

Abstract
Prostacyclin synthase (PCS) is an enzyme with antithrombotic, antiproliferative, and dilatory functions in the normal vasculature, and inactivation of PCS by tyrosine nitration may favor atherosclerotic processes. Here, we show that PCS is nitrated and inactivated in early stage atherosclerotic lesions (focal intimal thickenings). Immunoprecipitation with antibodies raised against nitrotyrosine yielded PCS as the main nitrated protein in blood vessels. Moreover, we identified two nitrated degradation products of PCS with molecular mass of 30 and 46 kd, which were selective for atherosclerotic tissue. Agonist (acetylcholine, angiotensin II)-induced prostacyclin formation was decreased in atherosclerotic vessels compared with normal tissue, whereas PGE2 formation was increased and cyclooxygenase activity remained unchanged. A selective loss of PCS activity was confirmed by direct measurement of enzymatic activity. In line with this, we observed defective relaxation of early atherosclerotic vessels following vasoconstrictive stimulation. This functional impairment was completely reversed by coincubation with an antagonist of the thromboxane/PGH2 receptor but not by a thromboxane synthase inhibitor. These data suggest that reduced PCS activity in atherosclerotic arteries prevents the rapid use of PGH2, which accumulates and acts as an agonist on the vasoconstrictive thromboxane receptor.
The main enzyme systems responsible for the vasodilatory function of the endothelium are prostacyclin synthase (PCS) and nitric oxide synthase (NOS). Functional impairment of one or both of these enzymes may predispose to various diseases, including atherosclerosis. 1,2 In fact, impaired endothelial vasodilation is the predominant mechanism underlying impaired vasoconstriction that precedes the development of atherosclerosis. 3,4 Moreover, the normally vasodilatory response to acetylcholine is converted to a constrictor response in patients with angiographic evidence of atherosclerosis with risk factors for coronary diseases or with congestive heart failure. 5-7
This abnormality cannot be attributed to a decreased nitric oxide (NO) production alone. On the contrary, the inducible isoform of NOS (NOS-2) with high NO output capacity, is strongly expressed in atherosclerotic lesions. 8 The resulting enhanced NO production largely compensates for impaired endothelial function with respect to vasodilation and antiaggregation. However, it also implies the risk of ONOO− formation and nitration of proteins when the production of .O2− is increased in parallel. Protein nitration is in fact a conspicuous feature of atherosclerotic plaques in human tissue, 8-10 but the functional implications have remained unknown.
We have previously shown that isolated PCS is nitrated and inactivated by ONOO− at nanomolar concentrations. 11,12 Using normal aortic vessel strips we have subsequently shown that ONOO− exposure leads to nitration of PCS, associated with impaired PGI2 production and a defective vasorelaxation. 13 Here, we extended our studies by correlating PCS nitration, PCS-inactivation, prostanoid production, and relaxation responses in atherosclerotic lesions. In addition, these studies provide new evidence that PCS inactivation in atherosclerosis not only leads to a decreased prostacyclin formation but also to accumulation of the active vasoconstrictor PGH2.
Materials and Methods
Materials
N-ω-mono-methyl-l-arginine (L-NMMA), 1S[1α, 2B(5Z),3b,4α]-7-[3-[2(phenylamino) carbonyl] hydrazino methyl [-7-oxabicyclo(2.2.1)hept-2-yl-5-heptenoic acid (SQ29548), and the enzyme-linked immunoassay kits for 6-keto-PGF1α, PGE2, and PGF2α, were obtained from Cayman SPI (Massy, France). Imidazol [1,5α] pyridine-5-hexanoic acid (CGS13080) was from Ciba-Geigy (Basel, Switzerland). Embedding medium (OCT compound) was from Miles (Elkhart, IN). Protein-A-sepharose CL-4B was obtained from Pharmacia (Freiburg, Germany). A monoclonal antibody against bovine macrophage CD11b (Mac-1) was obtained from Serotec Inc. (Eching, Germany). A monoclonal antibody against 3-nitrotyrosine was purchased from Upstate Biotechnology Incorporated (Hamburg, Germany). Rabbit anti-PCS antisera were produced in this laboratory as described previously. 14 Secondary antibodies were from Camon (Wiesbaden, Germany), Dianova (Hamburg, Germany), and Pierce (Freiburg, Germany). The ECL kits and nitrocellulose membranes (Hybond-C) were purchased from Amersham (Braunschweig, Germany). Other chemicals not indicated were acquired from Sigma (Deisenhofen, Germany) or from Merck (Darmstadt, Germany).
Isolation and Classification of Bovine Coronary Arteries
Bovine hearts were obtained from the local slaughterhouse. The epicardial coronary arteries of the left ventricle were quickly dissected and placed in an ice-cold Krebs-Ringer solution consisting of 118 mmol/L NaCl, 4.7 mmol/L KCl, 2.5 mmol/L CaCl2, 1.2 mmol/L MgSO4, 1.2 mmol/L KH2PO4, 25 mmol/L NaHCO3, 0.016 mmol/L EDTA, and 11 mmol/L glucose. Arteries were classified as atherosclerotic and included into the experiments when they displayed a slight yellow discoloration on the inner surface of the artery but no significant reduction of the lumen. Random samples were further processed for histological observation (n = 10). All atherosclerotic arteries displayed focal intimal thickenings without any sign of necrosis or rupture of plaques when stained with hematoxylin/eosin. Immunocytochemistry demonstrated a subendothelial accumulation of CD11 positive cells and basal membrane splitting. This was not observed in normal control vessels.
Immunohistochemistry
Arteries were fixed in 4% paraformaldehyde and cryoprotected in 30% sucrose/0.1 mol/L sodium cacodylate buffer. Tissues were then embedded in precooled, OCT-containing cups, frozen in isopentane (−80°C), cut (9-μm sections), and mounted on poly-l-lysine coated slides. After air-drying, they were blocked for 45 minutes in 4% fatty acid-free bovine serum albumin with either 10% goat serum or 10% horse serum and 3% Triton X-100 in phosphate-buffered saline, pH 7.2. This was followed by coincubation of the monoclonal anti-nitrotyrosine antibody (15 μg/ml) and a polyclonal PCS antibody (5 μg/ml) overnight at 4°C. Some sections were stained with the nitrotyrosine antibody prepared in 10 mmol/L 3-nitrotyrosine in 0.1 mol/L phosphate-buffered saline. Antibody binding was then visualized by coincubation of fluorescein isothiocyanate-conjugated anti-mouse IgG (1:50 dilution) with anti-rabbit IgG conjugated to Texas Red (1:100 dilution) for 1 hour. Sections were washed and examined under a Leica (Leica, Wetzlar, Germany) fluorescent microscope. Images were recorded with a ×40 lens by confocal microscopy (Leica TCS 4D) or conventional photography with constant settings of illumination and exposure. Negative controls were performed by eliminating either of the primary antibodies or by incubating sections with a nonspecific primary antibody. Immunohistochemical staining for CD11b was performed after pretreatment of tissue with 10% methanol/3% H202 in phosphate-buffered saline. The primary antibody was incubated at 4°C overnight. Antibody binding was visualized by incubation with a biotin-coupled secondary antibody, an avidin-biotin peroxidase complex (Vector, Burlingame, CA), and development with diaminobenzidine.
Immunoprecipation of Nitrotyrosine-Containing Proteins
The vessels were homogenized in a buffer consisting of 50 mmol/L Tris, 1 mmol/L EDTA, 0.1 mmol/L butylated hydroxytoluene, 1 mmol/L phenyl-methyl-sulfonylfluoride (PMSF), pH 7.4, and centrifuged (12,000 × g) for 1 hour. Supernatants were diluted in 50 mmol/L Tris buffer containing 1 mmol/L phenyl-methyl-sulfonylfluoride, 5 mmol/L EDTA, 150 mmol/L NaCl, and 0.5% Nonidet-40 (pH 8.0). Solubilized extracts were sonicated for 30 seconds in a Branson Sonifier 250 (Schwäbisch Gmünd, Germany) at 50% duty cycle and centrifuged (14,000 × g) at 4°C for 5 minutes. Protein concentrations were determined using the Bradford assay (Biorad, Germany). Solubilized poteins (3 mg in 500 μl) were precleared by addition of 40 μl of protein A sepharose CL-4B and centrifugation. The supernatant was incubated (18 hours, 4°C) with 10 μg of monoclonal anti-nitrotyrosine antibody (Upstate Biotechnology). Immune complexes were precipitated (1.5 hours, 4°C) with 30 μl of protein A CL-4B and washed with 0.5 ml of SNNTE (0.5% sucrose, 1% Nonidet P-40, 0.5 mol/L NaCl, 50 mmol/L Tris, 5 mmol/L EDTA, pH 7.4). Protein pellets were then precipitated by centrifugation (14,000 × g, 1 minute), resuspended in 80 μl of Laemmli sample buffer, heated at 95°C for 5 minutes, separated by 7.5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and blotted onto a nitrocellulose membrane. The membrane was blocked with 5% milk powder in phosphate-buffered saline/0.1% Tween 20 for 2 hours at room temperature and incubated with one of different polyclonal antibodies directed against PCS (1 μg/ml) at 4°C overnight. After washing, the membrane was further incubated with a goat anti-rabbit antibody at a dilution of 1:7500 for 45 minutes. Antibody binding was visualized by the ECL technique according to the supplier’s instructions. Immunoprecipitates from the same experiments were separated by 7.5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and the gel was silver stained as described. 15
Isometric Measurement of Tension in Bovine Coronary Arteries
A carefully cut spiral (20 × 5 mm) from bovine coronary arteries was suspended on a force-displacement TFT6V5 transducer (W. Fleck GmbH, Mainz, Germany) and bathed in 15 ml of Krebs-Ringer buffer, supplemented with 100 μmol/L L-NMMA and gassed with 95% O2/5% CO2 (37°C, pH 7.4). The vessels were allowed to equilibrate for 45 minutes at an isometric pretension of 2 g. Stimulation was performed by addition of agonists to the bath solution and recording of a full cycle of contraction and relaxation. With all agonists, an initial rapid phase of contraction was followed by relaxation of the vessel. The relaxation in this experimental setup was largely dependent on PCS activity because it was blocked by the cyclooxygenase inhibitor indomethacin (10 μmol/L) or a selective PCS-inhibitor U51605 (5 μmol/L). Relaxation was quantitated as percent reversibility compared with the initial contraction. In normal tissue, relaxation was always complete or even over-compensating (ie, >100%).
Prostanoid Determination
After stimulation, the organ bath medium was collected, acidified with HCl (pH 3.5) and extracted with ethylacetate. Prostanoids were analyzed by enzyme-linked immunosorbent assay according to the suppliers instructions. For the measurement of 14C-PGH2 metabolic conversion, 100 μmol/L was added to the vessel homogenate in a volume of 1 ml. After 3 minutes the reaction was stopped by acidification to pH 3.5. After ethylacetate extraction (3 volumes) the organic phase was evaporated under nitrogen. The residues were resuspended in 60 μl of ethyl acetate and subsequently separated by TLC. The prostanoids were quantified with a phosphoimaging system (Image Quant, Molecular Dynamics) as described. 16
Results
Tyrosine Nitration
Immunoprocipitation of proteins from homogenates of normal and atherosclerotic bovine arteries with an anti-PCS antibody yielded similar amounts of PCS (smooth muscle enzyme plus endothelial enzyme). Restaining of the blots with an anti-3-nitrotyrosine antibody showed heavy nitration of PCS from atherosclerotic vessels compared with that from normal tissue (Figure 1A) ▶ . To examine to what extent protein tyrosine nitration was selective for PCS in coronary arteries, we immunoprecipitated vessel homogenates with an anti-nitrotyrosine antibody. Separation of precipitated proteins and silver staining showed one band at 52 kd, which was identified as PCS by immunoblot. In addition, nitrated proteins with molecular mass of 30 and 46 kd were immunoprecipitated selectively from atherosclerotic vessels. Both proteins were recognized by several different PCS antibodies in an immunoblot and may represent PCS degradation products, resulting from increased turnover of the enzyme in diseased arteries (Figure 1A) ▶ . Some nitrated PCS was also precipitated from homogenates of normal vessels possibly because of postmortem nitration of this extremely susceptible enzyme. 12,13 However, the nitrated PCS degradation bands were highly selective for atherosclerotic vessels.
Figure 1.
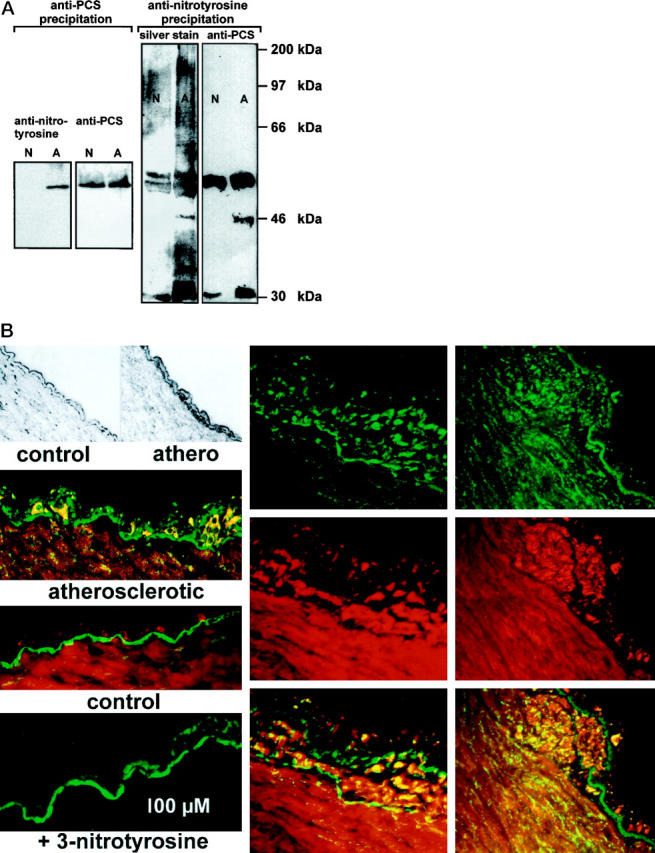
Nitrated protein and PCS in atherosclerotic bovine coronary arteries. A: Protein from normal (N) or atherosclerotic (A) bovine coronary arteries was immunoprecipitated either with an anti-PCS (left) or an anti-nitrotyrosine (right) antibody. Protein precipitated by anti-PCS was separated electrophoretically and immunostained with anti-nitrotyrosine or anti-PCS antibodies. Proteins precipitated by anti-nitrotyrosine were separated similarly and then either silver stained or analyzed by immunoblot with anti-PCS antibodies. Molecular mass of standard proteins run on the same gel are indicated. B: Top left, representative examples for normal (control) or early atherosclerotic vessels (athero) used in this study. Dark color in the thickened athero endothelium is due to CD11b immunoreactivity (macrophage marker); middle left, intimal thickening in atherosclerotic vessel that is strongly immunopositive for nitrotyrosine (green). PCS is stained in the intima and media (red). Below, control tissue is shown where nitrotyrosine staining is very weak and the endothelium is undisturbed. Bottom left, an intimal thickening was stained for nitrotyrosine in the presence of 10 mmol/L free 3-nitrotyrosine. Only the autofluorescence of the basal membrane is visible. Middle and right columns , more advanced lesions (top-green, protein-nitrotyrosine; middle-red, PCS; bottom, overlay image with yellow indicating colocalization). Nitrotyrosine is detectable also in the subintimal media below the strongly developed lesion shown to the right.
To gain further information on the localization of nitrated proteins in and around very early atherosclerotic lesions we used an immunohistochemical approach. Dense staining by anti-PCS was visible in the endothelium, and wave-like stainings were also seen in the vascular media. Weak nitrotyrosine immunoreactivity of the endothelium was sometimes observed in sections from normal vessels and was very pronounced in atherosclerotic lesions. At very early stages (mild intimal thickening) nitrotyrosine was found to be restricted mainly to the endothelial layer where it colocalized with PCS. More extended lesions were characterized by nitrotyrosine immunoreactivity both in the endothelium and in the adjacent subendothelial muscle layers (Figure 1B) ▶
Decreased PCS Activity
We tested whether the increased nitration of PCS in atherosclerotic vessels was associated with an altered prostanoid pattern. To minimize the effects of acetylcholine-induced nitric oxide production, 100 μmol/L L-NMMA was added before the stimulation with agonists. Stimulation of normal tissue with angiotensin II, acetylcholine, or arachidonic acid triggered a strong production of prostacyclin (PGI2) as measured by the accumulation of its degradation product 6-keto-PGF1α. In atherosclerotic vessels, 6-keto-PGF1α production was severely decreased. In parallel, production of PGE2, another metabolite of PGH2, was increased (Table 1) ▶ . The total amount of prostanoids formed was not significantly different in normal and atherosclerotic tissue and the amount of cyclooxygenase as determined by immunoblot was unchanged (not shown). The shift of the prostanoid pattern away from PGI2 was directly corroborated by measurements of PCS activity in vessel homogenates, using the cyclooxygenase product PGH2 as substrate. Also here the ratio of 6-keto-PGF1α/PGE2 formation was reversed in atherosclerotic vessels as compared with control tissue (Table 1) ▶ .
Table 1.
Shift of Prostanoid Synthesis Pattern in Atherosclerotic Vessels Because of Reduced Prostacyclin Synthase Activity
| Stimulus/substrate | Atherosclerotic | Metabolites formed: | ||
|---|---|---|---|---|
| 6-keto-PGF1α | PGE2 | PGF2α | ||
| Acetylcholine† | − | 121 ± 43 | 47 ± 13 | 31 ± 8 |
| (100 nmol/L) | + | 48 ± 34* | 129 ± 64* | 41 ± 11 |
| Angiotensin II† | − | 113 ± 61 | 57 ± 34 | 28 ± 11 |
| (50 nmol/L) | + | 43 ± 27* | 108 ± 38* | 45 ± 21 |
| Arachidonic acid† | − | 167 ± 67 | 115 ± 58 | 33 ± 8 |
| (10 μmol/L) | + | 63 ± 27* | 188 ± 54* | 45 ± 15 |
| 14C-PGH2‡ | − | 75 ± 11 | 19 ± 11 | 4.5 ± 0.9 |
| (100 μmol/L) | + | 31 ± 7* | 55 ± 14* | 5.4 ± 2.1 |
† Arteries were exposed to the stimuli for 30 minutes in an organ bath in the presence of 100 μmol/L N-methylarginine to inhibit NO formation. Prostanoids were extracted from the bath solution and quantitated by enzyme-linked immunosorbent assay. Data [in pg/mg wet tissue] represent means ± SEM from 10 independent experiments. Prostanoid release in unstimulated tissue was below the detection limit.
‡ Radiolabeled PGH2 was added to homogenized arteries. The percentage of conversion into different metabolites was determined after 3 minutes of incubation. Data [in percentage of 14C-PGH2 conversion] represent means ± SEM from seven independent experiments.
* P < 0.01 (atherosclerotic versus normal tissue).
Impaired Vasorelaxation
Finally we tested the functional implications of decreased PCS activity in early atherosclerotic lesions. Stimulation of normal arterial strips with acetylcholine or angiotensin II resulted in a rapid contraction, followed by a PGI2-dependent relaxation to the original length or even beyond. This relaxation was significantly impaired in atherosclerotic vessels (Figure 2A) ▶ . A similar impairment of the relaxation phase was observed when the contraction-relaxation cycle was triggered with arachidonic acid or the prostanoid precursor PGH2 (Figure 2B) ▶ . A defective relaxation was reproduced in control vessels when PCS was inhibited (not shown). Conversely, the relaxation deficiency was fully corrected in atherosclerotic vessels by inhibition of the thromboxane/PGH2 receptor with the selective antagonist SQ-29548 (Figure 2C) ▶ . The thromboxane synthase inhibitor CGS-13080 was ineffective in this model system (Figure 2C) ▶ .
Figure 2.
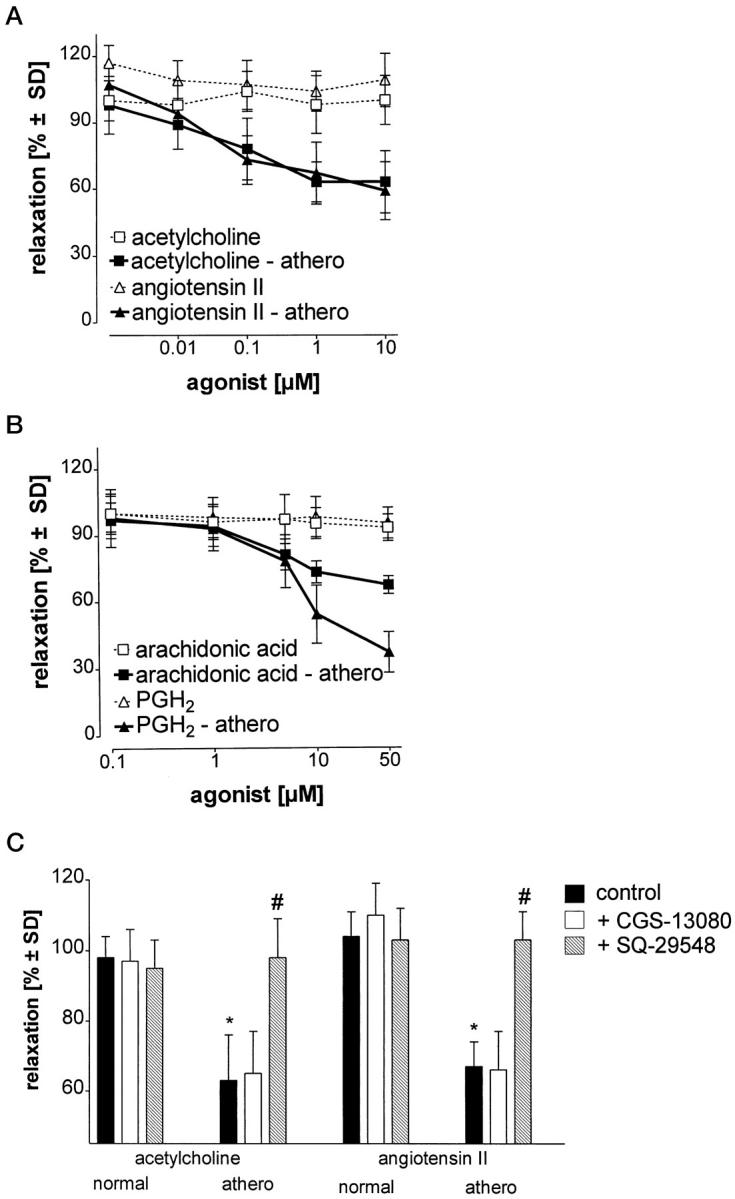
Defective relaxation of atherosclerotic aortic strips due to impaired PCS activity. A and B: Aortic strips were exposed to different concentrations of contractile stimuli. The extent of subsequent relaxation to the original length was quantitated in control tissue (open symbols) and atherosclerotic tissue (filled symbols). C: In some experiments, aortic strips were preincubated with CGS-13080 (10 μmol/L) or SQ-29548 (10 μmol/L) before stimulation with acetylcholine (10 μmol/L) or angiotensin II (10 μmol/L). Data represent means ± SD from five experiments. * P < 0.01 (atherosclerotic versus normal tissue); # P < 0.01 (SQ-29548 versus atherosclerotic).
Discussion
In the present study, we demonstrate that PCS is selectively nitrated in atherosclerotic vessels, and we provide new evidence suggesting that nitrative inactivation of PCS may contribute to the development of atherosclerotic lesions. Tyrosine nitration of PCS is most likely mediated by ONOO− formed endogenously from NO and superoxide anions, although contribution of additional mechanisms cannot be excluded. 17,18 The selectivity of ONOO− for PCS is explained by the exceptional sensitivity of PCS, which catalyzes its own nitration by ONOO−. 11 This exceptional sensitivity of PCS to nitration is further supported here by our findings that a basal amount of nitrated PCS was present in all normal coronary vessels, possibly because of rapid postmortem modification. This basal amount is strongly increased by direct treatment of the vessels with ONOO− (IC50 = 100 nmol/L) 13 or by triggering formation of endogenous ONOO− by lipopolysaccharide or hypoxia-reoxygenation (as shown in immunoprecipitation experiments with anti-nitrotyrosine antibodies; M. Zou, unpublished data). As shown here, the amount of nitrated PCS, and in particular of its 46- and 30-kd degradation products were also increased without experimental intervention in atherosclerotic vessels as compared with normal tissue.
All major cell types forming atherosclerotic lesions (endothelial cells, macrophages, and smooth muscle cells) may produce NO via NOS-3 or via cytokine-induced NOS-2. In addition, superoxide generation is increased by the activation of endothelial oxidases such as xanthine oxidase, NAD(P)H oxidase as well as by the contribution of oxidase systems from infiltrating leukocytes. Therefore, it is likely that relatively high steady state concentrations of ONOO− can be achieved in atherosclerotic lesions.
In the present study, we show that PCS is one of the main targets of ONOO− in atherosclerotic tissue. Nitration of the enzyme was associated with reduced enzymatic activity and, most importantly, with an impairment of vessel relaxation. Our results provide a potential explanation for the paradoxic effects of endothelium-dependent vasorelaxants such as acetylcholine, which trigger vasoconstriction in human or animal atherosclerotic arteries. 2-7 Even individuals with significant atherosclerotic risk factors but without clinically manifest atherosclerosis have a decreased vasodilator response in parallel with higher production of vasoconstricting prostaglandins. 2,6,19 Such abnormal responses are normalized by inhibition of cyclooxygenase. 2,6,16,19 Aspirin has no effect under normal conditions but improves/restores acetylcholine-mediated vasodilation in patients with atherosclerosis. 20
We have previously shown that treatment of normal vessels with ONOO− causes defective relaxation by a combination of two mechanisms: decreased production of vasorelaxant prostacyclin and accumulation of the vasoconstrictor PGH2, which stimulates the TxA2 receptor. Here we found that atherosclerotic vessels containing nitrated PCS display a similar functional behavior as arteries treated with ONOO−. These data suggest that endogenous nitration of PCS may contribute to the functional defects of the endothelium in pathological situations not only by a lack of the vasorelaxant prosta-cyclin but even more directly by causing accumulation of the prothrombotic and proconstrictive prostanoid PGH2 (Figure 3) ▶ .
Figure 3.

Regulation of proatherosclerotic events by arachidonic acid metabolites.
Acknowledgments
We thank Ms. Michelle Jendral for the technical support.
Footnotes
Address reprint requests to Dr. Volker Ullrich, University of Konstanz, Faculty of Biology, X910 - Sonnenbühl, D 78457 Konstanz, Germany. E-mail: volker.ullrich@uni-konstanz.de
Supported by DFG Forschergruppe We686/18-1 and Grant Ni518/2–1.
References
- 1.Ross R: The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature 1993, 362:801-809 [DOI] [PubMed] [Google Scholar]
- 2.Lüscher TF, Noll G: The pathogenesis of cardiovascular disease: role of the endothelium as a target and mediator. Atherosclerosis 1995, 118:S81-S90 [PubMed] [Google Scholar]
- 3.Selwyn AP, Kinlay S, Creager M, Libby P, Ganz P: Cell dysfunction in atherosclerosis and the ischemic manifestations of coronary artery disease. Am J Cardiol 1997, 79:17-23 [DOI] [PubMed] [Google Scholar]
- 4.Zeiher AM, Schächinger V: Coronary endothelial vasodilator dysfunction: clinical relevence and therapeutic implications. Z Kardiol 1994, 83:7-14 [PubMed] [Google Scholar]
- 5.Ludmer PL, Sewyn AP, Shook TL, Wayne RR, Mudge GH, Alexander RW, Ganz P: Paradoxical vasoconstriction induced by acetylcholine in atherosclerosis coronary arteries. N Engl J Med 1986, 315:1046-1051 [DOI] [PubMed] [Google Scholar]
- 6.Vita JA, Treasure CB, Nabel EG, McLenachan JM, Fish RD, Yeung AC, Vekshtein VI, Selwyn AP, Ganz P: Coronary vasomotor response to acetylcholine relates to risk factors for coronary artery disease. Circulation 1990, 81:491-497 [DOI] [PubMed] [Google Scholar]
- 7.Katz SD, Schwarz M, Yuen J, LeJemtel TH: Impaired acetylcholine-mediated vasodilation in patients with congestive heart failure: role of endothelium-derived vasodilating and vasoconstricting factors. Circulation 1993, 88:55-61 [DOI] [PubMed] [Google Scholar]
- 8.Buttery LDK, Springall DR, Chester AH, Evans TJ, Standfield N, Parums DV, Yacoub MH, Polak JM: Inducible nitric oxide synthase is present within human atherosclerostic lesions and promotes the formation and activity of peroxynitrite. Lab Invest 1996, 75:77-85 [PubMed] [Google Scholar]
- 9.Beckman JS, Ye YZ, Anderson G, Chen J, Accavitti MA, Tarpey MM, White CR: Extensive nitration of protein tyrosine in human atherosclerosis detected by immunohistochemistry. Biol Chem Hoppe-Seyler 1994, 375:81-88 [DOI] [PubMed] [Google Scholar]
- 10.Leeuwenburgh C, Hardy MM, Hazen SL, Wagner P, Oh-ishi S, Steinbrecher UP, Heinecke JW: Reactive nitrogen intermediates promote low density lipoprotein oxidation in human atherosclerotic intima. J Biol Chem 1997, 272:1433-1436 [DOI] [PubMed] [Google Scholar]
- 11.Zou M, Martin C, Ullrich V: Tyrosine nitration as a mechanism of selective inactivation of prostacyclin synthase by peroxynitrite. Biol Chem 1997, 378:707-713 [DOI] [PubMed] [Google Scholar]
- 12.Zou M, Ullrich V: Peroxynitrite formed by stimulaneous generation of nitric oxide and superoxide selectively inhibits bovine aortic prostacyclin synthase. FEBS Lett 1996, 382:101-104 [DOI] [PubMed] [Google Scholar]
- 13.Zou M, Jendral M, Ullrich V: Peroxynitrite induces prostaglandin endoperoxide-dependent vasospasm in bovine coronary arteries via tyrosine nitration of prostacyclin synthase. Br J Pharmacol 1999, 126:1283-1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siegle I, Nürsing R, Brugger R, Sprenger R, Zecher R, Ullrich V: Characterization of monoclonal antibodies generated against bovine and porcine prostacyclin synthase and quantitation of bovine prostacyclin synthase. FEBS Lett 1994, 347:221-225 [DOI] [PubMed] [Google Scholar]
- 15.Harlow E, Lane D: Antibodies: a laboratory manual. Edited by Harlow Lane. New York, Cold Spring Harbor Laboratory, 1988, pp 653
- 16.Klein T, Nürsing RM, Pfeilschifter J, Ullrich V: Selective inhibition of cyclooxygenase 2. Biochem Pharmacol 1994, 48:1605-1610 [DOI] [PubMed] [Google Scholar]
- 17.Sampson JB, Ye YZ, Rosen H, Beckman JS: Myeloperoxidase and horseradish peroxidase catalyze tyrosine nitration in proteins from nitrite and hydrogen peroxide. Arch Biochem Biophys 1998, 356:207-213 [DOI] [PubMed] [Google Scholar]
- 18.Eiserich JP, Hristova M, Cross CE, Jones AD, Freeman BA, Halliwell B, Van der Vilet A: Formation of nitric oxide-derived inflammatory oxidants by myeloperoxidase in neutrophils. Nature 1998, 391:393-397 [DOI] [PubMed] [Google Scholar]
- 19.Katz SD, Biasucci L, Sabba C, Strom JA, Jondeau G, Galvao M, Solomon S, Nikolic SD, Forman R, LeJemtel TH: Impaired endothelium-mediated vasodilation in the peripheral vasculture of patients with congestive heart failure. J Am Coll Cardiol 1992, 19:918-925 [DOI] [PubMed] [Google Scholar]
- 20.Husain S, Andrews NP, Mulcahy D, Panza JA, Quyyumi AA: Aspirin improves endothelial dysfunction in atherosclerosis. Circulation 1998, 97:716-720 [DOI] [PubMed] [Google Scholar]