

Abstract
Human islet amyloid polypeptide (hIAPP) is co-secreted with insulin from pancreatic islet β cells. This peptide spontaneously aggregates in the form of fibrils, and amyloid deposits are associated with dead or degenerating β cells, a hallmark of noninsulin-dependent diabetes mellitus. We demonstrated that COS-1 cells transfected with vectors expressing hIAPP exhibited intracellular amyloid deposits that were associated with cell death (O’Brien, Butler, Kreutter, Kane, Eberhardt, Am J Pathol 1995, 147:609–616). To establish the mechanism of cell death, we transfected COS-1 cells with vectors expressing amyloidogenic hIAPP or nonamyloidogenic rat IAPP and mutant hIAPP constructs and assayed them for markers characteristic of apoptosis and necrosis by fluorescence-activated cell sorting analysis. Amyloidogenic hIAPP-transfected COS cells contained up to threefold more apoptotic cells present at 96 hours after transfection compared with the nonamyloidogenic vector controls. The hIAPP-induced apoptosis was negligible at 24 and 48 hours after transfection and was maximal at 96 hours which parallels the time course of amyloidogenesis. Immunohistochemical staining and confocal microscopy showed that hIAPP is localized with distinct clustering in the endoplasmic reticulum and Golgi apparatus with no discernable extracellular staining. These experiments provide direct evidence that intracellular hIAPP amyloid causes cell death by triggering apoptotic pathways.
Islet amyloid polypeptide (IAPP, also designated amylin) is a 37-amino acid peptide that is produced in the β cells of the pancreas. 1-3 It is co-secreted with insulin, and its biological function is not known with certainty, although it has been implicated in regulating insulin and glucose metabolism. 1-3 Human (h) IAPP is highly amyloidogenic, and amyloid deposits are found in pancreata of >90% of patients with noninsulin-dependent diabetes mellitus (NIDDM). 3 The peptide spontaneously aggregates to form insoluble IAPP fibrils in vitro. 4 Intracellular deposition of IAPP has been shown to occur in the pancreata of diabetic macaques and in transgenic mice expressing hIAPP, which have been rendered insulin resistant via the administration of growth hormone and glucocorticoids. 5,6 Correlated with the presence of islet amyloid, there is a corresponding loss of up to 50% of β-cell mass in the pancreata of individuals with NIDDM. 3,7,8 Whereas a causative role of islet amyloid in the genesis of NIDDM has been considered highly unlikely, 9 the recent finding that families of individuals with premature onset NIDDM have a mutation within the IAPP gene (S20G), provides direct evidence for a causal link between the IAPP gene and the genesis of NIDDM. 10
Exogenously added hIAPP is cytotoxic and induces apoptosis when added to a variety of cultured cells, including primary rat pancreatic cells, hippocampal neurons, aortic endothelial cells, COS cells, and PC12 pheochromocytoma cells but neither fibroblasts nor H4 hepatoma cells. 11 We previously showed that intracellular amyloid generated from hIAPP was associated with cell death resembling apoptosis in COS-1 cells that have been transfected with an hIAPP expression plasmid. 12 The mechanism by which amyloid deposition leads to the loss of pancreatic β cells is not known. One hypothesis suggests that the IAPP monomers are secreted from the islet β cells and, at high local concentrations, will aggregate into insoluble fibrils in the extracellular spaces and islet capillaries. These amyloid deposits subsequently come into direct contact with the islet β cells, resulting in their death by an unknown mechanism. 11,13 However, we showed that the level of intracellular IAPP required to kill COS-1 cells was several orders of magnitude less than that required in the cell culture studies, 12 indicating that intracellular amyloid formation may be more detrimental.
To eventually understand the mechanism by which intracellular hIAPP causes cell death, we first sought to determine whether transfected COS-1 cells were dying by apoptosis or necrosis. In the current study we demonstrate that the intracellular accumulation of amyloido- genic hIAPP, but not nonamyloidogenic mutant forms of hIAPP or rat (r) IAPP, induces apoptosis in transfected COS-1 cells in a time-dependent manner. Our data suggest that hIAPP amyloid activates specific intracellular signaling pathways that result in apoptosis.
Materials and Methods
Plasmids
The cDNAs for hIAPP, its antisense construct (hIAPPanti), a mutant form of hIAPP (hIAPPmut), and rIAPP were cloned into the pMT2 expression plasmid 14 as described previously. 12 The pMT2 vector contains the adenovirus major late promoter with an SV40 enhancer. The IAPP cDNAs are cloned into an EcoRI site that attaches the adenovirus tripartite leader sequence allowing for efficient translation and positions the cDNA upstream of the SV40 early polyadenylation signal. The hIAPPmut was constructed via inverted polymerase chain reaction mutagenesis 15 using the oligonucleotides 5′-CCCGTTCTCCCACCTACCAACGTGGGATCC-3′ and 5′-ACCAAAGTTGTTGCTGG-3′ as primers with the pUC18-hIAPP plasmid 12 as template. This results in a cDNA, verified by direct didexoy sequencing, which converts the sequence GlyAlaIleLeuSerSer between amino acids 24–29 within the amyloidogenic domain to the sequence GlyProValLeuProPro, which corresponds to the rat sequence. The two additional differences between the human and rat IAPPs (His18 versus Arg18 and Phe23 versus Leu23) were preserved in hIAPPmut (see Figure 8 ▶ ). The pMT2 vector without insert served as an additional control plasmid.
Figure 8.

Amino acid sequences of hIAPP, hIAPPmut, and rIAPP. The amyloidogenic domain of hIAPP, GAILSS 4 , is indicated in bold.
Cell Culture and Transfections
COS-1 cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco-BRL, Gaithersburg, MD) supplemented with 10% FetalClone II (Hyclone, Logan, UT), 100 U/ml penicillin (Gibco-BRL), 100 U/ml streptomycin (Gibco-BRL), and 2 mmol/L l-glutamine (Gibco-BRL). Cells were maintained at 37°C in a humidified atmosphere containing 5% CO2 and passaged weekly. Subconfluent cells were harvested by trypsinization and 4 to 5 × 10 6 cells resuspended in 200 μl cold phosphate-buffered saline (PBS), 20 mmol/L HEPES, with 10 to 15 μg of the plasmid DNA. The cells were incubated on ice for 15 minutes, then electroporated at 900 μfarad and 250 volts in a BIO-RAD Gene Pulser (Richmond, CA) in standard cuvettes with a 4-mm electrode gap. The electroporated cells were cultured in 10 ml of medium in 10-cm tissue culture dishes. At 20 hours after transfection, the medium was replaced to remove nonadherent cells that had been killed or injured during electroporation. Positive controls for apoptosis were obtained by treating with 0.5 μg/ml tunicamycin, an inhibitor of protein glycosylation in the endoplasmic reticulum (ER), for 3 days.
Analysis for Plasma Membrane Alterations in Apoptosis
Cells were harvested by trypsinization at 24, 48, 72, or 96 hours and pooled with their culture medium so that cells that had lost their adherent properties during apoptosis (“floaters”) were included in the analysis. Cells were pelleted, washed, and resuspended in 400 μl of binding buffer (BB: 100 mmol/L HEPES (pH 7.4), 1.5 mol/L NaCl, 50 mmol/L KCl, 10 mmol/L MgCl2, 18 mmol/L CaCl2) and 100 μl of cells (1 × 106) aliquoted to 4-ml Falcon tubes for labeling and fluorescence-activated cell sorting (FACS) analysis. Cells were incubated with 1 μg of sample annexin-V-biotin conjugate (Trevigen, Inc, Gaithersburg, MD) at 4°C in the dark for 20 to 30 minutes, washed, and fluorescently labeled with streptavidin-phycoerythrin (PE) (Molecular Probes, Eugene, OR) at 1 μg of sample under the same conditions. Labeled cells were washed and resuspended in 400 μl of BB containing 6 μg/ml 7-amino actinomycin-d (7-AAD, Molecular Probes) and 2% formalin (Sigma, St. Louis, MO). Cells were analyzed on a fluorescence-activated cell sorter (FACSTAR, Becton Dickinson, San Jose, CA) within 2 hours of labeling. Data were analyzed using the PC Lysis program (Becton Dickinson). FACS gating based on forward scatter and side scatter was used to exclude cellular debris and doublets so that typically 14,000 ± 2000 out of 20,000 cells were selected for analysis. Every experiment included control samples that had been transfected with either the pMT2 vector or the pMT2-hIAPPanti gene.
Analysis for DNA Fragmentation by TUNEL
DNA fragmentation in transfected cells were analyzed using the FlowTACS® fluorescein isothiocyanate (FITC) kit from Trevigen and were performed in parallel with annexin-V labeling as described above. Briefly, 1 × 10 6 cells were fixed in 2% formalin for 10 minutes, washed with PBS, and incubated in Cytopore® (Trevigen) for 10 minutes. DNA fragments were end-labeled with biotinylated nucleotides and terminal deoxynucleotidyl transferase (TdT) for 60 minutes at 37°C, then the biotinylated ends fluorescently tagged with streptavidin-FITC for 20 minutes at room temperature. Cells were briefly fixed in 2% formalin and assayed by FACS within 2 hours. Data were analyzed by the PC Lysis program for cells staining positive for FITC (apoptotic) and those that are FITC negative (nonapoptotic).
Confocal Microscopy
Cells were maintained, harvested, and transfected as described above. Transfected cells (6 to 12 × 10 3 cells in 400 μl) were added to each well of 8-well culture chamber slides (LabTek, Naperville, IL) that had been precoated with poly-l-lysine. At 48 hours after transfection, the cells were labeled for: 1) intracellular or extracellular IAPP, 2) translocated phosphatidylserine (PS) residues with annexin-V, 3) fragmented DNA with FlowTACS®, 4) specific markers of subcellular organelles by immunohistochemistry, or 5) combinations of the above. Fluorescently labeled cells were analyzed on a dual-beam laser scanning confocal microscope (LSCM, model LSM310, Zeiss, Thornwood, NY).
Labeling of intracellular IAPP on adherent cells was accomplished by fixing the cells for 10 minutes in 3.7% formalin, washing, then blocking for 10 minutes in PBS containing 2 mg/ml goat globulin (NGG, Sigma) and 0.05% saponin (Sigma), designated NGG-Sap-PBS. Cells were incubated on ice for 30 to 60 minutes in 150 μl of a 1:2000 dilution of rabbit anti-amylin antiserum (Peninsula Laboratories, Belmont, CA), washed, and fluorescently labeled in a 1:2000 dilution of a goat anti-rabbit antibody conjugated with the Oregon Green fluorochrome (GAR-FITC, Molecular Probes). Cells were washed, fixed with 3.7% formalin, and mounted using the SlowFade® Light kit (Molecular Probes). Mounted slides were stored in the dark at 4°C and analyzed within 5 days. Labeling to detect extracellular IAPP was performed using the same protocols, except saponin was omitted from the buffer.
Dual immunohistochemical labeling to specifically determine the subcellular localization of amylin was accomplished by co-incubating blocked cells with a 1:2000 dilution of rabbit anti-amylin antiserum and diluted mouse monoclonal antibodies against the subcellular markers protein disulfide isomerase (PDI), an ER resident protein, p58, a Golgi resident protein, and Mann II, an ER/Golgi marker protein. Anti-PDI and anti-Mann II antibodies were diluted 1/500, and anti-p58 antibodies were diluted 1/200. Immunohistochemical labeling was in NGG-Sap-PBS on ice for 60 minutes. Sandwich labeling was done concurrently with GAR-FITC (for anti-amylin labeling) and goat-anti-mouse-Rhodamine-Red (GAM-RHOD, Molecular Probes) under the same conditions. Final fix, mount, and analysis were as described above.
To directly correlate the accumulation of intracellular amyloid with the appearance of apoptosis markers, transfected cells were double-labeled with anti-IAPP antiserum and either annexin-V or FlowTACS®. Extracellular PS residues were labeled first by incubating adherent cells with 0.2 μg/sample annexin-V in BB, fixing with ice-cold methanol for 20 minutes and blocking with NGG-Sap-PBS. Secondary labeling of extracellular annexin-V was accomplished by co-incubating with streptavidin-Rhodamine-X conjugate (0.2 μg/sample) and primary labeling of intracellular IAPP with a 1:2000 dilution of rabbit anti-IAPP antiserum in NGG-Sap-PBS. Secondary labeling of intracellular IAPP was done by incubating with a 1:2000 dilution of GAR-FITC. Labeled cells were fixed in 2% formalin, mounted (SlowFade®), and analyzed by LSCM, as described above.
To correlate intracellular IAPP with fragmented nuclear DNA characteristic of apoptosis, cells were first fixed in 3.7% formalin at room temperature, and the membranes were permeabilized by incubating in NGG-PBS (2 mg/ml) containing either a 50:50 dilution of Neuropore (Trevigen) or 0.05% saponin. Nuclear DNA was labeled first using TdT and biotinylated nucleotides (FlowTacs®). Biotin-labeled DNA fragments were fluorescently tagged concurrent with labeling for intracellular IAPP by a co-incubation with 1.5 μg/ml streptavidin-Rhodamine-X and a 1:2000 dilution of rabbit anti-amylin antibodies for 20 minutes at room temperature. Anti-amylin antibodies were then fluorescently labeled by incubation with a 1:2000 dilution of GAR-FITC. Cells were fixed in 3.7% formalin and mounted as described above. Mounted slides were stored at 4°C in the dark and analyzed within 5 days.
Statistical Analyses
In all cases the data from at least three independent experiments were subjected to multivariate analysis of variance using a post hoc Bonferroni t-test to evaluate the significance of individual variables in experiments containing multiple variables.
Results
We previously demonstrated that intracellular amyloid accumulation resulting from over expression of hIAPP in COS-1 cells was associated with ultrastructural changes, including pycnotic nuclei, suggesting that the cells were dying by apoptosis. 12 To establish the mechanism of cell death, we constitutively expressed hIAPP and several nonamyloidogenic forms of IAPP in COS-1 cells and analyzed the cells by FACS using markers characteristic of apoptosis (annexin-V labeling and TUNEL) and necrosis (7-AAD). These techniques for assessment of apoptosis are specific and reproducible and allows a quantitative sensitivity that is far higher than can be detected by other methods such as DNA-laddering techniques. 16,17 We also established the intracellular location of amylin accumulation by scanning confocal microscopy using fluorescently labeled antibodies directed against amylin and three markers of subcellular organelles. Transient expression of hIAPP in COS-1 cells was obtained by transfection with the pMT2-hIAPP gene. 12 As controls, pMT2-hIAPPanti, pMT2-hIAPPmut, and pMT2-rIAPP genes were used. 12 Transfection efficiency was assessed using green fluorescence protein expression plasmid (pGFP-N1, Clontech, Palo Alto, CA) and by both FACS and fluorescence microscopy at 48 or 72 hours after transfection. Transfection efficiency was found to range from 45% by fluorescent microscopy to 75% by FACS in which the differences are probably explained by differences in detection sensitivity (data not shown).
Because amyloid has been previously shown to accumulate in transfected COS-1 cells after 72 to 96 hours and the cells only exhibited pycnotic nuclei within this time period, 12 we first examined annexin-V and 7-AAD labeling by FACS of COS-1 cells that had been transfected with the pMT2, pMT2-hIAPP, and pMT2-hIAPPanti genes and cultured for 72 to 96 hours. Annexin-V binds to PS residues that have undergone a translocation from the cytoplasmic side of the plasma membrane to the extracellular surface early in the apoptotic pathway. 16 The PS-bound annexin-V-biotin is then fluorescently labeled with streptavidin-PE (λex = 488 nm, λem = 578 nm). The DNA-specific fluorescent stain 7-AAD (λex = 488 nm, λem = 647 nm) is excluded from live cells the membranes of which are intact but will leak through the compromised membranes of cells that have died from necrosis and those that are in the final stage of apoptosis. 18 This labeling technique allows separation of cells that are in the early and mid stages of apoptosis (annexin positive) from those in late apoptosis (positive for both annexin and 7-AAD), from necrotic (7-AAD positive only), and live cells (negative for both 7-AAD and annexin).
Representative dot plot profiles generated by FACS analysis of transfected COS-1 cells 96 hours after transfection are shown in Figure 1 ▶ in which annexin-V fluorescence is read on the x axis and 7-AAD on the y axis. Figure 1, A and B ▶ are typical for cells transfected with the control plasmids, pMT2 and pMT2-hIAPPanti, respectively and show that approximately 90% of the cells are viable (lower left panel) with 9.2 and 11.2% of the cells exhibiting positive staining for annexin (both upper and lower right panels), indicating they are apoptotic. These results are consistent with the expected amount of cell death in cultured cells and are virtually identical with those from untransfected cell cultures (data not shown). In contrast, cells that have been transfected with the amyloidogenic pMT2-hIAPP gene have approximately threefold more (29.9%) annexin positive (apoptotic) cells (Figure 1C) ▶ . In all experiments, the percentage of necrotic cells (upper left panel) in pMT2-, pMT2-hIAPPanti-, and pMT2-hIAPP-transfectants were virtually identical (approximately 3 to 5%), indicating that expression of neither amyloidogenic nor nonamyloidogenic forms of IAPP cause cell death by necrosis. It should be emphasized that because only approximately 50% of the cells are transfected, the number of cells undergoing hIAPP-induced apoptosis approaches 60% of the transfected cells that express hIAPP.
Figure 1.
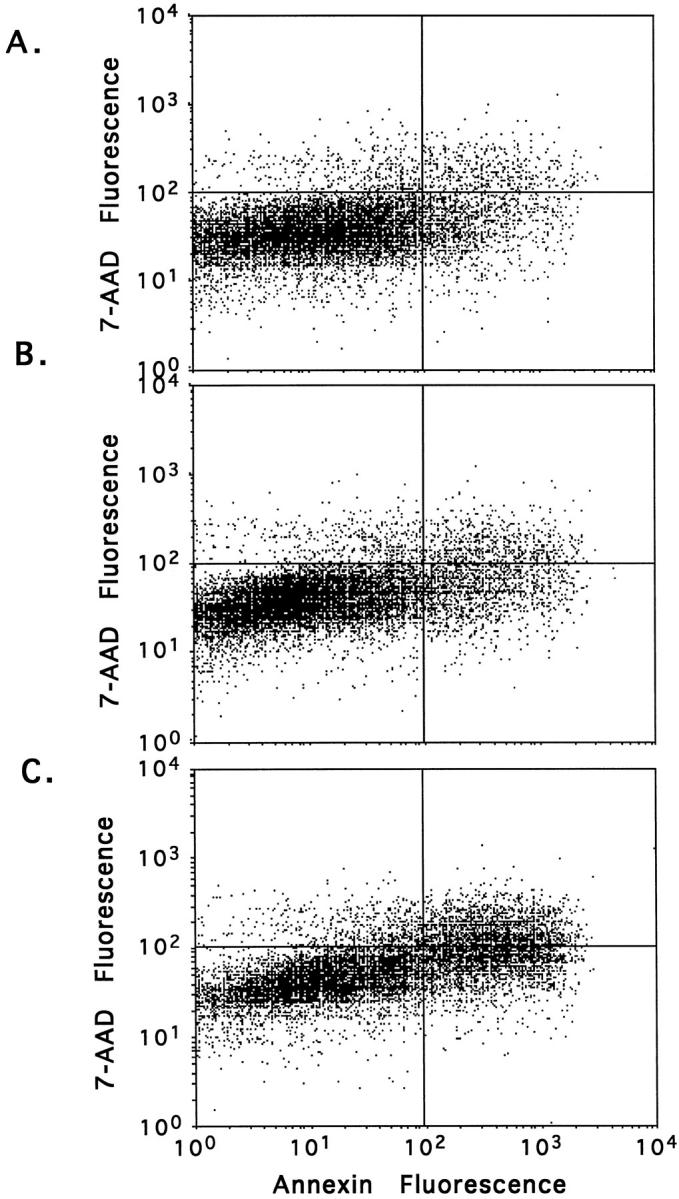
Expression of hIAPP induces apoptosis in transfected COS-1 cells. Cells were transfected with 15 μg of pMT2 control (A), pMT2-hIAPPanti (B), or pMT2-hIAPP (C) expression vectors as described (Materials and Methods) and cultured in complete medium for 96 hours. Apoptosis and necrosis were assessed by annexin-V labeling and 7-AAD staining, respectively, using FACS analysis. Dot plot profiles for annexin-V (x axis) and 7-AAD (y axis) for each treatment group are shown. The percentage of positive cells in the individual quadrants are shown (upper left panel, necrotic; upper right panel, late stage apoptosis; lower right panel, early to mid stage apoptosis).
We next examined the time course of apoptosis in pMT2-hIAPP- and pMT2-hIAPPanti-transfected COS-1 cells. Cells were transfected and cultured as described in Materials and Methods and were assayed for apoptosis by annexin-V labeling 24, 48, 72, and 96 hours later. The histogram profiles shown in Figure 2 ▶ compares cell number on the y axis with the relative fluorescence value, ie, annexin-V binding, on the x axis with apoptotic cells defined by bar M1. In Figure 2 ▶ there is a time-dependent increase in the number of apoptotic cells in pMT2-hIAPP transfectants (Figure 2A) ▶ but not pMT2-hIAPPanti (Figure 2B) ▶ . Apoptosis in both hIAPP (Figure 2A) ▶ and control (Figure 2B) ▶ transfectants is negligible at 24 and 48 hours transfection, but in hIAPP-expressing cells, apoptosis increased to two- to threefold greater than the control at 72 and 96 hours. These data correlate with the previously described time course for amyloid generation after transfection of COS-1 cells with the pMT2-hIAPP gene in which amyloid was only detectable by electron microscopy at 72 to 96 hours 12 and follows the intracellular expression and accumulation of hIAPP (discussed below).
Figure 2.

Time dependent induction of apoptosis in COS-1 cells expressing hIAPP. COS-1 cells were transfected with 15 μg of either the pMT2-hIAPP (A) or pMT2-hIAPPanti (B) expression vectors and cultured as described (Materials and Methods). At 24, 48, 72, and 96 hours after transfection, cells were assessed for apoptosis and necrosis by annexin-V and 7-AAD labeling with FACS analysis. Histogram profiles for annexin-V labeling for each group are shown with the apoptotic cells (annexin-V positive) delineated by the bar (M1).
We next co-transfected cells with equimolar concentrations of both the pMT2-hIAPP and pMT2-hIAPPanti genes and analyzed them for annexin-V and 7-AAD labeling. The rationale for these experiments was to block hIAPP-induced apoptosis by specifically blocking hIPP transcription with its own antisense RNA, thereby demonstrating a direct link between hIAPP expression and apoptosis. Inclusion of the antisense gene with the hIAPP-sense gene reduced apoptosis to that of the controls (Figure 3A) ▶ . Both the negative (pMT2-hIAPPanti) and positive (pMT2-hIAPPsense) controls in these experiments behaved as previously described in Figure 1 ▶ in which hIAPP expression results in high levels of apoptosis (25 to 30%) and control transfectants have only background levels (10 to 12%). Immunohistochemical detection of hIAPP showed only low level hIAPP expression in a much smaller population of cells in COS-1 cells co-transfected with both pMT2-hIAPPanti and pMT2-hIAPPsense vectors than with the pMT2-hIAPPsense vector alone (Figure 4D ▶ , discussed below). Thus, hIAPP expression is blocked by its antisense mRNA, thereby protecting the cells from hIAPP-induced apoptosis. These data indicate that the apoptotic events induced in the hIAPP transfected cells is dependent upon the specific expression of the hIAPP protein.
Figure 3.

Expression of the hIAPP polypeptide and its amyloidogenic domain are required for induction of apoptosis in COS-1 cells. A: Expression of the hIAPP antisense mRNA in COS-1 cells blocks hIAPP-induced apoptosis. COS-1 cells were transfected with the pMT2, pMT2-hIAPPanti, pMT2-hIAPP, or equimolar concentrations of both pMT2-hIAPP and pMT2-hIAPPanti expression vectors and cultured as described (Materials and Methods). After 96 hours the cells were assessed for apoptosis and necrosis by labeling with annexin-V (cross-hatched bars) and 7-AAD (shaded bars) and analyzed by FACS. B: Expression of hIAPP containing its amyloidogenic domain but not a mutant hIAPP or rIAPP, lacking the amyloidogenic domain, leads to apoptosis of transfected COS-1 cells. Cells were transfected with 15 μg of pMT2, pMT2-hIAPPanti, pMT2-hIAPP, pMT2-hIAPPmut, or pMT2-rIAPP expression vectors. Apoptosis was assessed by annexin-V binding and FACS analysis at 72 (open bars) or 96 hours (shaded bars). The data are from three independent experiments; data marked with asterisks were statistically different from control (P < 0.05) by multivariate analysis of variance and post hoc Bonferroni t-test.
Figure 4.
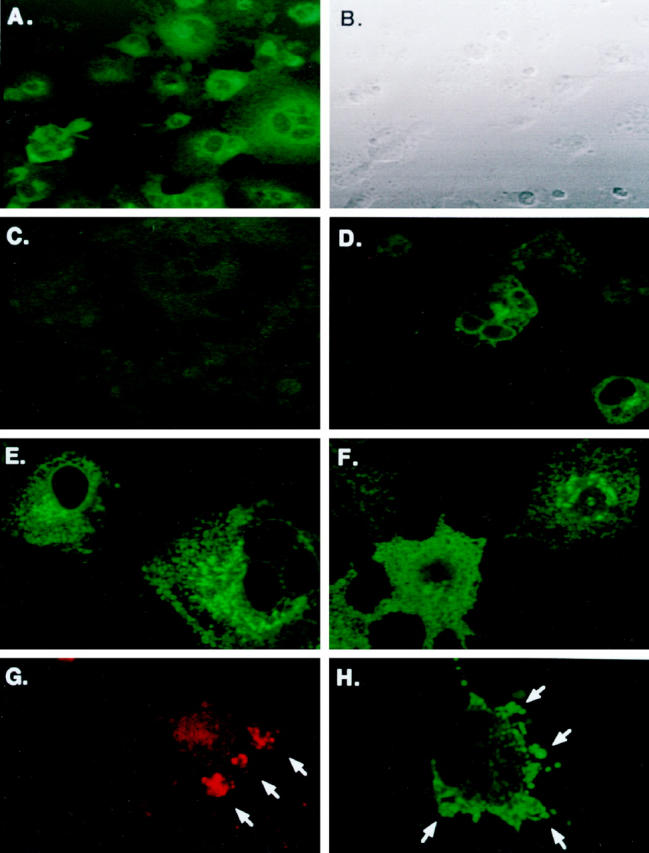
Immunofluorescent labeling of hIAPP, hIAPP variants, and rIAPP in COS-1 cells. Cells were transfected with the pMT2-hIAPP (A and B), pMT2-hIAPPanti (C), and co-transfected pMT2-hIAPP and pMT2-hIAPPanti expression vectors as described in Materials and Methods and cultured in 8-well chamber slides for 48 hours. Immunofluorescent labeling on permeabilized cells was performed with rabbit anti-amylin antibodies and FITC-labeled GAR secondary antibodies. Phase contrast (B) and immunoflouresence microscopy (A, C, and D) were performed concurrently on a Zeiss laser scanning confocal microscope. Intense immunofluoresence (green) is seen localized in clusters in the perinuclear regions of hIAPP-expressing cells only (A). Cells were also transfected with the nonamyloidogenic pMT2-hIAPPmut (E) or pMT2-rIAPP (F) expression vectors. After 48 hours, the permeabilized cells were labeled with rabbit anti-IAPP antibodies and GAR-FITC to detect intracellular IAPP (Materials and Methods) by LSCM. Apoptotic bodies (G) and membrane blebbing (H) extruding from cells undergoing apoptosis in pMT2-hIAPP-transfected COS-1 cells at 48 hours after transfection. Translocated extracellular phosphatidylserine residues were labeled by annexin-V and Rhodamine Red fluorescence and analyzed by LSCM. Annexin positive labeling on apoptotic cells (red staining) and formation of apoptotic bodies (arrows) are shown in G. Intracellular hIAPP is also evident in apoptotic blebs (green staining and arrows) as labeled with anti-amylin antibodies and FITC fluorescence in H.
Previously, we demonstrated that cells transfected with the pMT2-rIAPP gene exhibit immunoreactive labeling for rIAPP without evidence for fibril formation or cell death. 12 Therefore, we also examined the expression of rIAPP and hIAPPmut genes, as well as the hIAPPanti gene on cell viability. The percentage of annexin-V positive cells in pMT2-rIAPP or pMT2-hIAPPmut transfectants at either 72 or 96 hours after transfection is not significantly different than cells transfected with the control pMT2-hIAPPanti or pMT2 vectors (12%, Figure 3B ▶ ). Conversely, 32% of pMT2-hIAPP transfectants are apoptotic. These data indicate that only the amyloidogenic form of hIAPP will induce apoptosis, suggesting that a unique structure within this molecule is responsible for triggering the apoptosis signaling pathway.
To verify that the analysis for apoptosis using annexin-V labeling of translocated PS residues in hIAPP-transfected COS cells is consistent with other markers characteristic of apoptosis, we also used a technique in which the free ends of fragmented DNA are labeled with fluorescently tagged nucleotides (TUNEL) and analyzed by FACS. Although TUNEL does not strictly differentiate apoptotic cells from necrotic cells, this assay verifies that cells are undergoing DNA fragmentation, which is characteristic of apoptosis. Annexin-V labeling and TUNEL were performed in parallel on cells from the same transfection cultures and gave remarkably similar results (data not shown). The percentage of cells that were apoptotic in control cultures varied by less than 1% between TUNEL and annexin-V staining (9 to 10%), and in the hIAPP transfectants, the percentage of apoptotic cells averaged 31% by annexin-V staining and 29% by TUNEL at 96 hours. Because we analyzed necrosis by 7-AAD staining in parallel samples and showed that hIAPP expression does not increase background necrosis, we are confident that TUNEL is a reliable marker of apoptosis in these experiments. These data confirm that the expression of amyloidogenic hIAPP in transfected COS cells induces these cells to undergo programmed cell death by classic apoptotic mechanisms that include both translocation of PS residues and fragmentation of nuclear DNA. 16,17
We wanted to identify the cellular location of the expressed hIAPP in transfected COS cells to correlate cellular location with the onset of apoptotic events. Intracellular expression of hIAPP was confirmed using immunohistochemical labeling with antiamylin antiserum in the presence of the mild detergent saponin. As additional control, cells from the same transfections were also analyzed for apoptosis and necrosis by annexin-V and 7-AAD labeling and FACS to correlate IAPP expression with cell death. Figure 4, A and B ▶ shows pMT2-hIAPP-transfected cells in both phase contrast and immunofluorescent microscopy. Figure 4A ▶ shows the intracellular accumulation of amylin immunoreactivity in distinct granular structures in the immediate perinuclear region of pMT2-hIAPP transfected cells (green fluorescence) and that approximately one-half of the cells are labeled (compare with Figure 4B ▶ ). Conversely, cells that have been transfected with the pMT2-hIAPPanti plasmid (Figure 4C) ▶ or mock-transfected controls (data not shown) exhibit no amylin immunoreactivity. When cells were co-transfected with pMT2-hIAPP and pMT2-hIAPPanti plasmids, the number of cells exhibiting immunoreactive amylin was greatly reduced, and the level of intracellular hIAPP expression was decreased (Figure 4D) ▶ , demonstrating that co-transfection of the hIAPP antisense expression vector reduced hIAPP expression significantly. Intracellular hIAPP immunoreactivity could be detected as early as 24 hours transfection (data not shown) and is optimal at 48 hours. By 72 hours the very high immunoreactive signal and cellular destruction by apoptosis limits the use of immunohistochemical labeling and LSCM in these studies.
We also transfected COS-1 cells with the two nonamyloidogenic variants of IAPP mentioned previously, rat IAPP (pMT2-rIAPP) and a mutant form of human IAPP (pMT2-hIAPPmut), and analyzed them for expression of intracellular amylin. Both rat IAPP (Figure 4E) ▶ and mutant hIAPP (Figure 4F) ▶ show a pattern of immunoreactivity that is comparable with that of hIAPP (Figure 4A) ▶ with localization and clustering in the perinuclear region, whereas the antisense control exhibits no amylin immunoreactivity (Figure 4C) ▶ . It is very interesting that although both the rat and mutant forms of IAPP are being synthesized and accumulate in subcellular organelles at levels comparable with the amyloidogenic hIAPP, they do not induce apoptosis as indicated by annexin-V labeling (Figure 3B ▶ , discussed above). This data indicate that the amyloidogenic region (amino acids 20–29) within the hIAPP protein is unique, making it capable of triggering the apoptotic signaling pathways, whereas the nonamyloidogenic forms do not despite the high levels of accumulation within subcellular organelles. Further evidence that the mechanism of cell death proceeds by apoptosis was indicated by the finding of cells exhibiting immunoreactive apoptotic bodies (Figure 4G) ▶ and membrane blebbing (Figure 4H) ▶ . These structures are hallmark morphological features of apoptosis that represent different stages of the organized breakdown of cells, whereby the cellular contents are packaged in membrane-bound apoptotic bodies that are phagocytosed and destroyed by neighboring cells without eliciting an immune reaction. 19,20
To determine that the expressed IAPP is intracellular and not a secreted extracellular form, we performed immunohistochemical labeling of control pMT2-hIAPPanti- versus pMT2-hIAPP-transfected COS cells in the presence and absence of the detergent saponin. Cells were fixed so that insoluble, extracellular IAPP, if present, would remain in place. In Figure 5 ▶ there is no evidence of extracellular immunoreactivity in nonpermeabilized pMT2-hIAPP- or pMT2-hIAPPanti-transfected cells (Figure 5, A and C ▶ , respectively) or intracellular immunoreactivity in permeabilized control cells (Figure 5D) ▶ . However, in permeabilized pMT2-hIAPP transfectants there is a high concentration of localized intracellular immunoreactivity in the immediate perinuclear region (Figure 5B) ▶ clearly demonstrating the intracellular accumulation of hIAPP. Combined with the previous studies demonstrating that apoptosis correlates with hIAPP expression, these data demonstrate that apoptosis is initiated by the intracellular accumulation of hIAPP as opposed to its secretion and extracellular aggregation. Consistent with this interpretation, we had earlier shown that the secreted levels of hIAPP were insufficient to kill COS-1 cells. 12 To test this experimentally, we incubated COS-1 cells that had been transfected with the control pMT2 and pMT2-hIAPP vector for 96 hours in either normal medium or conditioned medium from COS-1 cells that had been transfected with the pMT2-hIAPP expression vector for 96 hours. As shown in Figure 5E ▶ , incubating COS-1 cells transfected with pMT2 vector in conditioned medium did not induce apoptosis. Similarly, the conditioned medium did not increase the amount of apoptosis in cells that had been transfected with the pMT2-hIAPP plasmid (Figure 5E) ▶ .
Figure 5.
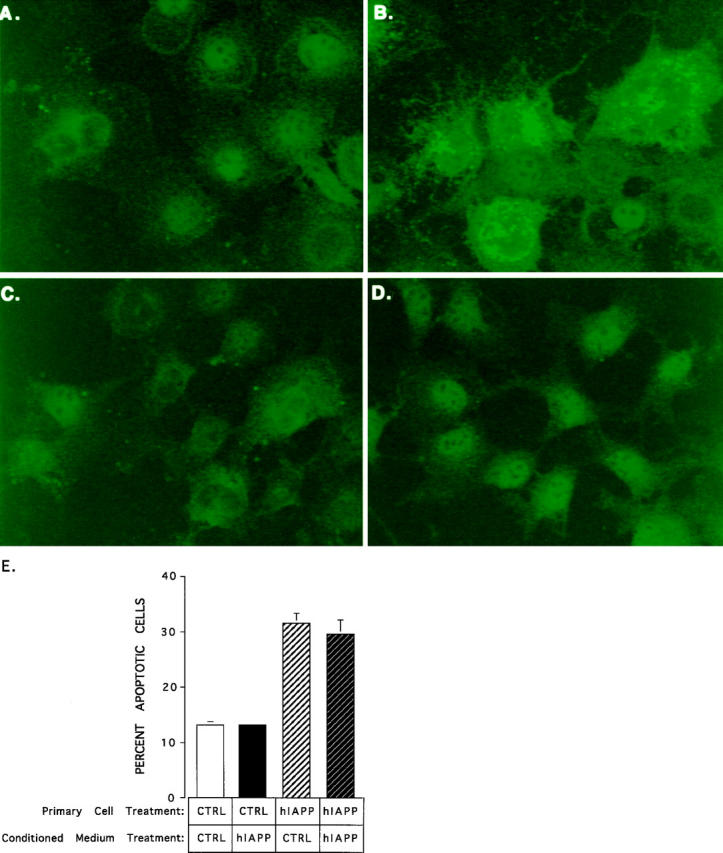
Immunofluorescent labeling of amylin in permeabilized and nonpermeabilized hIAPP-expressing COS-1 cells. COS-1 cells were transfected with the pMT2-hIAPP (A and B) or pMT2-hIAPPanti (C and D) expression vectors and cultured in multiwell chamber slides for 72 hours. Fixed cells were either permeabilized with 0.05% saponin (B and D) or left untreated (A and C) prior to immunofluorescent staining with anti-amylin antibodies. Immunofluorescent labeling (green) is seen only in permeabilized hIAPP-expressing cells (B). Nonpermeabilized cells show no evidence of amylin immunolabeling (A) above the background levels (C and D). E depicts a conditioned media experiment in which the medium from pMT2 control (CTRL) or pMT2-hIAPP (hIAPP) transfected cells was added to COS-1 cells that were freshly transfected with pMT2 (CTRL) or pMT2-hIAPP (hIAPP) plasmids and then incubated for 96 hours to determine whether extracellular amylin/amyloid was cytotoxic. The data represent the average results from two independent experiments and the error bars reflect the average deviation from the mean in the two experiments.
Additional evidence that the intracellular accumulation of amyloidogenic hIAPP is directly linked to apoptosis is seen in Figure 6 ▶ , which demonstrates strong amylin immunoreactivity (Figure 6, A and C ▶ , green) in combination with either annexin-V labeling (Figure 6A ▶ , red) or intense TUNEL staining of fragmented nuclear DNA (Figure 6C ▶ , red) in pMT2-hIAPP-transfected COS-1 cells. Conversely, cells that have been transfected with the pMT2-hIAPPanti plasmid have only very low levels of background staining with either annexin-V or TUNEL (Figure 6, B and D ▶ , respectively). These data link the accumulation of intracellular amylin with distinct markers characteristic of apoptosis in the same cell.
Figure 6.

Double labeling of intracellular hIAPP and specific markers of apoptosis is found only in hIAPP-expressing COS-1 cells. A: Intracellular accumulation of hIAPP is associated with extracellular annexin-V labeling indicative of apoptosis in pMT2-hIAPP-transfected COS-1 cells. Extracellular annexin was fluorescently labeled with streptavidin-rhodamine (red) and intracellular IAPP with FITC (green). The majority (>90%) of control pMT2-hIAPPanti-transfected cells (B) have only background levels of annexin labeling; the single brightly labeled cell in B is undergoing apoptosis. Intracellular accumulation of hIAPP (green) is also associated with TUNEL labeling of fragmented DNA (red) in the nucleus of pMT2-hIAPP- (C) but not control pMT2-transfected cells (D). Double labeling of adherent COS-1 cells was performed as described in Materials and Methods.
To establish the subcellular location of hIAPP accumulation in pMT2-hIAPP-transfected cells, we performed dual immunolabeling with antiamylin antiserum in combination with antibodies against three subcellular marker proteins: protein disulfide isomerase (PDI) in the ER, Mann II in the ER-Golgi, and p58 in the Golgi. The yellow immunofluorescence (Figure 7B) ▶ demonstrates colocalization of amylin (Figure 7C ▶ , green) with the Golgi marker p58 (Figure 7A ▶ , red) in pMT2-hIAPP-transfected cells. Intracellular amylin is not detected in either control or nontransfected cells, although p58 immunoreactivity is present (not shown). Likewise in Figure 7E ▶ there is clear co-localization (yellow) of amylin immunoreactivity (Figure 7F ▶ , green) with the ER-Golgi marker Mann II (Figure 7D ▶ , red). Amylin immunoreactivity is not present in nontransfected cells, an example of which can be seen just above dual stained cells in Figure 7, D and E ▶ . Amylin also co-localizes (Figure 7H ▶ , yellow) with the endoplasmic reticulum marker, PDI (Figure 7G ▶ , red) and amylin (Figure 7I ▶ , green). These data confirm that the expression of hIAPP in pMT2-hIAPP-transfected cells results in the accumulation of amylin immunoreactivity within the ER and Golgi apparatus. Because the immunolabeling was performed on cells 48 hours after being transfected with the hIAPP expression vector, the subcellular accumulation of amylin within the ER and Golgi apparatus precedes the onset of apoptosis (72 to 96 hours).
Figure 7.

Intracellular accumulation of hIAPP in COS-1 cells is found co-localized with both Golgi- and ER-specific markers. Dual immunolabeling with antibodies against the Golgi p58K marker protein (A; red), Mann II ER-Golgi marker (D; red), or the ER marker, PDI (G; red), and anti-amylin (green in C, F, and I), demonstrates a co-localization with both ER and Golgi markers (yellow in B, E, and H). Immunolabeling was performed on COS-1 cells 48 hours after transfection with pMT2-hIAPP and pMT2-hIAPPanti control plasmids. Permeabilized cells were concurrently labeled with rabbit anti-IAPP and mouse anti-marker antibodies, then sandwich labeled with GAR-FITC and GAM-RHOD, and analyzed by LSCM (Materials and Methods).
Discussion
We previously demonstrated that over-expression of hIAPP in transfected COS-1 cells led to intracellular accumulation of amyloid fibrils, which was associated with cell death. 12 In the present study, using fluorescent markers of apoptosis and/or necrosis coupled with FACS analysis in conjunction with immunohistochemical labeling and LSCM analysis of amylin and subcellular marker proteins, we demonstrate that hIAPP-transfected COS-1 cells are dying by apoptosis triggered by intracellular hIAPP. These data suggest that intracellular amyloid initiates a cascade of intracellular signaling events that trigger the apoptotic pathway.
Significant amounts of hIAPP immunoreactivity were detected in both the ER and Golgi apparatus by 48 hours after transfection of COS-1 cells with the pMT2-hIAPP gene (Figure 7) ▶ , although electron microscopy revealed that the cells exhibited intact plasma membranes and intracellular organelle structures with normal chromatin patterns and lacked evidence for amyloid formation. 12 Conversely, at 96 hours the hIAPP-transfected cells exhibited intense intracellular hIAPP immunogold labeling associated with intracellular amyloid, highly condensed chromatin and disrupted plasma membranes, all characteristic of apoptotic cells. 12 In the current study, significant apoptosis was induced at 72 to 96 hours but not at earlier time points (Figures 1 to 3) ▶ ▶ ▶ , indicating that amyloid formation is correlated with and precedes the induction of apoptosis. Although the expression and accumulation of hIAPPmut and rIAPP within the ER is comparable with that of the amyloidogenic hIAPP (Figure 4) ▶ , 12 neither of these proteins is cytotoxic as indicated by annexin-V and 7-AAD labeling (Figure 3) ▶ . In addition, hIAPPmut reverts the amyloidogenic domain of hIAPP between amino acids 20 and 29 4 (Figure 8 ▶ , GAIL), to the nonamyloidogenic domain of the rIAPP peptide while preserving the two other amino acid differences between these two species. Thus the amyloidogenic domain of hIAPP must be responsible for activation of the apoptotic signaling events. Our data provide evidence that the accumulation of amyloid within the ER and/or Golgi apparatus provides a specific stimulus for the induction of apoptosis. Whereas further work will be required to ascertain the nature of this signal, a likely starting point might well be the various ER quality control mechanisms that insure the orderly trafficking of correctly folded polypeptides. Such mechanisms include the unfolded protein response 21,22 and ER overload response pathways. 23,24,25 Both of these pathways provide for the induction of ER chaperones, including Bip/Grp78, Grp94, calnexin, calreticulin, peptidyl prolyl isomerase, and protein disulfide isomerase that are involved in protein folding. 26 In addition, the degradation of malfolded proteins by the ubiquitin-proteosome pathway is an essential component of the ER quality control systems, and the regulation of this pathway also involves molecular chaperones. 27,28 Some preliminary evidence for the involvement of these quality control pathways in the regulation of amyloid already exists. For example, in the studies of human insulinomas, which provided the first evidence for intracellular IAPP amyloid formation, ubiquitin immunoreactivity was observed as punctate intracellular labeling in association with intracellular as well as extracellular amyloid deposits. 29 In addition, we have demonstrated previously that small heat shock proteins inhibit the in vitro formation of amyloid by the Alzheimer Aβ1–42 polypeptide, suggesting that chaperones might be involved in inhibiting the formation of certain peptide conformations that lead to amyloid formation. 30
While the exact mechanism of β-cell death in NIDDM is unknown and the role of amyloid in β-cell death is controversial, 3,9 the present studies suggest that intracellular amyloid-induced apoptosis may contribute to the loss of β-cell mass in NIDDM. Recent evidence that strongly supports a direct role of IAPP in the genesis of NIDDM is the finding that a missense mutation of the IAPP gene (S20G) is associated with early onset (≥35 years) NIDDM in a high proportion of individuals carrying the mutation. 10 Thus NIDDM bears similarities to Alzheimer disease in which rare mutations of the APP gene are associated with early onset disease and increased amyloid deposition in the brains of afflicted individuals. 31-33
A number of investigators have examined hIAPP expression in transgenic mice and have observed conflicting results. Janson et al 34 reported that transgenic mice homozygous for the hIAPP gene (RHF-hIAPP+/+) express high levels of hIAPP and spontaneously develop diabetes mellitus. In the RHF-hIAPP+/+ mice amyloid deposits are not observed, but intra- and extra-cellular amorphous aggregates of hIAPP are present, suggesting that structural forms of hIAPP in addition to or other than amyloid may be cytotoxic. 34 Treatment of the hemizygous RHF-hIAPP+/− mice with growth hormone and dexamethasone to induce insulin resistance for 4 weeks did cause islet amyloidosis, including intra- and extra-cellular deposits that preceded β-cell dysfunction; 6 however, the untreated RHF-hIAPP+/− mice did not spontaneously develop any signs of pancreatic dysfunction or glucose intolerance. 6,34 D’Alessio et al 35 reported initially that their transgenic mice lacked islet amyloid deposits but had higher levels of islet insulin and proinsulin. 36 Interestingly, when these animals were moved to another institution and subjected to a change in diet, extensive islet amyloid deposits were observed in male transgenic mice that were older than 13 months but were only observed in 11% of the female mice. 13 In these animals, half of those that had islet amyloid deposits were hyperglycemic with plasma glucose levels >11 mmol/L. In younger animals (6 to 9 months), islet amyloid deposits were rarely observed; however, their occurrence was associated with severe hyperglycemia with plasma glucose levels >22 mmol/L. 13 Two other studies of transgenic animals expressing hIAPP were largely negative with failure to demonstrate islet amyloid or effect changes in circulating glucose levels. 37,38 In one of these studies, some amyloid in secretory granules was observed. 38 In the other study, when the islets were cultured in vitro in relatively high concentrations of glucose (11 to 22 mmol/L), amyloid was observed to develop within 7 days. 37 More recently, Soeller et al, 39 crossed RHF-hIAPP+/− with the insulin-resistant agouti viable yellow (Avy/a) mice, and the resultant hybrids developed a slow onset form of diabetes that was associated with the accumulation of islet amyloid and a reduction of β-cell mass. Taken together, these studies support the hypothesis that islet amyloid plays a role in β-cell dysfunction that leads to hyperglycemia; however, other factors, including the absolute levels of IAPP expression, the specific conformation of the IAPP aggregates, diet, and insulin resistance appear to play important conditional roles in the development of these abnormalities.
Because the cell destruction that occurs in NIDDM appears to be confined solely to the β cells, the concept that the cells are killed through an intracellular apoptotic mechanism is attractive, as apoptosis would be expected to minimize inflammatory effects that could damage the surrounding tissue. Whereas much work remains to unravel the mysteries of hIAPP-induced cell death, this COS cell model will be useful in establishing the intracellular signaling pathway that leads to apoptosis and provide insights about the mechanisms by which intracellular amyloid exerts its cytotoxic action.
Acknowledgments
The authors thank James Tarara, Holly Lamb, and Teresa Halsey for assistance with the flow cytometry and laser confocal microscopy studies, Dr. Y. S. Prakash for assistance with the fluorescence microscopic evaluation of green fluorescent protein expression in transfected COS-1 cells, Cheri Mueske and Dr. Andrew Arnold for preparing and sequencing the hIAPPmut cDNA, and Dr. Mark McNiven for supplying ER and Golgi marker antibodies. The authors thank Ruth Kiefer for editorial and secretarial assistance.
Footnotes
Address reprint requests to Dr. Norman L. Eberhardt, 4-407 Alfred, Mayo Clinic, Rochester, MN 55905. E-mail: eberhardt@mayo.edu.
Supported by grants from the National Institutes of Health (AG08031 and AG14522, NLE), a gift from the Quade Amyloidosis Research Fund (NLE), and Mayo Foundation Research Funds.
References
- 1.Clark A, Charge SB, Badman MK, MacArthur DA, de Koning EJ: Islet amyloid polypeptide: actions and role in the pathogenesis of diabetes. Biochem Soc Trans 1996, 24:594-599 [DOI] [PubMed] [Google Scholar]
- 2.Lorenzo A, Yankner BA: Amyloid fibril toxicity in Alzheimer’s disease and diabetes. Ann NY Acad Sci 1996, 777:89-95 [DOI] [PubMed] [Google Scholar]
- 3.O’Brien TD, Butler PC, Westermark P, Johnson KH: Islet amyloid polypeptide: a review of its biology and potential roles in the pathogenesis of diabetes mellitus. Vet Pathol 1993, 30:317-332 [DOI] [PubMed] [Google Scholar]
- 4.Westermark P, Engstrom U, Johnson KH, Westermark GT, Betsholtz C: Islet amyloid polypeptide: pinpointing amino acid residues linked to amyloid fibril formation. Proc Natl Acad Sci USA 1990, 87:5036-5040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Brien TD, Wagner JD, Litwak KN, Carlson CS, Cefalu WT, Jordan K, Butler PC: Islet amyloid and islet amyloid polypeptide in cynomolgus macaques (Macaca fascicularis): an animal model of human non-insulin-dependent diabetes mellitus. Vet Pathol 1996, 33:479-485 [DOI] [PubMed] [Google Scholar]
- 6.Couce M, Kane LA, O’Brien TD, Charlesworth J, Soeller W, McNeish J, Roche P, Butler PC: Treatment with growth hormone and dexamethasone in mice transgenic for human islet amyloid polypeptide causes islet amyloidosis and beta-cell dysfunction. Diabetes 1996, 45:1094-1101 [DOI] [PubMed] [Google Scholar]
- 7.Westermark P, Wilander E: The influence of amyloid deposits on the islet volume in maturity onset diabetes mellitus. Diabetologia 1978, 15:417-421 [DOI] [PubMed] [Google Scholar]
- 8.Clark A, Wells CA, Buley ID, Cruickshank JK, Vanhegan RI, Matthews DR, Cooper GJ, Holman RR, Turner RC: Islet amyloid, increased A-cells, reduced B-cells and exocrine fibrosis: quantitative changes in the pancreas in type 2 diabetes. Diabetes Res 1988, 9:151-159 [PubMed] [Google Scholar]
- 9.Rewers M, Hamman RF: Diabetes in America. 1995:pp 179-220 National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD,
- 10.Sakagashira S, Sanke T, Hanabusa T, Shimomura H, Ohagi S, Kumagaye KY, Nakajima K, Nanjo K: Missense mutation of amylin gene (S20G) in Japanese NIDDM patients. Diabetes 1996, 45:1279-1281 [DOI] [PubMed] [Google Scholar]
- 11.Lorenzo A, Razzaboni B, Weir GC, Yankner BA: Pancreatic islet cell toxicity of amylin associated with type-2 diabetes mellitus. Nature 1994, 368:756-760 [DOI] [PubMed] [Google Scholar]
- 12.O’Brien TD, Butler PC, Kreutter DK, Kane LA, Eberhardt NL: Human islet amyloid polypeptide expression in COS-1 cells: a model of intracellular amyloidogenesis. Am J Pathol 1995, 147:609-616 [PMC free article] [PubMed] [Google Scholar]
- 13.Verchere CB, D’Alessio DA, Palmiter RD, Weir GC, Bonner-Weir S, Kahn SE: Islet amyloid formation associated with hyperglycemia in transgenic mice with pancreatic beta cell expression of human islet amyloid polypeptide. Proc Natl Acad Sci USA 1996, 93:3492-3496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wong GG, Witek JS, Temple PA, Wilkens KM, Leary AC, Luxenberg DP, Brown EL, Kay RM, Orr EC, Shoemaker C, Golde DW, Kaufman RJ, Hewick RM, Wang EA, Clark SC: Human GM-CSF: molecular cloning of the complementary DNA and purification of the natural and recombinant proteins. Science 1985, 228:810-815 [DOI] [PubMed] [Google Scholar]
- 15.Hemsley A, Arnheim N, Toney MD, Cortopassi G, Galas DJ: A simple method for site-directed mutagenesis using the polymerase chain reaction. Nucleic Acids Res 1989, 17:6545-6551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martin SJ, Reutelingsperger CP, McGahon AJ, Rader JA, van Schie RC, Green DR: Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J Exp Med 1995, 182:1545-1556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gavrieli MJ, Sherman Y, Ben-Sasson SA: Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol 1992, 119:493-501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmid I, Krall WJ, Uittenbogaart CH, Braun J, Giorgi JV: Dead cell discrimination with 7-amino-actinomycin D in combination with dual color immunofluorescence in single laser flow cytometry. Cytometry 1992, 13:204-208 [DOI] [PubMed] [Google Scholar]
- 19.Alison MR, Sarraf CE: Apoptosis: a gene-directed programme of cell death. J R Coll Physicians Lond 1992, 26:25-35 [PMC free article] [PubMed] [Google Scholar]
- 20.Guchelaar HJ, Vermes A, Vermes I, Haanen C: Apoptosis: molecular mechanisms and implications for cancer chemotherapy. Pharm World Sci 1997, 19:119-125 [DOI] [PubMed] [Google Scholar]
- 21.Sidrauski C, Chapman R, Walter P: The unfolded protein response: an intracellular signaling pathway with many surprising features. Trends Cell Biol 1998, 8:245-249 [DOI] [PubMed] [Google Scholar]
- 22.Cox JS, Chapman RE, Walter P: The unfolded protein response coordinates the production of endoplasmic reticulum protein and endoplasmic reticulum membrane. Mol Biol Cell 1997, 8:1805-1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brewer JW, Cleveland JL, Hendershot LM: A pathway distinct from the mammalian unfolded protein response regulates expression of endoplasmic reticulum chaperones in non-stressed cells. EMBO J 1997, 16:7207-7216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pahl HL, Baeuerle PA: The ER-overload response: activation of NF-kappa B. Trends Biochem Sci 1997, 22:63-67 [DOI] [PubMed] [Google Scholar]
- 25.Pahl HL, Sester M, Burgert HG, Baeuerle PA: Activation of transcription factor NF-kappa B by the adenovirus E3/19K protein requires its ER retention. J Cell Biol 1996, 132:511-522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wei J, Hendershot LM: Protein folding and assembly in the endoplasmic reticulum. EXS 1996, 77:41-55 [DOI] [PubMed] [Google Scholar]
- 27.Sommer T, Wolf DH: Endoplasmic reticulum degradation: reverse protein flow of no return. FASEB J 1997, 11:1227-1233 [DOI] [PubMed] [Google Scholar]
- 28.Sherman MY, Goldberg AL: Involvement of molecular chaperones in intracellular protein breakdown. EXS 1996, 77:57-78 [DOI] [PubMed] [Google Scholar]
- 29.O’Brien TD, Butler AE, Roche PC, Johnson KH, Butler PC: Islet amyloid polypeptide in human insulinomas: evidence for intracellular amyloidogenesis. Diabetes 1994, 43:329-336 [DOI] [PubMed] [Google Scholar]
- 30.Kudva YC, Hiddinga HJ, Butler PC, Mueske CS, Eberhardt NL: Small heat shock proteins inhibit in vitro A beta(1-42) amyloidogenesis. FEBS Lett 1997, 416:117-121 [DOI] [PubMed] [Google Scholar]
- 31.Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, Lieberburg I, Selkoe DJ: Mutation of the beta-amyloid precursor protein in familial Alzheimer’s disease increases beta-protein production. Nature 1992, 360:672-674 [DOI] [PubMed] [Google Scholar]
- 32.Tanzi RE, George-Hyslop PS, Gusella JF: Molecular genetics of Alzheimer disease amyloid. J Biol Chem 1991, 266:20579-20582 [PubMed] [Google Scholar]
- 33.Joachim CL, Selkoe DJ: The seminal role of beta-amyloid in the pathogenesis of Alzheimer disease. Alzheimer Dis Assoc Disord 1992, 6:7-34 [DOI] [PubMed] [Google Scholar]
- 34.Janson J, Soeller WC, Roche PC, Nelson RT, Torchia AJ, Kreutter DK: Spontaneous diabetes mellitus in transgenic mice expressing human islet amyloid polypeptide. Proc Natl Acad Sci USA 1996, 93:7283-7288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.D’Alessio DA, Verchere CB, Kahn SE, Hoagland V, Baskin DG, Palmiter RD, Ensinck JW: Pancreatic expression and secretion of human islet amyloid polypeptide in a transgenic mouse. Diabetes 1994, 43:1457-1461 [DOI] [PubMed] [Google Scholar]
- 36.Verchere CB, D’Alessio DA, Palmiter RD, Kahn SE: Transgenic mice overproducing islet amyloid polypeptide have increased insulin storage and secretion in vitro. Diabetologia 1994, 37:725-728 [DOI] [PubMed] [Google Scholar]
- 37.de Koning EJ, Morris ER, Hofhuis FM, Posthuma G, Hoppener JW, Morris JF, Capel PJ, Clark A, Verbeek JS: Intra- and extracellular amyloid fibrils are formed in cultured pancreatic islets of transgenic mice expressing human islet amyloid polypeptide. Proc Natl Acad Sci USA 1994, 91:8467-8471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yagui K, Yamaguchi T, Kanatsuka A, Shimada F, Huang CI, Tokuyama Y, Yamamura K, Miyazaki J, Mikata A, Yoshida S, Hideichi M: Formation of islet amyloid fibrils in beta-secretory granules of transgenic mice expressing human islet amyloid polypeptide/amylin. Eur J Endocrinol 1995, 132:487-496 [DOI] [PubMed] [Google Scholar]
- 39.Soeller WC, Janson J, Hart SE, Parker JC, Carty MD, Stevenson RW, Kreutter DK, Butler PC: Islet amyloid-associated diabetes in obese Avy/a mice expressing human islet amyloid polypeptide. Diabetes 1998, 47:743-750 [DOI] [PubMed] [Google Scholar]