

Abstract
A major complication in sepsis is progressively impaired lung function and susceptibility to intrapulmonary infection. Why sepsis predisposes the lung to injury is not clear. In the current studies, rats were rendered septic by cecal ligation/puncture and evaluated for increased susceptibility to injury after a direct pulmonary insult (deposition of IgG immune complexes or airway instillation of lipopolysaccharide). By itself, cecal ligation/puncture did not produce evidence of lung injury. However, after a direct pulmonary insult, lung injury in septic animals was significantly enhanced. Enhanced lung injury was associated with increased accumulation of neutrophils in lung, enhanced production of CXC chemokines (but not tumor necrosis factor-α) in bronchoalveolar lavage fluids, and increased expression of lung vascular intercellular adhesion molecule-1 (ICAM-1). Complement depletion or treatment with anti-C5a abolished all evidence of enhanced lung injury in septic animals. When stimulated in vitro, bronchoalveolar lavage macrophages from septic animals had greatly enhanced CXC chemokine responses as compared with macrophages from sham-operated animals or from septic animals that had been complement depleted. These data indicate that the septic state causes priming of lung macrophages and suggest that enhanced lung injury in the septic state is complement dependent and related to increased production of CXC chemokines.
The successful treatment of sepsis in humans continues to be a substantial clinical challenge. 1,2 In intensive care units, a major complication in septic patients is impaired respiration, often leading to adult respiratory distress syndrome and onset of multiple organ failure. 3-5 It is thought in many cases of sepsis that there is loss of the gut-blood barrier, resulting in translocation of gram-negative bacteria to subepithelial areas and ultimately entry of bacteria or bacterial lipopolysaccharide (LPS) into the vascular compartment. 5-7 It has been demonstrated both experimentally and clinically that sepsis causes the appearance in plasma of a series of cytokines, such as interleukin (IL)-1, tumor necrosis factor (TNF)-α, or IL-6. 1,2,8-10 This phenomenon has been termed the systemic inflammatory response syndrome. This condition appears to place organs (liver, lung, and kidneys) at risk of injury and failure. It has been postulated that during sepsis the lung is especially susceptible to injury in the presence of a direct intrapulmonary insult (the so-called second hit), 11 such as ischemia, blunt thoracic injury, intrapulmonary presence of bacteria, or ventilator-induced pulmonary injury, to name only a few examples. Why the liver and kidneys also become targets of injury during sepsis is poorly understood.
Although a septic-like state has been induced experimentally by infusion of LPS or live bacteria, cecal ligation/puncture (CLP) in rodents seems to mimic many features of the septic state in humans. Animals develop progressive bacteremia, appearance of multiple cytokines and chemokines in plasma, hypermetabolism, fever, and other clinical features similar to those found in humans with sepsis. 8 In the present study CLP was studied in rats. The experimental data indicate that during sepsis a direct intrapulmonary insult (induced by LPS or deposition of immune complexes) results in augmented accumulation of neutrophils, higher levels of CXC chemokines, and evidence of enhanced lung injury. The studies also indicate that these events are complement dependent and associated with a priming of lung macrophages.
Materials and Methods
Cecal Ligation and Puncture and Lung Injury
Male Long-Evans specific-pathogen-free rats (275 to 300 mg; Harlan, Indianapolis, IN) were used in all studies. Anesthesia was induced by intraperitoneal administration of ketamine (20 mg/100 mg body weight). After shaving the abdomen and application of a topical disinfectant, a 2-cm midline incision was made, and the cecum was identified and ligated below the ileocecal valve, with care being taken not to occlude the bowel. The cecum was then subjected to a single through-and-through perforation with a 23-gauge needle. After repositioning the bowel, the abdominal incision was closed in layers with plain gut surgical suture 4-0 (Ethicon, Somerville, NJ) and metallic clips. Sham animals underwent the same procedure except for ligation and puncture of the cecum. Before and after surgery, animals had unlimited access to food and water. After an interval of up to 36 hours, lung injury was induced either by intratracheal instillation of 100 μg of bacterial LPS (Sigma Chemical Co., St. Louis, MO) or by intrapulmonary deposition of IgG immune complexes as described elsewhere. 12,13 For the latter, 10 mg of bovine serum albumin (BSA; Sigma Chemical Co.) were given intravenously (with trace amounts, 0.5 μCi, of 125I-labeled BSA) after intratracheal administration of 2.5 mg of polyclonal rabbit anti-BSA IgG (Organon Teknika Corp., West Chester, PA) in a total volume of 300 μl. In the LPS model of lung injury, 125I-labeled BSA was also given intravenously. Animals were sacrificed 6 hours after instillation of LPS and 4 hours after IgG immune-complex-induced alveolitis. These intervals selected for sacrifice represent times of peak lung injury, as determined in previous experiments. 12,13 The pulmonary circulation was then flushed with 10 ml of phosphate-buffered saline (PBS). Lung vascular permeability indices were determined by the ratio of extravasated 125I-labeled BSA present in lung parenchyma to the amount present in 1.0 ml of blood obtained from the posterior vena cava at the time of sacrifice. In other sets of animals, bronchoalveolar lavage (BAL) fluids were collected at the times indicated, using repetitive (three times) instillation and withdrawal of 5 ml of saline via an intratracheal cannula. After addition of a protease inhibitor cocktail (1 μg/ml leupeptin, 1 μg/ml aprotinin, 10 μg/ml trypsin inhibitor, and 1 μg/ml pepstatin), samples were centrifuged at 3000 rpm for 10 minutes, and supernatant fluids were subsequently used for chemokine quantitation. Cell pellets from centrifuged BAL fluids were assessed for differential cell counts. 14 Animals receiving antibody treatment were injected intravenously at the time of the CLP procedure with 300 μg of goat anti-rat C5a antibody (purified and characterized as described previously 15 ) or with 300 μg of preimmune goat IgG, similar to an earlier protocol. 15 For another set of experiments, anti-C5a was administered at 6, 12, 18, or 24 hours after CLP. Complement depletion was performed by three serial intraperitoneal injections of 25 U of purified cobra venom factor (CVF) at 12-hour intervals as described elsewhere. 12 In some animals, a carotid artery catheter (PE-50, Becton-Dickinson Co., Sparks, MD) was placed through an anterior cervical incision and, after subcutaneous tunneling, externalized at the posterior neck. The animals were allowed to recover for 24 hours with free access to food and water and then subjected to the CLP procedure. Blood samples were obtained in 12-hour intervals and subsequently used for measuring serum hemolytic complement activity (CH 50 assay) as reported. 15 For each group, n ≥ 4.
Measurement of Lung Vascular Intercellular Adhesion Molecule-1 and Quantitation of Cytokines
Quantitation of lung vascular intercellular adhesion molecule (ICAM)-1 was determined by binding of an 125I-labeled antibody to ICAM-1 (1A29, a gift from Dr. M. Miyasaka, Osaka University, Japan) as described in previous studies. 16 The ELISA techniques for measurement of cytokine-induced neutrophil chemoattractant (CINC) and macrophage inflammatory protein (MIP)-2 in rat BAL fluids are described elsewhere. 14 Cell culture supernatants and BAL fluids were evaluated for TNF-α activity using a standard WEHI cell cytotoxicity assay as previously reported. 17
Isolation and In Vitro Stimulation of Alveolar Macrophages
Alveolar macrophages were isolated at the times indicated by BAL of lungs of anesthetized rats of CLP or sham-treated groups. After centrifugation of lavage fluids, cells were resuspended in culture medium and plated into 48 microtiter well plates (Corning, New York, NY) at a concentration of 1 × 10 6 cells/well. Immune complexes were formed by addition to anti-BSA of BSA at the point of antigen equivalence. Complexes were added to wells over a concentration range of 0.8 to 100 μg/ml. After an incubation period of 4 hours at 37°C, supernatant fluids from macrophages were collected and evaluated for chemokine content.
Statistical Analysis
All values were expressed as mean ± SEM. Significance was assigned where P < 0.05. Data sets were analyzed employing one-way analysis of variance, and individual group means were then compared with the Student-Newman-Keuls multiple comparison test. To calculate percentage change between groups, values obtained from negative controls were subtracted from each data point.
Results
Clinical Evidence of Sepsis after CLP
Animals subjected to CLP developed the typical clinical signs of sepsis: decreased physical activity, piloerection, cessation in grooming behavior, glazed eyes with crusting exudate, diarrhea, lethargy, and occasionally death. The overall mortality rate 36 hours after the CLP procedure under the conditions used (two punctures with a 23-gauge needle) was 12%. Bacteremia was regularly detectable in blood samples drawn 36 hours after CLP (data not shown). To assess the extent of systemic complement activation, serial blood samples were collected in 12-hour intervals via an intra-arterial catheter and examined for serum complement hemolytic activity (using sensitized sheep erythrocytes). Absorbance values (541 nm) were determined and values expressed as percentage of the hemolytic activity present in serum obtained before CLP. At 12 hours after induction of sepsis, complement hemolytic activity was reduced to 86.3 ± 4.4% (P < 0.05) of that found in pre-sepsis serum, with a further decline at 24 hours and 36 hours, to 69.5 ± 5.7% and 68.8 ± 5.6% (for both, P < 0.05), respectively (Figure 1) ▶ . Thus, consumptive depletion of complement occurred in animals undergoing CLP.
Figure 1.
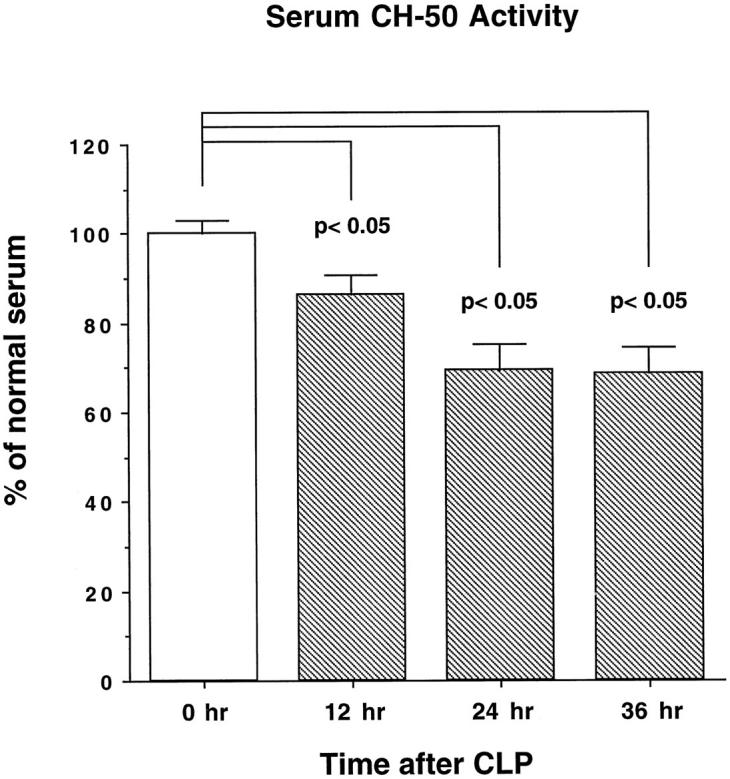
Serum complement hemolytic activity (CH50) of animals undergoing CLP-induced sepsis. Blood samples were collected via an intra-arterial catheter. Sensitized sheep erythrocytes were exposed to various dilutions of serum samples, and hemolysis was detected with a spectrophotometer at 541 nm. For easier comparability, absolute values of the measured absorbance were calculated to the percentage of normal serum (100%); n = 4. See text for details.
Enhanced Lung Injury during Sepsis
The extent of lung injury was assessed in septic and nonseptic animals after a direct intrapulmonary insult (4 hours after deposition of IgG immune complexes or 6 hours after airway instillation of LPS). Lung vascular permeability relative to time and experimental manipulation is shown in Figure 2 ▶ . The vascular permeability index, as determined by the ratio of 125I-labeled albumin in lung and blood, rose from a value of 0.21 ± 0.02 in sham-operated animals to 0.46 ± 0.06 in nonseptic rats with immune-complex-induced alveolitis. Compared with the sham control groups, there was no detectable evidence of increased lung permeability 36 hours after onset of CLP in the absence of a primary intrapulmonary insult (CLP, 36 hours, Figure 2 ▶ ), the permeability index being 0.25 ± 0.02. Lung injury caused by a direct intrapulmonary insult (immune complexes) 12 and 24 hours after induction of sepsis was associated with permeability indices of 0.39 ± 0.03 and 0.41 ± 0.01, respectively, which statistically were not different from the values in the nonseptic group receiving immune complexes alone. However, in CLP animals at 36 hours, immune-complex-induced lung injury resulted in a permeability index increase by 106% to a value of 0.73 ± 0.1 (P < 0.05 when compared with injury caused by immune complex deposition alone). Increased lung injury was also found in septic (CLP) animals receiving airway instillation of LPS. Intratracheal instillation of 100 μg of LPS into normal animals resulted in a permeability index of 0.34 ± 0.02 (as compared with the value obtained in sham lungs receiving PBS, 0.22 ± 0.02). At 36 hours after CLP in animals receiving LPS, the permeability index rose by 105% (P < 0.05) to a value of 0.47 ± 0.02 (Figure 2) ▶ . Thus, in animals receiving a direct intrapulmonary insult, sepsis caused an increased lung permeability index.
Figure 2.

Enhanced lung vascular leakage 12 to 36 hours after CLP in the presence or absence of a direct lung insult (intrapulmonary deposition of IgG immune complexes (IC) or intratracheal instillation of LPS). The lung vascular permeability index, referred to in this and in subsequent figures as vascular leakage, was assessed by measuring the extravasation of intravenously administered 125I-labeled BSA for 4 hours in the immune complex model and 6 hours in the LPS model. Open bars represent negative control groups; hatched bars represent positive controls. Calculations of percent change were determined by first subtracting the negative control values from the positive control groups. See text for details. For this and all subsequent groups, n ≥ 5.
To determine whether complement was required for the sepsis-induced increase of vascular permeability after a direct intrapulmonary insult, animals were either complement depleted by serial intraperitoneal injections with CVF before induction of CLP or treated with anti-C5a blocking antibody (300 μg), which was infused intravenously just before induction of CLP. In complement-depleted, nonseptic animals with immune complex deposition, there was a significant decrease (by 54%, P < 0.05) in vascular leakage of albumin, with an injury index falling from 0.44 ± 0.04 to 0.32 ± 0.01 (Figure 3) ▶ . In the presence of sepsis, the direct intrapulmonary insult caused the vascular leakage index to rise to 0.74 ± 0.06, but the value fell by 50% (P < 0.05) to 0.47 ± 0.04 in complement-depleted rats and by 57% (P < 0.05) to 0.43 ± 0.08 in rats treated with anti-C5a (Figure 3) ▶ . As the vascular leakage indices of the complement-depleted and anti-C5a-treated groups did not statistically differ from the values obtained in the immune-complex-injured group in the absence of sepsis, the complement-depleting or blocking procedures completely abolished the injury-enhancing effects of sepsis in lung. To investigate when during sepsis in vivo blockade of C5a (with antibody) would be effective, treatment with anti-C5a was done 6, 12, 18, and 24 hours after CLP. The results are shown in Figure 4 ▶ . In this set of experiments the intra-alveolar deposition of IgG immune complexes resulted in a lung vascular permeability index of 0.47 ± 0.01, whereas the same direct intrapulmonary insult in septic animals caused a value of 0.76 ± 0.05. As expected, intravenous administration of anti-C5a at the time of CLP resulted in a complete abrogation of the enhancing effect of sepsis on direct intrapulmonary insult, with a vascular leakage index of 0.49 ± 0.07. Systemic infusion of anti-C5a at 6 hours proved to be as effective in inhibiting the priming effect of CLP on lung, with a value of 0.49 ± 0.02. Administration of anti-C5a at 12 hours led to significant but somewhat reduced protection from lung injury enhancement, with a permeability index of 0.57 ± 0.02 (P < 0.05, when compared with animals receiving both hits in the presence of nonspecific IgG). Delaying treatment with anti-C5a to 18 hours and 24 hours after CLP did not demonstrate protective effects in lung injury, with lung vascular leakage indices of 0.69 ± 0.02 and 0.7 ± 0.06 (for both, the P value was not significant when compared with the values in the positive control groups treated with preimmune IgG), respectively. Thus, C5a blockade was effective in suppressing the inflammatory-enhancing effects of sepsis only in the early stages of the sepsis model.
Figure 3.
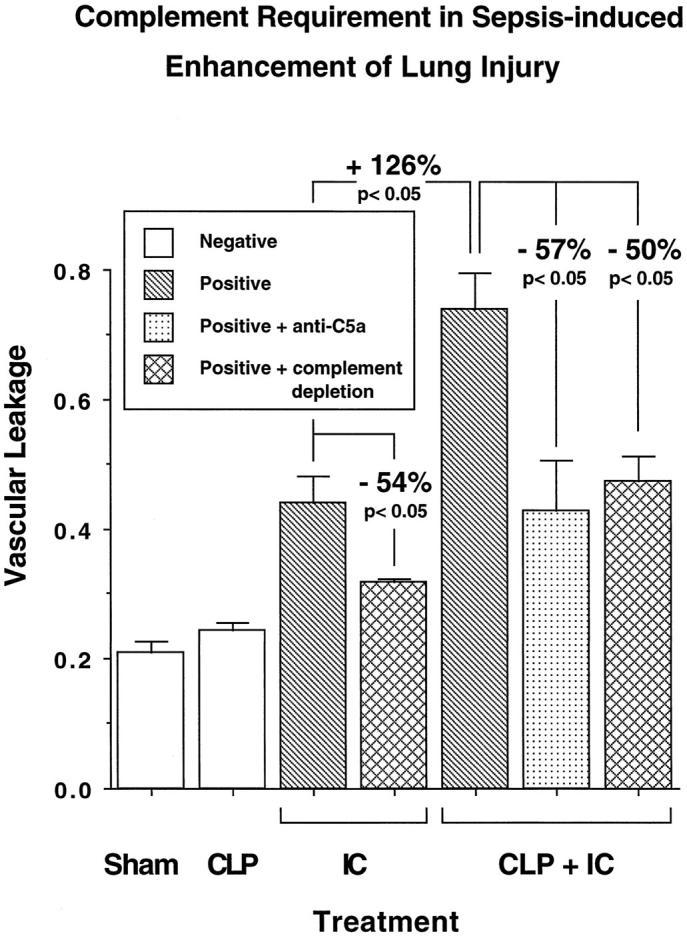
Complement requirements for enhanced lung injury in septic animals. Anti-C5a was administered intravenously at the time of CLP (36 hours before IgG immune complex-induced lung injury (IC)). Positive and negative control groups received preimmune IgG instead. Complement depletion was achieved by repetitive intraperitoneal injections of CVF before CLP.
Figure 4.
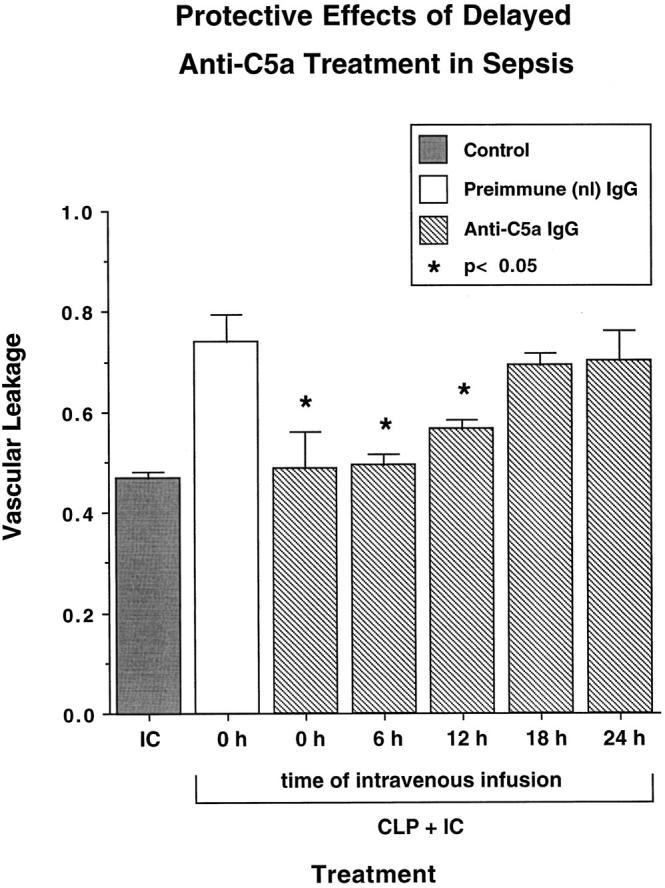
Protective effects of delayed anti-C5a treatment until after induction of sepsis. Animals were subjected to CLP, followed by a single intravenous administration of 300 μg of anti-C5a after indicated intervals. Control animals received 300 μg of preimmune goat IgG at the time of CLP. At 36 hours after CLP a direct pulmonary insult by intra-alveolar IgG immune complex (IC) deposition was induced, and lung vascular permeability was assessed 4 hours later by detection of extravasation of albumin (vascular leakage). In one control group, immune complexes were instilled intratracheally in the absence of sepsis. Statistical differences shown are in comparison with the septic group treated with immune complexes in the presence of preimmune IgG.
Enhanced Neutrophil Recruitment during Sepsis
As lung injury in the immune complex alveolitis model is neutrophil dependent, 12 we also investigated neutrophil content in BAL fluids from animals with CLP alone or CLP together with a direct lung injury induced by deposition of immune complexes. The enhancement of lung vascular permeability in septic animals receiving a direct intrapulmonary insult (described in Figures 2 and 3 ▶ ▶ ) was also reflected by increased numbers of neutrophils in BAL fluids (Figure 5) ▶ . The neutrophil counts (expressed as × 104/ml BAL fluid) were 24.3 ± 1.79/ml in sham-operated animals and 17.7 ± 1.20/ml in CLP animals at 36 hours. Immune complex deposition in lungs of otherwise normal animals caused BAL neutrophil values to rise to 40.7 ± 2.96/ml. However, 36 hours after CLP and 4 hours after immune complex deposition, there was a 3.4-fold increase in neutrophil content in BAL fluids rising to a value of 95.0 ± 16.5/ml (P < 0.05 when compared with BAL neutrophils after immune complex deposition in nonseptic animals), representing a 340% increase. In a companion set of experiments, BAL neutrophils were assessed in animals receiving intravenous administration of anti-C5a or preimmune goat IgG, each being given as 300 μg before CLP and induction of lung injury. Whereas the neutrophil content in BAL fluids from animals treated with preimmune goat IgG followed by CLP and immune-complex-induced alveolitis was 205 ± 22.6/ml, treatment of rats under the same protocol with anti-C5a led to a dramatic drop to a value of 101 ± 23.5/ml (P < 0.05). There was no statistically significant difference between BAL levels of neutrophils in nonseptic animals with immune complex deposition and in anti-C5a-treated septic animals with co-existent immune-complex-induced alveolitis (P > 0.05). Thus, CLP-induced sepsis at 36 hours was associated with greatly increased accumulation of neutrophils after exposure of lung to a direct insult. This, in turn, was followed by a significant increase in lung vascular permeability, both effects being completely suppressible by complement depletion or systemic treatment with anti-C5a.
Figure 5.

Neutrophil content in BAL fluids. BAL samples were centrifuged, and pellets were resuspended for differential cell counts. For matters of simplicity, data are expressed as percentage of values obtained in nonseptic positive controls. Absolute values are described in the text. As indicated, measurements were made 4 hours after initiation of IC reactions and 36 hours after initiation of CLP. When used, goat anti-C5a or preimmune goat IgG (each at 300 μg) was injected intravenously.
Morphological Features of Lungs
Morphological evaluation of lungs was performed. In keeping with vascular leakage indices and BAL neutrophil content (Figures 2 to 4) ▶ ▶ ▶ , normal lungs and lungs from CLP animals at 36 hours had normal morphological features (Figure 6, A and B ▶ , respectively). Lungs directly injured with immune complexes in nonseptic animals had the expected evidence of hemorrhage and neutrophil accumulation (Figure 6C) ▶ , whereas in the co-presence of CLP, the intensity of hemorrhage and neutrophil influx was accentuated (Figure 6D) ▶ .
Figure 6.
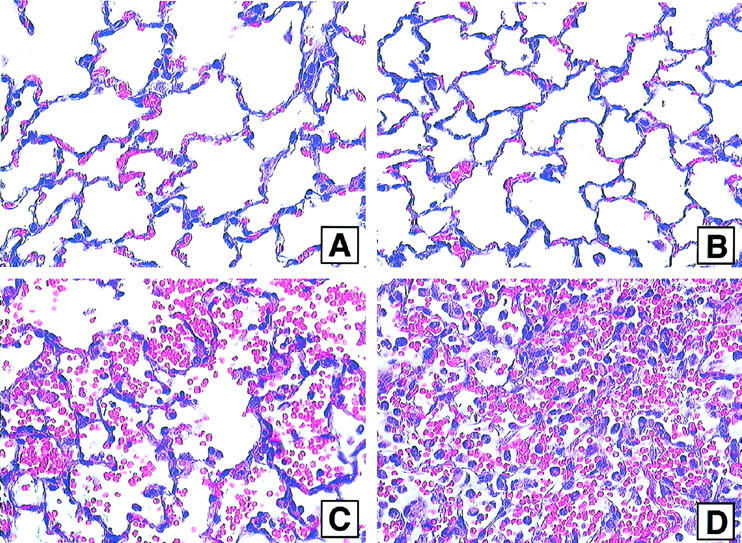
Lung sections obtained from animals undergoing sham surgery (A), CLP alone (B) or intra-alveolar deposition of IgG immune complexes alone (C) or in combination with CLP induced 36 hours earlier (D). H&E; magnification, ×40.
Enhanced CXC Chemokine Generation in Lungs of Septic Animals
To explore the causes for enhanced lung injury and increased neutrophil accumulation in septic animals subjected to a direct lung insult (immune complex deposition or LPS instillation), BAL fluids were assessed for content of the CXC chemokines MIP-2 and CINC. These CXC chemokines were selected because, in the setting of lung injury induced by LPS or deposition of IgG immune complexes, these chemokines have been shown to play a major role in neutrophil recruitment and development of lung injury. 13,14 MIP-2 levels were undetectable in BAL fluids from sham-operated animals and from animals 36 hours after CLP alone (Figure 7A) ▶ . In BAL fluids, direct lung injury induced by immune complex deposition resulted in MIP-2 content of 301 ± 30.9 ng/ml. Lungs of animals injured with immune complexes 36 hours after CLP had significantly increased (38%, P < 0.05) levels of MIP-2 in BAL fluids, 414.9 ± 12.5 ng/ml. This enhancement above and beyond levels found in immune-complex-injured lung of nonseptic animals was completely abolished when animals were pretreated with anti-C5a, causing MIP-2 levels to fall to 294 ± 15.9 ng/ml. Expression of CINC in BAL fluids was very low in sham-operated animals (12 ± 6.9 ng/ml) and undetectable in BAL fluids of CLP animals at 36 hours. In lungs from nonseptic rats after IgG immune complex deposition, CINC levels rose to 2060 ± 162 ng/ml. In animals 36 hours after CLP and with immune-complex-induced lung injury, CINC levels in BAL fluids were further increased (by 81%, P < 0.05) to 3726 ± 497 ng/ml (Figure 7B) ▶ . In companion rats, treatment with anti-C5a at the time of induction of sepsis also prevented the increased levels of CINC, reducing the values to 2131.2 ± 190.4 ng/ml BAL fluid.
Figure 7.

BAL content of CXC chemokines (A and B) and lung vascular ICAM-1 (C) 36 hours after CLP-induced sepsis in the presence or absence of an intrapulmonary insult (deposition of IC). Quantitation of chemokines was achieved using sandwich ELISA techniques; up-regulation of lung vascular ICAM-1 was determined by binding of 125I-labeled anti-ICAM-1 as described in the text. Sham-operated animals underwent surgery without CLP.
The same enhancing effects of CLP on pulmonary chemokine production were observed when LPS was used as a trigger of direct lung injury. In sham-treated rats or in CLP rats, BAL levels of MIP-2 and CINC were <13 ng/ml (Table 1) ▶ . When LPS was instilled into otherwise normal lungs, MIP-2 and CINC levels rose to 139 ± 6.2 and 1906 ± 128 ng/ml, respectively. The combination of LPS instillation into CLP animals treated intravenously with 300 μg of preimmune IgG caused increases in MIP-2 and CINC levels by 25% and 88%, respectively. In animals treated with anti-C5a, these increases in levels of CXC chemokines were diminished, such that the levels were the same as those found in otherwise normal rats with airway instillation of LPS. Thus, in CLP rats undergoing direct injury by LPS instillation, there was a C5a-dependent increase in MIP-2 and CINC, similar to findings in CLP animals undergoing direct lung injury by IgG immune complex deposition (Figure 7) ▶ .
Table 1.
BAL Content of Chemokines in LPS-Injured Lung
| Condition | MIP-2 | CINC | ||
|---|---|---|---|---|
| Sham | <1 ng/ml | 12± 6.9 | ||
| CLP | <1 ng/ml | <1 ng/ml | ||
| LPS | 139 ± 6 | 1906± 128 | ||
| P < 0.05 | P < 0.05 | |||
| CLP+ LPS, normal IgG (300 μg) | 271± 19 | 3566± 122 | ||
| NS | NS | |||
| CLP+ LPS, anti-C5a (300 μg) | 174± 12 | 2286± 33 |
Numbers represent nanograms of MIP-2 or CINC/ml in BAL fluids. For each condition, n = 4 animals. Animals with CLP were sacrificed at 36 hours. Animals receiving 100 μg of LPS intratracheally were sacrificed 6 hours later. ] ] ] ]
Levels of TNF-α in BAL Fluids
TNF-α is known to be expressed in immune-complex-induced lung injury and to regulate the expression of vascular ICAM-1 in the inflamed lung. 16 TNF-α levels were measured in BAL fluids in the presence or absence of CLP-induced sepsis and in the presence or absence of a direct lung insult (immune complexes). In agreement with other reports, 18,19 CLP alone did not cause increased levels of TNF-α in BAL fluids at 24 or 36 hours. The levels of TNF-α were similar to those in sham-operated animals, with values consistently below 1.5 ng/ml. The presence or absence of sepsis did not affect the levels of TNF-α in BAL fluids after direct lung injury. Immune-complex-induced lung injury in the presence or absence of sepsis was associated with BAL levels of TNF-α of 70.6 ± 4.7 and 60.7 ± 5.10 ng/ml, respectively (P value not significant). Thus, the enhancement of lung injury in septic rats cannot be linked to changes in BAL levels of TNF-α.
Enhanced Expression of Lung Vascular ICAM-1 in Lungs of Septic Animals
As lung vascular ICAM-1 is known to be involved in intrapulmonary recruitment of neutrophils, in a variety of conditions, we measured this adhesion molecule by lung vascular fixation of 125I-labeled anti-ICAM-1 (1A29, a murine IgG1). The binding index was calculated by the ratio of radioactivity in lung to radioactivity in blood, adjusted by subtraction of nonspecific binding values of irrelevant 125I-labeled control IgG1 (MOPC-21, Sigma Chemical Co.). Results are shown in Figure 7C ▶ . Sham-operated and animals with CLP alone (at 36 hours) demonstrated very small binding indices, 0.005 ± 0.002 and 0.004 ± 0.002, respectively (P value not significant). After immune-complex-induced lung injury in otherwise normal animals, the binding index rose nearly 10-fold to a value of 0.033 ± 0.007. In septic animals with lungs also subjected to the immune complex insult, there was a further increase (by 118%, P < 0.05) in the binding index for ICAM-1, rising to a value of 0.066 ± 0.006. Thus, sepsis sets the stage for accentuated up-regulation of lung vascular ICAM-1 in the presence of a direct intrapulmonary insult, although this is not reflected in changes in BAL levels of TNF-α. Increased lung vascular ICAM-1 expression is associated with intensified neutrophil recruitment and lung injury.
Generation of CXC Chemokines by Alveolar Macrophages from Septic and Nonseptic Rats
Alveolar macrophages were obtained from sham-operated and from septic rats (36 hours after CLP) and placed into tissue culture wells, with 1 × 10 6 cells/well. The resulting monolayers were then stimulated for 4 hours with increasing doses of IgG immune complexes (0.8 to 100 μg/ml). Cell culture supernatant fluids were assessed for chemokine content. As expected, there was dose-dependent expression of MIP-2 and CINC in cultures of alveolar macrophages obtained from sham-operated animals (Figure 8) ▶ . Macrophages from septic animals consistently demonstrated significantly higher levels of MIP-2 and CINC production when compared with macrophages from nonseptic animals, indicating that priming of these cells had occurred in vivo during sepsis (Figure 8, A and B) ▶ . These data are consistent with the results obtained in BAL fluids (Table 1 ▶ ; Figure 7 ▶ ). Macrophages from septic animals that had been complement depleted failed to show enhanced in vitro generation of CXC chemokines (Figure 8) ▶ , supporting the concept that complement is required for the in vivo priming process. These results extend the in vivo findings of enhanced pulmonary chemokine production in complement-intact, septic animals that have received a direct intrapulmonary insult. Complete abrogation of this enhancement occurred with C5a blockade.
Figure 8.

Dose responses for in vitro chemokine generation by rat alveolar macrophages obtained from sham-operated animals or 36 hours after CLP in the presence or absence of complement depletion. A total of 10 6 macrophages were plated in tissue culture wells followed by addition of a range of concentrations of immune complexes. Supernatant fluids were collected 4 hours later for ELISA analysis of MIP-2 and CINC. Results are shown using macrophages from CLP animals, from CLP animals with complement depletion, and from sham-operated animals. *Statistical difference between the CLP group and the two accompanying groups at the same dose of stimulus.
Discussion
To the extent that the CLP model of sepsis relates to the pathogenesis of events occurring in humans during sepsis, the data in the current report may have clinical relevance. In agreement with other reports, 18,19 under the sublethal conditions used, we did not detect evidence of an inflammatory response or morphological changes in lungs of animals with CLP-induced sepsis alone. However, after a direct intrapulmonary insult there was enhanced recruitment of neutrophils into lungs of septic animals, associated with significantly increased evidence of intrapulmonary injury. This was directly correlated with increased levels in BAL fluids of the CXC chemokines MIP-2 and CINC but not in BAL levels of TNF-α. The reason for enhanced levels of these CXC chemokines appears to be linked to the fact that the septic state primes alveolar macrophages for exaggerated chemokine responses. The in vivo priming effect of sepsis on lung was time dependent, as the increase in lung permeability index and in neutrophil accumulation were not detectable until 36 hours after CLP. An accentuated lung inflammatory response continued beyond 36 hours, as enhancement of lung injury after CLP was also seen at 48 hours (data not shown). All of these events were complement dependent and specifically required the role of C5a. In the face of complement depletion or blockade of C5a, all increments in chemokines, in neutrophil accumulation, and in lung vascular leakage of albumin in septic animals subjected to a direct intrapulmonary insult were abrogated. It is perhaps not too surprising that a role for C5a was found, as C5a plays a role in vivo in lung vascular ICAM-1 up-regulation of rats undergoing acute inflammatory injury after intrapulmonary deposition of IgG immune complexes. 15,16 Furthermore, in acute lung injury after systemic activation of complement, C5a has been shown to be critically required for up-regulation of lung vascular P-selectin and subsequent recruitment of neutrophils. 20 It seems likely that, in CLP-induced sepsis, C5a is produced locally (in the peritoneum) or systemically (in the plasma compartment), as described by Nakae et al, 21 and that it ultimately gains access to the distal airway compartment where it reacts with C5a receptors to bring about macrophage priming. These cells then become hyperresponsive to a subsequent stimulus. In the IgG immune-complex-induced lung injury model, a compartmentalized role for C5a has been demonstrated by the superior protective effects of intratracheally administered anti-C5a when compared with intravenous delivery of anti-C5a. 15 In the current report we hypothesize that C5a present in the plasma gains access to lung macrophages. This is supported by the finding of progressive systemic complement activation during sepsis (Figure 1) ▶ and by the ability of intravenously administered anti-C5a to block the priming phenomenon in lung during the first 12 hours of sepsis (Figure 4) ▶ .
There is precedent for the role of complement in the septic syndrome and in the response to the presence of bacteria or LPS. Dogs genetically deficient in C3 showed decreased clearance of LPS and enhanced shock and organ damage after infusion of LPS. 22 LPS-induced lethality was greatly increased in C4-deficient guinea pigs, 23 in C6-deficient rabbits, 24 and in mice that were made genetically deficient in C3 or C4. 25 Also, C3-deficient mice had delayed clearance of group B streptococci and poorer survival. 26 Curiously, C5-deficient mice seem resistant to the effects of LPS, a finding that was associated with a diminished TNF-α response. 27 In septic humans, nonsurvivors had significantly higher blood levels of C5a, 21 suggesting the failure to regulate generation of C5a. In LPS-infused rats, C5a blockade by antibody attenuated changes in hemodynamic parameters, 28 protected against secondary lung injury, and improved short-term survival. 29,30 C5-deficient mice had improved survival times after CLP. 31 Finally, in LPS-infused pigs, blockade of C5a reduced blood levels of IL-6 by 75%, although it was not apparent that other parameters were affected by this treatment. 32 All of these data suggest that, after LPS infusion and in some bacteremic and septic states, complement activation products may be produced in excess (or may not be adequately regulated), contributing to undesirable outcomes.
It was somewhat surprising to note that TNF-α levels in BAL fluids obtained from septic rats with immune-complex-injured lungs were the same as in immune-complex-induced lungs from sham-operated rats. This was in contrast to increased neutrophil accumulation occurring in septic rats (Figure 5) ▶ , as in earlier studies using the immune complex model a linkage was established between BAL levels of TNF-α and up-regulation of lung vascular ICAM-1. 16 However, recently it has been shown that, when TNF-α is present together with the complement-derived membrane attack complex (C5b-9), synergistic up-regulation in vitro of endothelial ICAM-1 and E-selectin occurs. 33 It is possible in the context of the in vivo model of lung injury in septic rats that such an interaction might be occurring, but this remains to be demonstrated. The in vivo relationship between C5a, TNF-α, and up-regulation of vascular ICAM-1 in the lung 15 might also suggest synergistic interactions in vivo between these mediators and lung vascular ICAM-1.
The priming of lung macrophages obtained from septic rats appears to be linked to a requirement for C5a based on the data in Figure 8 ▶ . Whether C5a can per se prime alveolar macrophages has not been evaluated. An alternate possibility might be that in septic rats there has been a C5a-dependent release in lung of cytokines such as TNF-α or IL-1, both of which are known to have the ability to prime phagocytic cells for enhanced responses to other stimuli. In the priming process biochemical events such as tyrosine phosphorylation and NF-κB activation are thought to be involved. 34 Again, whether C5a can induce tyrosine phosphorylation of proteins or whether it can cause NF-κB activation in the lung remains to be determined.
The priming of lung macrophages in the CLP model in rats as well as enhanced injury after a direct lung insult appear to be dependent on C5a. The advantage of direct blockade of C5a over other blocking strategies such as with anti-C5 is that anti-C5a may preserve the protective roles of C5b-9 in bacterial lysis. Protection against lung injury afforded by infusion of anti-C5a occurred even when infusion of anti-C5a was delayed (Figure 4) ▶ . This might be relevant in septic humans, providing a longer therapeutic window.
Acknowledgments
We thank Mrs. Robin G. Kunkel for preparation of lung tissue sections and Mrs. Beverly Schumann for secretarial support.
Footnotes
Address reprint requests to Dr. Peter A. Ward, Department of Pathology, University of Michigan Medical School, Room 7520 MSRB I, 1301 Catherine Road, Ann Arbor, MI 48109-0602. E-mail: pward@umich.edu.
Supported by National Institutes of Health grants GM-29507 and HL-31963.
References
- 1.Young LS: Gram-negative sepsis. Mandell GL Douglas SD Bennett JC eds. Principles and Practice of Infectious Diseases. 1979, :pp 571-608 John Wiley and Sons, New York [Google Scholar]
- 2.Bone RC: The sepsis syndrome: definition and general approach to management. Clin Chest Med 1996, 17:175-181 [DOI] [PubMed] [Google Scholar]
- 3.Kaplan RL, Sahn SA, Petty TL: Incidence and outcome of respiratory distress syndrome in gram-negative sepsis. Arch Intern Med 1979, 139:867-869 [PubMed] [Google Scholar]
- 4.Moore FA, Moore EE, Read RA: Postinjury multiple organ failure: role of extrathoracic injury and sepsis in adult respiratory distress syndrome. New Horizons 1993, 1:538-549 [PubMed] [Google Scholar]
- 5.Polk HC, Shields CL: Remote organ failure: a valid sign of occult intra-abdominal infection. Surgery 1977, 81:310-331 [PubMed] [Google Scholar]
- 6.Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, Schein RM, Sibbald WJ: Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Chest 1992, 101:1644-1655 [DOI] [PubMed] [Google Scholar]
- 7.Border JR, Hasett J, LaDuca J, Seibel R, Steinberg S, Mills B, Losi P, Border D: The gut origin sepsis state in blunt multiple trauma (ISS = 40) in the ICU. Ann Surg 1987, 206:427-448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wichtermann KA, Bauer AE, Chaudry IH: Sepsis and shock: a review of laboratory models and a proposal. J Surg Res 1980, 29:189-201 [DOI] [PubMed] [Google Scholar]
- 9.Walley KR, Lukacs NW, Standiford TJ, Strieter RM, Kunkel SL: Balance of inflammatory cytokines related to severity and mortality of murine sepsis. Infect Immun 1996, 64:4733-4738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ertel W, Morrison MH, Wang P, Zheng F, Ayala A, Chaudry IH: The complex pattern of cytokines in sepsis. Ann Surg 1991, 214:141-148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moore FA, Moore EE: Evolving concepts in the pathogenesis of multiple organ failure. Surg Clin North Am 1995, 75:257-277 [DOI] [PubMed] [Google Scholar]
- 12.Johnson KJ, Ward PA: Acute immunologic pulmonary alveolitis. J Clin Invest 1974, 54:349-357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmal H, Shanley TP, Jones ML, Friedl HP, Ward PA: Role for MIP-2 in LPS-induced lung injury in rats. J Immunol 1996, 156:1963-1972 [PubMed] [Google Scholar]
- 14.Shanley TP, Schmal H, Warner RL, Schmid E, Friedl HP, Ward PA: Requirement for the C-X-C chemokines: MIP-2 and CINC in IgG immune complex-induced lung injury. J Immunol 1997, 158:3439-3448 [PubMed] [Google Scholar]
- 15.Mulligan MS, Schmid E, Beck-Schimmer B, Till GO, Friedl HP, Brauer RB, Hugli TE, Miyasaka M, Warner RL, Johnson KJ, Ward PA: Requirement and role of C5a in acute lung inflammatory injury in rats. J Clin Invest 1996, 98:503-512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mulligan MS, Vaporciyan AA, Miyasaka M, Tamatani T, Ward PA: Tumor necrosis factor-α regulates in vivo intrapulmonary expression of ICAM-1. Am J Pathol 1993, 142:1739-1749 [PMC free article] [PubMed] [Google Scholar]
- 17.Espevik T, Nissen-Meyer J: A highly sensitive cell line, WEHI 164 clone 13, for measuring cytotoxic factor, tumor necrosis factor from human monocytes. J Immunol Methods 1986, 95:99-105 [DOI] [PubMed] [Google Scholar]
- 18.Ayala A, Perrin MM, Kisala JM, Ertel W, Chaudry IH: Polymicrobial sepsis selectively activates peritoneal but not alveolar macrophages to release inflammatory mediators interleukins-1 and -6 and tumor necrosis factor. Circ Shock 1992, 36:191-199 [PubMed] [Google Scholar]
- 19.McMasters KM, Cheadle WG: Regulation of macrophage TNF-α, IL-1β, and Ia (I-Aα) mRNA expression during peritonitis is site dependent. J Surg Res 1993, 54:426-430 [DOI] [PubMed] [Google Scholar]
- 20.Mulligan MS, Schmid E, Till GO, Hugli TE, Friedl HP, Roth RA, Ward PA: C5a-dependent up-regulation in vivo of lung vascular P-selectin. J Immunol 1997, 158:1857-1861 [PubMed] [Google Scholar]
- 21.Nakae H, Endo S, Inada K, Yoshida M: Chronological changes in the complement system in sepsis. Jpn J Surg. 1996, 26:225-229 [DOI] [PubMed] [Google Scholar]
- 22.Quezado ZM, Hoffman WD, Winkelstein JA, Yatsiv I, Koev CA, Cork LC, Elin RJ, Eichacker PQ, Natanson C: The third component of complement protects against Escherichia coli endotoxin-induced shock and multiple organ failure. J Exp Med 1994, 179:569-578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.May JE, Kane MA, Frank MM: Host defense against bacterial endotoxemia: contribution of the early and late components of complement to detoxification. J Immunol 1972, 109:893-895 [PubMed] [Google Scholar]
- 24.Johnson KJ, Ward PA: Protective function of C6 in rabbits treated with bacterial endotoxin. J Immunol 1971, 106:1125-1127 [PubMed] [Google Scholar]
- 25.Fischer MB, Prodeus AP, Nicholson-Weller A, Ma M, Murrow J, Reid RR, Warren HB, Lage AL, Moore FD, Rosen FS, Carroll MC: Increased susceptibility to endotoxin shock in complement C3- and C4-deficient mice is corrected by C1 inhibitor replacement. J Immunol 1997, 159:976-982 [PubMed] [Google Scholar]
- 26.Wessels MR, Butko P, Ma M, Warren HB, Lage AL, Carroll MC: Studies of group B streptococcal infection in mice deficient in complement component C3 or C4 demonstrate an essential role for complement in both innate and acquired immunity. Proc Natl Acad Sci USA 1995, 92:11490-11494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barton PA, Warren JS: Complement component C5 modulates the systemic tumor necrosis factor response in murine endotoxic shock. Infect Immun 1993, 61:1474-1481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smedegård G, Cui L, Hugli TE: Endotoxin-induced shock in the rat: a role for C5a. Am J Pathol 1989, 135:489-497 [PMC free article] [PubMed] [Google Scholar]
- 29.Stevens JH, O’Hanley P, Shapiro JM, Mihm FG, Satoh PS, Collins JA, Raffin TA: Effects of anti-C5a antibodies on the adult respiratory distress syndrome in septic primates. J Clin Invest 1986, 77:1812-1816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mohr M, Höpken U, Oppermann M, Mathes C, Goldmann K, Siever S, Götze O, Burchardi H: Effects of anti-C5a monoclonal antibodies on oxygen use in a porcine model of severe sepsis. Eur J Clin Invest 1998, 28:227-234 [DOI] [PubMed] [Google Scholar]
- 31.Olsen LM, Moss GS, Baukus O, Das Gupta TP: The role of C5a in septic lung injury. Ann Surg 1985, 202:771-776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Höpken UE, Mohr M, Strüber A, Montz H, Burchardi H, Götze O, Oppermann M: Inhibition of interleukin-6 synthesis in an animal model of septic shock by anti-C5a monoclonal antibodies. Eur J Immunol 1996, 26:1103-1109 [DOI] [PubMed] [Google Scholar]
- 33.Kilgore KS, Shen JP, Miller BF, Ward PA, Warren JS: Enhancement by the complement membrane attack complex of tumor necrosis factor-α induced endothelial cell expression of E-selectin and ICAM-1. J Immunol 1995, 155:1434-1441 [PubMed] [Google Scholar]
- 34.Bassal S, Liu YS, Thomas RJ, Philips WA: Phosphotyrosine activity in the macrophage is enhanced by lipopolysaccharide, tumor necrosis factor α, and granulocyte/macrophage-colony stimulating factor: correlation with priming of the respiratory burst. Biochim. Bioophys. Acta 1997, 1355:343-352 [DOI] [PubMed] [Google Scholar]