

Abstract
Loss of the long arm of chromosome 4 has been identified previously as a common occurrence in adenocarcinomas of the esophagus and gastroesophageal junction by relatively low resolution genetic surveys. To better define the extent of 4q deletion in these neoplasms we isolated DNA from 29 primary carcinomas using microdissection, and used DNA obtained from xenografts of 14 carcinomas grown in immunodeficient mice in an assay of loss of heterozygosity of 25 polymorphic microsatellite markers distributed along the chromosomal arm. Two carcinomas exhibited widespread microsatellite instability and were excluded from deletion mapping. In the remaining 41 carcinomas, loss of heterozygosity was detected in 33 (80%). Twenty-three cancers showed complete or extensive reduction to homozygosity along the length of the long arm. Ten cancers had smaller discrete areas of loss and were principally useful in discerning three non-overlapping areas of consensus genetic deletion. Area 1 centered on marker D4S1534 at 4q21.1–22, area 2 centered on marker D4S620 at 4q32–33, and area 3 centered on marker D4S426 at 4q35. No known tumor suppressor genes map to these loci, but the frequent deletion of these areas in gastroesophageal carcinomas and in other carcinomas suggests that undiscovered tumor suppressor genes may reside here.
Adenocarcinomas of the esophagus and gastroesophageal junction are common human malignancies which have had an unexplained 350% increase in incidence over the past few decades. 1 Adenocarcinomas of the esophagus arise in a metaplastic change of the esophageal mucosal epithelial layer known as Barrett esophagus, in which the normal stratified squamous epithelium is replaced by mucinous epithelium of gastric and/or intestinal morphology. 2 There is evidence that tumors of the gastroesophageal junction arise in similar metaplastic epithelium, which has been termed “short segment Barrett esophagus” by some investigators. 3 Barrett esophagus is thought to be due primarily to uncontrolled reflux of gastric contents into the esophagus and is not strongly linked to the risk factors for metaplasia and carcinoma of the distal stomach. However, this etiology alone is insufficient to explain the rising incidence of this highly lethal form of cancer.
To better understand the pathogenesis of esophageal adenocarcinomas, the molecular genetic changes that occur in these neoplasms have been under active investigation. Only two specific changes, mutation of the p53 tumor suppressor gene and methylation inactivation of the p16 tumor suppressor gene, have been found to occur in the majority of these neoplasms. 4,5 However, there are other areas of consensus genetic loss which suggest other, as yet unidentified, tumor suppressor genes may be involved. The techniques of comparative genomic hybridization and microsatellite allelotyping have been used to survey these tumors for chromosomal deletion, and several chromosomes have been identified as areas of common deletion. 6-12 Among these is chromosome 4, which contains no known tumor suppressor gene. To determine the incidence of genetic deletion on the long arm of chromosome 4 in esophageal and gastroesophageal adenocarcinomas, and to define novel genetic loci of tumor suppression, we have analyzed primary tumor specimens and tumor xenografts for loss of heterozygosity (LOH) of polymorphic microsatellite markers.
Materials and Methods
Specimen Selection
Hematoxylin and eosin (H&E)-stained histological sections of surgical resection specimens of adenocarcinoma of the esophagus or gastroesophageal junction were examined. Thirty-six resected esophageal tumors were selected which were either confined to the distal esophagus or which involved the gastroesophageal junction with adjacent Barrett mucosa in the lower esophagus. After microdissection and DNA extraction, 7 cases were omitted due to the poor performance of material in polymerase chain reaction (PCR) assays, leaving 29 cases of primary tumors in our study.
Xenografts
Samples of fresh tumors were implanted subcutaneously into immunodeficient mouse strains, as described previously. 9 Following tumor growth and harvesting, tumor samples were cryostat sectioned and examined histologically for verification of growth of adenocarcinoma cells. Additional cryostat sections were processed for DNA using proteinase K-SDS extraction. Corresponding normal DNA was extracted from non-neoplastic gastric mucosa obtained from the surgical resection specimens.
Microdissection and DNA Preparation
For the primary carcinomas, a series of fresh 7-μm sections were made of archival formalin-fixed, paraffin-embedded tissue and mounted on plain glass slides. Depending on the growth pattern of the carcinoma, and the degree to which surrounding non-neoplastic tissue encroached on the tumor cells, samples of tumor were either microdissected manually using a scalpel or by laser capture microdissection. 13 Histological sections for manual microdissection were prepared as described previously, 14 while sections for laser capture microdissection were prepared as directed by the manufacturer of the apparatus (Arcturus Engineering Inc., Mountain View, CA). Genomic DNA was extracted from microdissected cells with a buffer containing nonionic detergent and proteinase K, as described previously. 14
Microsatellite Marker Selection
An initial panel of microsatellite markers was selected from the CHLC chromosome 4 sex averaged recombination minimization linkage map (GDB:4263357) to provide markers that spanned 4q at approximately 10 cM intervals. Additional makers were obtained from the Genome Database (http://www.gdb.org/) to provide more uniform coverage of markers based on cytogenetic location (directly assessed or inferred). The ordering of the markers was independently verified on the CEPH/Genethon chromosome 4 linkage map (GDB:1103650), the Marshfield chromosome 4 sex averaged linkage map (GDB:9800494), and the Stanford Human Genome Center YAC STS-content map of chromosome 4 (http://www-shgc.stanford.edu/Mapping/phys_map/Chr4YAC.html).
Polymerase Chain Reaction
MapPairs primers for chromosome 4 microsatellite markers were obtained from Research Genetics (Huntsville, AL). PCR amplification of microsatellite markers was performed in 20-μl reaction volumes using 10 μl of prepared genomic DNA, 2 units of Taq polymerase (Gibco/BRL), and appropriate primers at a final concentration of 0.3 mmol/L. Reactions were performed in the following buffer conditions: 67 mmol/L Tris-HCl (pH 8.8), 16 mmol/L (NH4)2SO4, 10 mmol/L β-mercaptoethanol, 4 mmol/L MgCl2, 5% formamide, 0.2 mmol/L dATP, 0.2 mmol/L dTTP, 0.2 mmol/L dGTP, 5.0 μmol/L dCTP, and 1.0 μCi of [α32P]dCTP. The reaction components were heated to 94°C for 5 minutes and cooled to 78°C before addition of the Taq polymerase and radiolabeled nucleotide. Amplification then proceeded for 40 cycles using these parameters: denaturation at 94°C for 30 seconds, annealing at 50°C for 30 seconds, extension at 72°C for 60 seconds. All reactions concluded with a final extension step of 72°C for 5 minutes.
Autoradiography and Interpretation of Loss of Heterozygosity
PCR amplification products were treated with 20 μl of stop buffer (99.5% formamide, 1 mmol/L EDTA, 0.05% bromophenol blue, 0.05% xylene cyanol) and heated to 98°C for 3 minutes. Normal and tumor PCR products were then loaded into adjacent lanes and electrophoresed on a 6% denaturing polyacrylamide gel. Completed gels were then dried and exposed to autoradiography film. Normal and tumor lanes were compared for each case, and tumors were designated at each marker as homozygous (non-informative), heterozygous with no loss, heterozygous having undergone allelic loss (LOH), or having undergone an allelic shift (microsatellite instability) based on visual inspection, using criteria previously outlined. 15 A tumor was classified as having undergone LOH at a particular locus only if the predominant band(s) associated with one allele showed a diminution in intensity of 50% or more in the tumor relative to normal. Assays exhibiting microsatellite instability were not used to score loss of heterozygosity. All assays were independently scored by two investigators (C.A.R. and C.A.M.). All assays scored as LOH were repeated for confirmation, as were assays in which there were differences in interpretation between the readers (<5% of assays) and cases which exhibited allelic imbalance which did not meet the criteria of clear cut LOH. For the latter cases, tissue microdissection and LOH analysis was repeated. Cases which continued to show imbalance (typically an increased level of one allele in the tumor sample with no diminution of the second allele) were scored as heterozygous for the purposes of this study.
Results
Tissue was microdissected and DNA extracted from 36 esophageal and gastroesophageal junction adenocarcinomas. Of these, 29 provided DNA of sufficient quality to amplify products in a panel of microsatellite PCR reactions. One primary carcinoma exhibited microsatellite instability in over half of the markers tested and was omitted from the analysis of deletion boundaries. Fourteen tumor xenografts of esophageal and gastroesophageal junction adenocarcinomas were successfully grown and harvested. One xenograft tumor displayed widespread microsatellite instability and was also dropped from analysis of deletion boundaries.
Both primary and xenografted tumors showed a high rate of genetic deletion on chromosome 4q. Of the 41 carcinomas remaining in the analysis, 33 (80%) showed reduction to homozygosity of at least one heterozygous microsatellite marker when tumor and normal DNA samples were compared (Figure 1) ▶ . Twenty three cancers showed complete or extensive reduction to homozygosity along the length of the long arm (Figure 2) ▶ . In the latter group several cancers exhibited a patchy pattern of chromosomal loss, with areas of retained heterozygosity punctuated by areas of LOH. Ten tumors had smaller discrete areas of loss.
Figure 1.
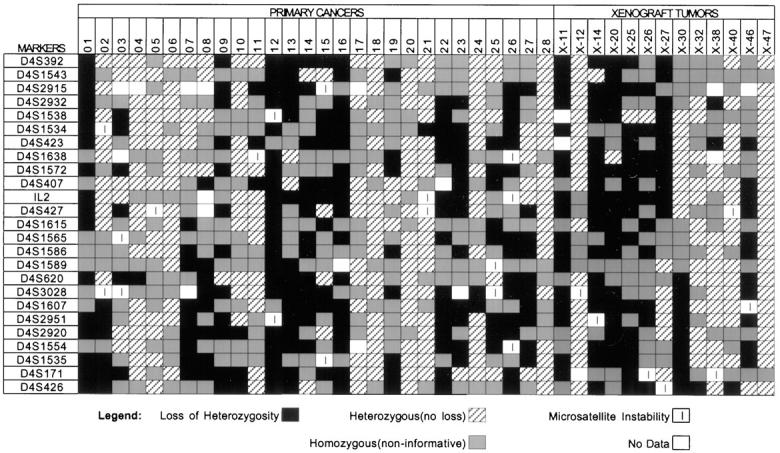
Results of microsatellite LOH analysis. The microsatellite markers are listed in the right hand column in sequential order, with the marker most proximal to the centromere (D4S392) on top. The results of the primary cancer cases (designated by number) are listed on the left 28 columns. The results of the xenograft tumors (designated with an X) are listed in the right 13 columns.
Figure 2.

Diagram of genetic loss on 4q. An ideogram of 4q is shown, with cytogenetic bands designated on the extreme left side. The location of the microsatellite markers is shown on the right side of the ideogram. Markers which have specific cytogenetic localization are shown with the darker horizontal lines connected to the vertical lines that show the limits of the localization. The extent of LOH for the cancers in this study is denoted by the vertical lines to the right of the chromosome, shown with the cancer designation immediately above the line. The beads on the lines show the location of LOH and are horizontally aligned with the marker name. The vertical lines are interrupted by areas on the chromosome with retained heterozygous markers. In the absence of informative markers, regions of loss have been joined to provide the most conservative inference of the extent of the deletions. The three regions of non-overlapping consensus areas, provided by localized deletions, are shown by the brackets and designated by number.
From the pattern of chromosomal loss, non-overlapping areas of consensus chromosomal deletion can be discerned (Figure 2) ▶ . Three areas defined by cancers with small isolated deletions were considered the strongest candidates for putative loci of tumor suppressor genes. Area 1 was defined by the isolated deletion in tumor 21 at D4S1534 and bounded by heterozygous markers at D4S423 and D4S1538. This corresponds to the cytogenetic location 4q21.1–4q22. In addition to tumor 21, two other cancers (11 and X38) also had deletions isolated to this portion of the chromosome with no deletion in other consensus areas. Examples of the microsatellite assays in this region are shown in Figure 3 ▶ . Five other tumors (3, 13, 15, 19, 25) with extensive but discontinuous patterns of LOH had deletions that included this locus. In total 13 of 23 cancers (56%) informative at this locus had LOH (Figure 1) ▶ , and 23 of the 41 cancers (56%) in this study had deletion boundaries that potentially involve this locus.
Figure 3.

Example of microsatellite PCR assays in deletion region 1. The markers are listed at the top of the diagram in relative order, with the centromeric position to the right. Autoradiographs of informative microsatellite PCR assays are shown in boxes to the right of the cancer designation (N, non-neoplastic tissue; T, tumor). Below the boxes the designation H stands for retention of heterozygosity, L stands for loss of heterozygosity in tumor samples. Arrows point to alleles lost in tumor samples. The horizontal lines below the results of each case designate the deletion boundaries inferred from the LOH results and correspond to those in Figure 2 ▶ . The deletion in case 21 is confined between markers D4S423 and D4S2538. The potential region of deletion overlap in this group of cancers is between markers D4S1534 and D4S1538.
The second area of consensus deletion is defined by the isolated deletion of marker D4S620 in tumor 4, and is bounded by heterozygous markers at D4S1586 and D4S1607. This corresponds to the cytogenetic location 4q32–33 (Figure 2) ▶ . Another cancer (24) showed a similar small, isolated deletion. Thirteen of 27 cancers (48%) informative at this locus had LOH, and 26 of the 41 cancers (63%) in this study had deletion boundaries that potentially involve this locus. Interestingly, 3 cancers with extensive deletion of this portion of the chromosome had retention of heterozygosity exclusively at D4S620 in an otherwise uniform area of deletion. Such a finding has been suggested to correspond to areas of homozygous deletion in impure tumor samples. 16 We investigated this possibility by meticulously procuring microdissected tumor specimens by laser capture microdissection and performed duplex PCR reactions of D4S620 and markers known to be retained in the tumors. We found no discernible differences between the amplification level of the D4S620 and the control markers (data not shown), a finding not compatible with the presence of a homozygous deletion in this area.
The third area of consensus deletion is present at the distal most portion of the chromosome, and was defined by minimal deletions in two tumors (X32 and X40) by our most distal marker D4S426. These deletions were bounded by heterozygosity at D4S1535; the corresponding cytogenetic location is 4q35 (Figure 2) ▶ . Another tumor (10) showed a similar isolated small deletion, and 2 tumors (3 and 15) with discontinuous patterns of LOH had isolated areas of deletion at this locus. In total, 16 of 26 cancers (62%) informative at this locus had LOH, and 22 of the 41 cancers (54%) in our study had deletion boundaries that potentially involved this locus.
Discussion
We had previously performed genetic surveys of gastroesophageal adenocarcinomas using comparative genomic hybridization and found that genetic loss on chromosome 4 was a common occurrence. 7 In the current study, we included primary tumors and tumor xenografts from our previous work, and enlarged the sample size with additional primary tumors to confirm with a second method, microsatellite allelotyping, that chromosome 4 deletions occur in 80% of such neoplasms. These results are in agreement with a previous lower resolution allelotyping study that detected chromosome 4 loss in more than half of esophageal adenocarcinomas. 8
We used a sufficient number of markers to map the extent of chromosomal deletion. In tumors exhibiting chromosome 4q deletion, over half showed total or near total loss of heterozygosity. However, the remaining neoplasms showed localized deletion or exhibited a patchy distribution of chromosomal loss. These localized deletions centered on three non-overlapping regions of the chromosome. This finding suggests that more than one tumor suppressor gene may be present on 4q.
There is precedence for multiple targets of tumor suppression on a single chromosomal arm. 18q loss is common in pancreatic and colorectal carcinoma. The tumor suppressor gene Smad4/DPC4 appears to be one target of inactivation by such deletions but does not explain all such genetic events. In the case of pancreatic carcinoma, only about half of all such deletions have been found to be explained as inactivating events of Smad4/DPC4. 17 Some of these deletions involve the putative tumor suppressor gene DCC, 18 but there may be other targets of genetic inactivation as well. In the case of colorectal carcinoma, Smad4/DPC4 is the target of only a minority of 18q deletions. 19,20 However, an adjacent tumor suppressor gene, Smad2/MADR2, has also been shown to undergo biallelic inactivation in colorectal tumors with 18q deletion. 19,21 Since there are 2000–5000 genes per chromosome, it is perhaps not unexpected that in some cases mutated tumorigenic loci will reside on the same chromosomal arm.
Some of the deletion regions we have identified in esophageal adenocarcinoma have been found in other neoplasms. Loss of chromosome 4 has been found in the majority of breast carcinomas, 22,23 hepatocellular carcinomas, 24,25 and several forms of squamous carcinoma. 26,27 A deletion mapping study of hepatocellular carcinomas showed multiple non-overlapping regions of deletion in the distal portion of the chromosome. 24 One of these consensus area of deletion centers on D4S620, which is contained in deletion area 2 of our study. Deletion of the same locus is correlated with the immortalization of human keratinocytes in tissue culture. 28 Since cellular immortalization is one characteristic of malignant transformation, it is possible that a gene involved in the regulation of cell senescence is a target of inactivation in cancers with genetic deletion at this locus.
The region we identified as deletion area 3 appears to play a role in human bladder carcinoma. One study found that loss of the distal portion of the chromosome (assayed by loss of D4S426, as in our study) occurred in 24% of tumors. 29 Loss of this region was associated with increased malignant behavior of the cancer, as it correlated with advanced clinical stage and high histological grade. The shared areas of genetic deletion among these cancers suggest that genes inactivated at these loci have regulatory effects on cell behavior in numerous states of differentiation.
The mapping of genetic deletions in tumor samples, xenografts and cell lines has been the major pathway to the finding of new tumor suppressor genes. The p16INK4a/CDKN2/MTS1, 30 Smad4/DPC4, 17 PTEN/MMAC1, 31,32 and PPP2R1B 33 genes were principally discovered in this way. Additionally, although the MEN1 gene was initially mapped by linkage analysis, it was the analysis of somatic deletions in the chromosomal region in neuroendocrine tumors that provided the map coordinates for the positional cloning of this gene. 15,34 The genetic loci identified in the current work have characteristics of tumor suppressor genes, given the high rate of loss in gastroesophageal carcinomas, but do not correspond to any known tumor suppressor genes. Additional mapping of these loci with subsequent positional cloning attempts are warranted to discover the genes inactivated by these deletion events. The elucidation of the biochemical properties of these gene products will be an important step in understanding the biology of these highly lethal and increasingly common neoplasms.
Footnotes
Address reprint requests to Chris Moskaluk, Dept. of Pathology, Box 214, University of Virginia Health Sciences Center, Charlottesville, VA 22908. E-mail: cam5p@virginia.edu.
Supported by National Cancer Institute Grant 5K08CA74431–02.
References
- 1.Devesa SS, Blot WJ, Fraumeni JF: Changing patterns in the incidence of esophageal and gastric carcinoma in the United States. Cancer 1998, 83:2049-2053 [PubMed] [Google Scholar]
- 2.Haggitt RC: Barrett’s esophagus, dysplasia, and adenocarcinoma. Hum Pathol 1994, 25:982-993 [DOI] [PubMed] [Google Scholar]
- 3.Hamilton SR, Smith RR, Cameron JL: Prevalence and characteristics of Barrett esophagus in patients with adenocarcinoma of the esophagus or esophagogastric junction. Hum Pathol 1988, 19:942-948 [DOI] [PubMed] [Google Scholar]
- 4.Wong DJ, Barrett MT, Stoger R, Emond MJ, Reid BJ: p16INK4a promoter is hypermethylated at a high frequency in esophageal adenocarcinomas. Cancer Res 1997, 57:2619-2622 [PubMed] [Google Scholar]
- 5.Souza RF, Meltzer SJ: The molecular basis for carcinogenesis in metaplastic columnar-lined esophagus. Gastroenterol Clin North Am 1997, 26:583-597 [DOI] [PubMed] [Google Scholar]
- 6.Huang Y, Boynton RF, Blount PL, Silverstein RJ, Yin J, Tong Y, McDaniel TK, Newkirk C, Resau JH, Sridhara R, Reid BJ, Meltzer SJ: Loss of heterozygosity involves multiple tumor suppressor genes in human esophageal cancers. Cancer Res 1992, 52:6525-6530 [PubMed] [Google Scholar]
- 7.Moskaluk CA, Hu J, Perlman EJ: Comparative genomic hybridization of esophageal and gastroesophageal adenocarcinomas shows consensus areas of DNA gain and loss. Genes Chromosomes Cancer 1998, 22:305-311 [PubMed] [Google Scholar]
- 8.Hammoud ZT, Kaleem Z, Cooper JD, Sundaresan RS, Patterson GA, Goodfellow PJ: Allelotype analysis of esophageal adenocarcinomas: evidence for the involvement of sequences on the long arm of chromosome 4. Cancer Res 1996, 56:4499-4502 [PubMed] [Google Scholar]
- 9.El-Rifai W, Harper JC, Cummings OW, Hyytinen ER, Frierson HF, Jr, Knuutila S, Powell SM: Consistent genetic alterations in xenografts of proximal stomach and gastro-esophageal junction adenocarcinomas. Cancer Res 1998, 58:34-37 [PubMed] [Google Scholar]
- 10.Dolan K, Garde J, Gosney J, Sissons M, Wright T, Kingsnorth AN, Walker SJ, Sutton R, Meltzer SJ, Field JK: Allelotype analysis of oesophageal adenocarcinoma: loss of heterozygosity occurs at multiple sites. Br J Cancer 1998, 78:950-957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moskaluk CA, Rumpel CA: Allelic deletion in 11p15 is a common occurrence in esophageal and gastric adenocarcinoma. Cancer 1998, 83:232-239 [PubMed] [Google Scholar]
- 12.Wu TT, Watanabe T, Heitmiller R, Zahurak M, Forastiere AA, Hamilton SR: Genetic alterations in Barrett esophagus and adenocarcinomas of the esophagus and esophagogastric junction region. Am J Pathol 1998, 153:287-294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Emmert-Buck M, Bonner R, Smith P, Chuaqui R, Zhuang Z, Goldstein S, Weiss R, Liotta L: Laser capture microdissection. Science 1996, 274:998-1001 [DOI] [PubMed] [Google Scholar]
- 14.Moskaluk CA, Kern SE: Microdissection and polymerase chain reaction amplification of genomic DNA from histological tissue sections. Am J Pathol 1997, 150:1547-1552 [PMC free article] [PubMed] [Google Scholar]
- 15.Emmert-Buck MR, Lubensky IA, Dong Q, Manickam P, Guru SC, Kester MB, Olufemi SE, Agarwal S, Burns AL, Spiegel AM, Collins FS, Marx SJ, Zhuang Z, Liotta LA, Chandrasekharappa SC, Debelenko LV: Localization of the multiple endocrine neoplasia type I (MEN1) gene based on tumor loss of heterozygosity analysis. Cancer Res 1997, 57:1855-1858 [PubMed] [Google Scholar]
- 16.Cairns P, Polascik TJ, Eby Y, Tokino K, Califano J, Merlo A, Mao L, Herath J, Jenkins R, Westra W, Rutter JL, Buckler A, Gabrielson E, Tockman M, Cho KR, Hedrick L, Bova GS, Isaacs W, Koch W, Schwab D, Sidransky D: Frequency of homozygous deletion at p16/CDKN2 in primary human tumours. Nat Genet 1995, 11:210-212 [DOI] [PubMed] [Google Scholar]
- 17.Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E, Weinstein CL, Fischer A, Yeo CJ, Hruban RH, Kern SE: DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996, 271:350-353 [DOI] [PubMed] [Google Scholar]
- 18.Hahn SA, Hoque AT, Moskaluk CA, da Costa LT, Schutte M, Rozenblum E, Seymour AB, Weinstein CL, Yeo CJ, Hruban RH, Kern SE: Homozygous deletion map at 18q21.1 in pancreatic cancer. Cancer Res 1996, 56:490-494 [PubMed] [Google Scholar]
- 19.Riggins GJ, Thiagalingam S, Rozenblum E, Weinstein CL, Kern SE, Hamilton SR, Willson JK, Markowitz SD, Kinzler KW, Vogelstein B: Mad-related genes in the human. Nat Genet 1996, 13:347-349 [DOI] [PubMed] [Google Scholar]
- 20.Thiagalingam S, Lengauer C, Leach FS, Schutte M, Hahn SA, Overhauser J, Willson JK, Markowitz S, Hamilton SR, Kern SE, Kinzler KW, Vogelstein B: Evaluation of candidate tumour suppressor genes on chromosome 18 in colorectal cancers. Nat Genet 1996, 13:343-346 [DOI] [PubMed] [Google Scholar]
- 21.Eppert K, Scherer SW, Ozcelik H, Pirone R, Hoodless P, Kim H, Tsui LC, Bapat B, Gallinger S, Andrulis IL, Thomsen GH, Wrana JL, Attisano L: MADR2 maps to 18q21, and encodes a TGFβ-regulated MAD-related protein that is functionally mutated in colorectal carcinoma. Cell 1996, 86:543-552 [DOI] [PubMed] [Google Scholar]
- 22.Schwendel A, Richard F, Langreck H, Kaufmann O, Lage H, Winzer KJ, Petersen I, Dietel M: Chromosome alterations in breast carcinomas: frequent involvement of DNA losses including chromosomes 4q and 21q. Br J Cancer 1998, 78:806-811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tirkkonen M, Johannsson O, Agnarsson BA, Olsson H, Ingvarsson S, Karhu R, Tanner M, Isola J, Barkardottir RB, Borg A, Kallioniemi OP: Distinct somatic genetic changes associated with tumor progression in carriers of BRCA1 and BRCA2 germ-line mutations. Cancer Res 1997, 57:1222-1227 [PubMed] [Google Scholar]
- 24.Piao Z, Park C, Park JH, Kim H: Deletion mapping of chromosome 4q in hepatocellular carcinoma. Int J Cancer 1998, 79:356-360 [DOI] [PubMed] [Google Scholar]
- 25.Chou YH, Chung KC, Jeng LB, Chen TC, Liaw YF: Frequent allelic loss on chromosomes 4q and 16q associated with human hepatocellular carcinoma in Taiwan. Cancer Lett 1998, 123:1-6 [DOI] [PubMed] [Google Scholar]
- 26.Bockmuhl U, Wolf G, Schmidt S, Schwendel A, Jahnke V, Dietel M, Petersen I: Genomic alterations associated with malignancy in head and neck cancer. Head Neck 1998, 20:145-151 [DOI] [PubMed] [Google Scholar]
- 27.Hampton GM, Larson AA, Baergen RN, Sommers RL, Kern S, Cavenee WK: Simultaneous assessment of loss of heterozygosity at multiple microsatellite loci using semi-automated fluorescence-based detection: subregional mapping of chromosome 4 in cervical carcinoma. Proc Natl Acad Sci USA 1996, 93:6704-6709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Loughran O, Clark LJ, Bond J, Baker A, Berry IJ, Edington KG, Ly IS, Simmons R, Haw R, Black DM, Newbold RF, Parkinson EK: Evidence for the inactivation of multiple replicative lifespan genes in immortal human squamous cell carcinoma keratinocytes. Oncogene 1997, 14:1955-1964 [DOI] [PubMed] [Google Scholar]
- 29.Polascik TJ, Cairns P, Chang WY, Schoenberg MP, Sidransky D: Distinct regions of allelic loss on chromosome 4 in human primary bladder carcinoma. Cancer Res 1995, 55:5396-5399 [PubMed] [Google Scholar]
- 30.Kamb A, Gruis NA, Weaver-Feldhaus J, Liu Q, Harshman K, Tavtigian SV, Stockert E, Day R, Johnson BE, Skolnick MH: A cell cycle regulator potentially involved in genesis of many tumor types. Science 1994, 264:436-440 [DOI] [PubMed] [Google Scholar]
- 31.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R: PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275:1943-1947 [DOI] [PubMed] [Google Scholar]
- 32.Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, Frye C, Hu R, Swedlund B, Teng DH, Tavtigian SV: Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet 1997, 15:356-362 [DOI] [PubMed] [Google Scholar]
- 33.Wang SS, Esplin ED, Li JL, Huang LY, Gazdar A, Minna J, Evans GA: Alterations of the Ppp2r1b gene in human lung and colon cancer. Science 1998, 282:284-287 [DOI] [PubMed] [Google Scholar]
- 34.Chandrasekharappa SC, Guru SC, Manickam P, Olufemi SE, Collins FS, Emmert-Buck MR, Debelenko LV, Zhuang Z, Lubensky IA, Liotta LA, Crabtree JS, Wang Y, Roe BA, Weisemann J, Boguski MS, Agarwal SK, Kester MB, Kim YS, Heppner C, Dong Q, Spiegel AM, Burns AL, Marx SJ: Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science 1997, 276:404-407 [DOI] [PubMed] [Google Scholar]