: Abnormal merosin in adults - a new form of late onset muscular dystrophy not linked to chromosome 6q2. Brain 1998, 121:581-588 [DOI] [PubMed] [Google Scholar]
- 7.Anderson LVB, Davison K, Moss JA, Richard I, Fardeau M, Tomé FMS, Hübner C, Lasa A, Colomer J, Beckmann JS: Characterization of monoclonal antibodies to calpain 3 and protein expression in muscle from patients with limb-girdle muscular dystrophy type 2A. Am J Pathol 1998, 153:1169-1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anderson LVB: Multiplex western blot analysis of the muscular dystrophy proteins. Muscular Dystrophy: Methods and Protocols. Edited by KMD Bushby, LVB Anderson. From The Methods in Molecular Medicine Series edited by JM Walker. Totowa, NJ, Humana Press, 1999. In press
- 9.Nicholson LVB, Davison K, Falkous G, Harwood C, O’Donnell E, Slater CR, Harris JB: Dystrophin in skeletal muscle: I. Western blot analysis using a monoclonal antibody. J Neurol Sci 1989, 94:125-136 [DOI] [PubMed] [Google Scholar]
- 10.Otter T, King SM, Witman GB: A two-step procedure for efficient electrotransfer of both high-molecular-weight (>400,00), and low-molecular-weight (<20,000) proteins. Anal Biochem 1987, 162:370-377 [DOI] [PubMed] [Google Scholar]
- 11.Nicholson LVB, Johnson MA, Gardner-Medwin D, Bhattacharya S, Harris JB: Heterogeneity of dystrophin expression in patients with Duchenne and Becker muscular dystrophy. Acta Neuropathol 1990, 80:239-250 [DOI] [PubMed] [Google Scholar]
- 12.Laemmli UK: Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227:680-685 [DOI] [PubMed] [Google Scholar]
- 13.Towbin H, Staehelin T, Gordon J: Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA 1979, 76:4350-4354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sewry C, Taylor J, Anderson LVB, Ozawa E, Pogue R, Piccolo F, Bushby KMD, Dubowitz V, Muntoni F: Abnormalities in α-, β-, and γ-sarcoglycan in patients with limb-girdle muscular dystrophy. Neuromuscul Disord 1996, 6:467-474 [DOI] [PubMed] [Google Scholar]