

Abstract
Keratinocyte growth factor (KGF) has been used successfully to prevent alveolar damage induced by oxygen exposure in rodents. However, this treatment was used intratracheally and before oxygen exposure, which limited its clinical application. In the present study, mice were treated with the recombinant human KGF intravenously before (days −2 and −1) or during (days 0 and +1) oxygen exposure. In both cases, lung damage was attenuated. KGF increased the number of cells incorporating bromodeoxyuridine (BrdU) in the septa and in bronchial epithelium of air-breathing mice but not of oxygen-exposed mice, indicating that the protective effect of KGF is not necessarily associated with proliferation. Oxygen-induced damage of alveolar epithelium and, unexpectedly, of endothelium was prevented by KGF treatment as seen by electron microscopy. We investigated the effect of KGF on different mechanisms known to be involved in oxygen toxicity. The induction of p53, Bax, and Bcl-x mRNAs during hyperoxia was to a large extent prevented by KGF. Surfactant proteins A and B mRNAs were not markedly modified by KGF. The anti-fibrinolytic activity observed in the alveoli during hyperoxia was to a large extent prevented by KGF, most probably by suppressing the expression of plasminogen activator inhibitor-1 (PAI-1) mRNA and protein. As PAI-1 −/− mice are more resistant to hyperoxia, KGF might act, at least in part, by decreasing the expression of this protease inhibitor and by restoring the fibrinolytic activity into the lungs.
Exposure to pure oxygen leads to diffuse alveolar damage and exudation of plasma into the alveolar space and respiratory distress. Alveolar leak occurs when both endothelial and epithelial cells are damaged, suggesting that these cells are crucial to maintain the alveolar-capillary barrier. 1
Keratinocyte growth factor (KGF) produced by mesenchymal cells has been shown to promote growth of different epithelia in vitro and in vivo and in particular of type II pneumocytes. 2-5 It acts on a receptor whose expression is limited to epithelial cells. 6 Administration of KGF is also known to increase the resistance to a variety of injuries induced by oxygen, chemotherapy, and irradiation. 4,5,7 Thus, intratracheal KGF administration before oxygen exposure protects rats from oxygen-induced lung injury. 4 The mode of action of KGF is, however, poorly understood. KGF treatment of rats does not increase the antioxidant defenses of the lung. 4 As it increases the number of alveolar and bronchiolar epithelial cells, this led to the suggestion that a larger capital of epithelial cells before the injury would increase the resistance to oxygen-induced injury. This interpretation is also in accord with the observations showing that in most cases KGF needs to be administered before the injury to be effective. Additional possible modes of action have been suggested by the study of cell lines in vitro, such as the decrease of apoptosis in epithelial cells, 8 an increase in the synthesis of surfactant proteins, 9 and an increase of plasminogen activator activity. 10
In the present study, we administered KGF intravenously (i.v.) before and after the beginning of oxygen exposure and examined different lung cellular and extracellular constituents. Protection of epithelial cells and, surprisingly, of endothelial cells was achieved when KGF was administered either before or after the beginning of exposure. As the proliferative effect of KGF is abolished in oxygen-breathing mice, this suggests that protection is not limited to proliferation only. We then examined the influence of KGF on the expression of other mediators involved in oxygen toxicity. We and others have previously shown that hyperoxic alveolar damage is associated with an increased expression of factors involved in cell death, such as p53, p21, bax, and bcl-x. 11-13 We found that the expression of these factors was attenuated in the lung of KGF-treated mice. As the absence of plasminogen activator inhibitor-1 (PAI-1) delays oxygen toxicity, 14 we also examined the induction of the plasminogen activators (urokinase-type (uPA) and tissue-type (tPA)) and PAI-1. Our results indicate that KGF might act, at least in part, by decreasing the production of PAI-1 during hyperoxic injury.
Materials and Methods
Mice
C57BL/6 female mice were purchased from Iffa Credo (Labresle, France) and bred in our animal facility for two generations. CBA/j female mice, purchased from Jackson Laboratories (Bar Harbor, ME), were used in experiments performed at Amgen (Thousand Oaks, CA). Experiments were performed with 2- to 3-month-old mice weighing 20 to 25 g.
Reagents
Recombinant human (rh)KGF expressed and purified in Escherichia coli was prepared at Amgen as previously described, 2 diluted with saline, and injected i.v. twice at 10 mg/kg/dose. Control mice received saline only.
Hyperoxia Exposure and in Vivo Treatment
Mice were placed in a sealed Plexiglas chamber and exposed to 100% oxygen as described. 15 Mice exposed to room air in the same conditions served as controls. Food and water were available ad libitum. Recombinant human KGF (total dose 20 mg/kg) was administered i.v. either on day −2 and day −1 or on day 0 and day 1 from the beginning of oxygen exposure. This study protocol was approved by the ethical committee on animal experiments (Office Vétérinaire Cantonal of Geneva). Pulmonary edema was determined by measuring the wet lung weight as previously described. 15
Light and Electron Microscopy
Lungs were fixed by the intratracheal instillation of glutaraldehyde (electron microscopy (EM)) or Carnoy (immunohistochemistry) under a 20-cm hydrostatic pressure. Sections across the hilar regions of left and right lobes were examined. To label cells in S phase, bromodeoxyuridine (BrdU, 100 mg/kg; Sigma Chemical Co., St. Louis, MO) was injected intraperitoneally (i.p.) 2 hours and 30 minutes before sacrifice. Nuclei of lung cells containing BrdU were detected by immunohistochemistry using alkaline-phosphatase-conjugated anti-BrdU (1:20; Boehringer Mannheim, Mannheim, Germany) and revealed with a fast red colorimetric kit (Dako, Copenhagen, Denmark). The immunostained sections were scanned with a high-sensitivity Phototonic Science Coolview color camera (Carl Zeiss, Oberkochen, Germany) as described previously. 14 The whole section was analyzed (20 fields) for the BrdU or for fibrin staining and the same number in the adjacent section for the control staining. Three to four different mice were analyzed for each condition, and the percentage of positive cells was calculated for each condition. Fibrin detection was performed with rabbit anti-mouse fibrin(ogen) IgGs 14 and analyzed as above. The results were expressed after having subtracted the values of the control staining (performed simultaneously without anti-BrdU or with the preimmune serum).
For EM, sections were embedded in Epon and then examined with a Philips CM10 (Philips SA, Zurich, Switzerland) 400 electron microscope at 70 kV. Quantification of the alveolar damage was performed in a section across the left lobe of four to six mice for each condition examined. Two blocks per mice were examined, and pictures (40 per block) were acquired in a randomized manner. Pictures (original magnification, ×2800) were projected and overlaid with a lattice of 84 bars and 168 points. All points over epithelial and endothelial interstitial cells as well as the extracellular interstitial space were counted by two different investigators. Corresponding volumes were evaluated by dividing the number of points by the number of intersections with the alveolar basal membrane. 16-18 To evaluate more precisely the epithelial injury, and in particular type I cell damage, we evaluated type I cell lesion by grade. These cells were considered damaged if they presented the following morphological characteristics: blebbing of the surface and/or disruption of the plasma membrane. For each EM image (25 per animal, n = 4)) an injury score was determined according to the following scale: 0, no injury; 1, 0% to 10% of the epithelial surface damaged; 2, 10% to 50%; and 3, >50% of the epithelial surface damaged. This score, established independently by two different investigators, compared the KGF-treated group (days 0 and 1) and the NaCl-treated group during oxygen exposure.
RNA Analyses
After removal, lungs were immediately frozen in liquid nitrogen and stored at −80°C. After homogenization in guanidium/thiocyanate, total RNA was isolated by cesium chloride centrifugation as described previously. 19 Nitrocellulose blots of total RNA were hybridized with the following complementary 32P-labeled UTP RNA probes (specific activity, 400 Ci/mmol; Amersham International, Little Chalfont, UK) corresponding to the complete coding sequences of human bax and bcl-x 11 (kind gifts of J. C. Martinou, Glaxo Wellcome, Geneva, Switzerland) and of mouse surfactant proteins A and B (SPA and SPB; kind gifts of J. A. Whitsett, Cincinnati, OH). RNAse protection assays were performed as described for detection of mouse uPA, tPA, and PAI-1 mRNA. 20,21 Quantification of RNAse protection assays and Northern blots was achieved by PhosphorImager analysis (Molecular Dynamics, Sunnyvale, CA) using the ImageQuant Software (Molecular Dynamics). To evaluate gel loading and membrane transfer, the blots were stained with methylene blue. These blots were analyzed by densitometry, and small differences in loading were normalized by the density of the 18 S rRNA bands. Results of the normalized mRNA abundance are expressed as the mean increase ± SD compared with control mice.
Detection of Internucleosomal DNA Fragmentation
Lungs were homogenized by polytronic disruption in PBS/10 mmol/L EDTA (10 mg of tissue in 0.5 ml). The homogenate was then centrifuged at 13,000 rpm for 20 minutes at 4°C. The supernatant was kept and treated for 30 minutes at 37°C with 20 μg/ml RNAse A and another 30 minutes with 200 μg/ml proteinase K. After three phenol/chloroform extractions, 1/10 sodium acetate 3 mol/L, pH 4.8, was added and DNA was precipitated with 1 vol of isopropanol. The pellet was centrifuged at 13,000 rpm for 10 minutes at 4°C, washed with 70% ethanol, dried, and resuspended in 10 mmol/L Tris, 0.1 mmol/L EDTA, pH 8.0. The samples were run in 1% agarose gel, and DNA fragmentation was revealed with ethidium bromide.
Protein Analysis
Lungs were homogenized at 4°C for 30 minutes in 1 ml of lysis buffer (50 mmol/L Tris, pH 8, 250 mmol/L NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS) containing 1 μg/ml aprotinin and 100 μg/ml phenylmethylsulfonyl fluoride (PMSF). After centrifugation at 15,000 × g, 40 μg of supernatant protein was loaded per lane onto 12% polyacrylamide gels and electrophoresed. Proteins were transferred to nitrocellulose membranes, and nonspecific binding was blocked with 5% nonfat milk in TBS (20 mmol/L Tris/HCl, 500 mmol/L NaCl, pH 7.5) overnight at 4°C. Membranes were washed with TBS/Tween (0.025%) and incubated for 1 hour at 20°C with a rabbit anti-mouse p53 (AB-5, Calbiochem, La Jolla, CA) in gelatin buffer (0.24% gelatin, 50 mmol/L Tris/HCl, pH 7.4, 5 mmol/L EDTA, 0.15 mol/L NaCl, 0.05% Tween 20) at a concentration of 2 μg/ml. For PAI-1 detection, the membranes were first preincubated with a goat anti-mouse IgG 5 for 30 minutes, washed three times with TBS-T, and then incubated for 1 hour at 20°C with a mouse anti-human PAI-1 in gelatin buffer. 22 Membranes were washed three times with TBS/Tween 0.025%. A goat anti-rabbit IgG or a goat anti-mouse horseradish-peroxidase-conjugated (Santa Cruz Biotech, Santa Cruz, CA) were used as second antibody at a concentration of 400 ng/ml. Membranes were washed again with TBS-T and revealed with an enhanced chemiluminescence detection reagent (ECL) kit, Amersham International) at room temperature and before being exposed to Hyperfilm-MP films (Amersham International). When necessary, results were quantified as described above using subsaturated emulsions on x-ray film. 11
Bronchoalveolar lavage (BAL) was performed by instilling into the trachea 3 ml of saline solution containing 0.2 mol/L EDTA, 5 mmol/L 2-iodoacetamide (Sigma), and 250 U/ml aprotinin (Bayer, Switzerland). BAL fluid was recovered immediately on ice under negative hydrostatic pressure, and the supernatant was collected after centrifugation. BAL protein concentration was determined by Bradford’s method, and zymography was performed as described. 23,24
Statistical Analysis
For each parameter measured or calculated, the values for all animals of the experimental groups were averaged, and the SD of the mean was calculated. The significance of differences between the values of one of the experimental groups and those of the control group was determined using either the unpaired Student’s t-test or with the nonparametric Mann-Whitney U test when indicated. For multiple group comparisons, one-way analysis of variance with post hoc Bonferroni correction was used. Significance level was set at P < 0.05
Results
KGF Prevents Alveolar Damage When Administered before and after Oxygen Exposure
Oxygen exposure increased markedly the lung weight secondary to alveolar leak. KGF administered i.v. prevented the increase in lung weight on the 4th day. Moreover, the efficacy of KGF administration on day 0 and on day 1 was similar as when given 2 days and 1 day before oxygen exposure (Table 1) ▶ .
Table 1.
Lung Weight of Mice Injected with Saline and KGF during Hyperoxia (90 Hours)
| Lung weight (g) | ||
|---|---|---|
| Air | Hyperoxia | |
| Saline | 0.17 ± 0.02 | 0.31 ± 0.10 |
| KGF (days 0 and 1) | 0.19 ± 0.01 | 0.24 ± 0.04* |
| KGF (days−2 and−1) | 0.21 ± 0.04* |
*P < 0.05 compared with saline.
Effect of KGF on the Proliferation of Alveolar and Epithelial Cells in Air- or Oxygen-Breathing Mice
As oxygen exposure is known to arrest cell growth and to block cells at the G1/S interface, 25 we explored the effect of KGF on cell proliferation during hyperoxia. BrdU was injected 2 hours and 30 minutes before sacrifice. Hyperoxia decreased significantly BrdU uptake within the lung compared with room air (Figure 1) ▶ .
Figure 1.

Mice were injected intraperitoneally with 100 mg/kg BrdU 2 hours and 15 minutes before sacrifice. BrdU uptake of the nuclei was revealed by immunohistochemistry, and quantification was performed. The whole section was analyzed (20 fields) for the BrdU staining and the same number in the adjacent section for the control staining. Three to four different mice were analyzed for each condition, and the percentage of positive cells was calculated for each condition. The results are the mean ± SD of three to four mice for each condition. KGF treatment given 72 hours before sacrifice increased significantly the number of positive cells in air-breathing mice. When mice were exposed to oxygen, the number of positive cells was diminished compared with air-breathing mice with (P < 0.0001) or without KGF treatment (P < 0.01).
In air-breathing mice, KGF treatment increased 20-fold the number of positive nuclei per field, whereas in oxygen-breathing mice, KGF did not increase significantly the number of positive nuclei (Figure 1) ▶ . This number was, however, still below the number of positive cells seen in air-breathing mice. These results demonstrate a marked reduction of cell proliferation during hyperoxia, which could not be reverted by KGF administration.
Effect of Hyperoxia and KGF on Different Lung Cellular and Extracellular Volumes: Evaluation by Semiquantitative Electron Microscopy
Exposure to oxygen leads after 72 hours to an increase of interstitial volume (cellular and extracellular). No change in epithelial or endothelial volume was seen at this time (Figure 2) ▶ . At 90 hours of exposure, marked alterations of epithelial and endothelial cells were observed (Figure 3) ▶ . The increase in interstitial volume was significant and concerned the extracellular volume. At this time, there was a significant loss in endothelial volume compared with control (P < 0.01; Figure 2 ▶ ).
Figure 2.

Morphometric analysis of lung volumes during hyperoxia. Results are the mean ± SD of five to six mice. Endothelial volume decreased and total interstitial volume increased (cellular and extracellular) significantly at 90 hours of hyperoxia (*P < 0.05), whereas epithelial volume did not change.
Figure 3.
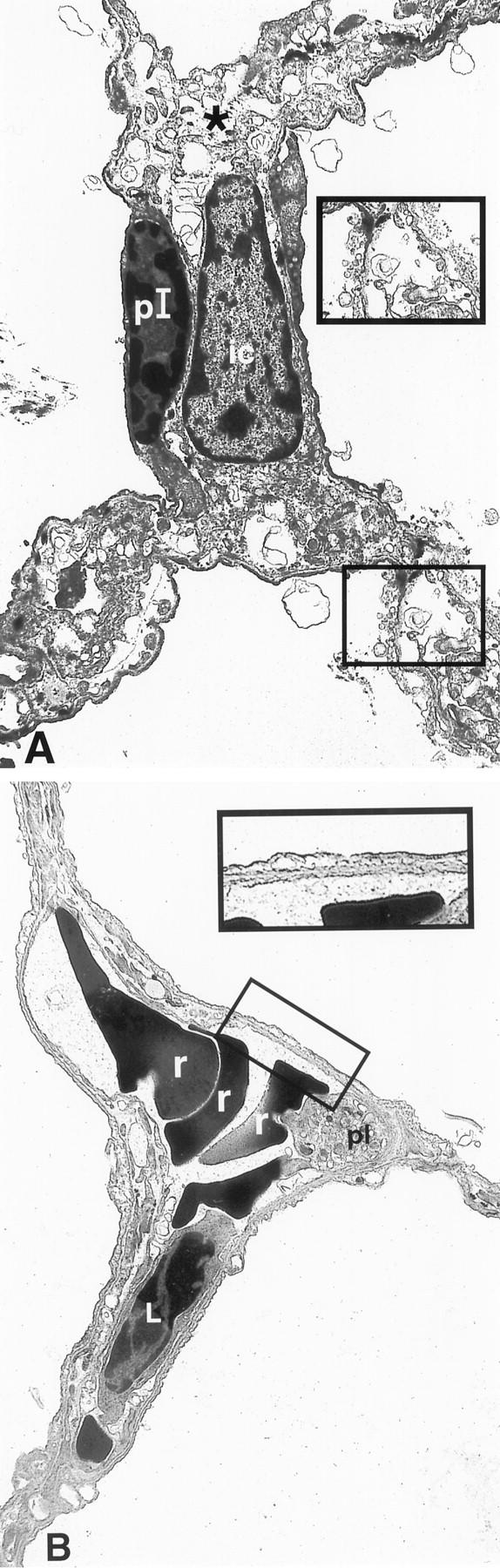
Alveolar septa from mice exposed to oxygen for 90 hours in a saline-treated mouse (A) and in a KGF-treated mouse (B). KGF was injected on day 0 and day 1 of oxygen exposure. A: Note the very significant interstitial edema and the condensed chromatin of the nucleus of the type I epithelial cell. Higher magnification of the damaged epithelium and endothelium. B: Presence of red blood cells and probably of a leukocyte within the capillary. Note the minimal changes of the epithelium and endothelium (original magnification, ×2800 and ×5200, respectively). pI, type I epithelial cell; ic, interstitial cell; r, red blood cell; L, leukocyte; pl, platelet.
In air-breathing mice, KGF increased only the epithelial volume as early as 24 hours after injection. In Figure 4 ▶ , the different volumes were measured 72 hours after the last KGF injection. This effect lasted at least until 90 hours after injection (not shown).
Figure 4.

Effect of KGF treatment on different lung volumes. Results are the mean ± SD of five to six mice. *P < 0.05; **P < 0.001.
In oxygen-breathing mice, when KGF was injected before oxygen exposure, the increase in epithelial volume was still detectable at 90 hours (P < 0.001 compared with saline-injected mice), whereas when KGF was injected during exposure, the increase in epithelial volume was less evident (P value was nonsignificant compared with saline-injected mice; Figure 4 ▶ ). KGF-treated mice (during oxygen exposure) showed minimal epithelial and endothelial damage as seen in a representative picture (Figure 3) ▶ . Interestingly, the endothelial volume, which was markedly affected by hyperoxia, was protected by KGF treatment either administered before or after the beginning of oxygen exposure (P < 0.05; Figure 4 ▶ ). There was a trend to reduce the extracellular interstitial volume in KGF-treated mice exposed to hyperoxia, but this was not significant with the number of mice examined (n = 5).
As the measurement of volume index does not take into account whether epithelial cells are injured or intact, we determined an injury score. Epithelial cells of the NaCl-treated group showed a mean score of 2.20 ± 0.39 compared with 0.46 ± 0.29 for the KGF-treated group (P = 0.003).
Effect of KGF Treatment on Apoptosis and Expression of Apoptosis-Related Genes and Proteins
Apoptosis has been shown to occur in hyperoxic lungs. 11,26 We examined by agarose electrophoresis the incidence of nucleosomal DNA fragmentation of lung DNA, a hallmark feature of apoptosis. Hyperoxic injury was associated with the appearance of nucleosomal ladders, but this was not significantly attenuated by KGF treatment in five mice per group (not shown). This suggests that the cytoprotective effect of KGF is not associated with a decrease in apoptosis.
p53 is a factor associated with the regulation of cell proliferation and cell death. By Western blot analysis, we reported a significant increase in p53 expression during hyperoxia. 11 This was attenuated in KGF-treated mice. p53 was below the detectable level on subsaturated films in the lung of air-breathing mice with or without KGF treatment. The quantitative analysis showed a threefold reduction in KGF-pretreated mice and a fourfold reduction in KGF-post-treated mice compared with the level induced by hyperoxia (P < 0.05; Figure 5 ▶ ). We also analyzed the expression of bax and bcl-x mRNAs by Northern blots. A quantitative analysis showed that although bax mRNA expression was not affected by KGF treatment, the bcl-x mRNA level was decreased significantly in KGF-treated mice during hyperoxia exposure (Figure 6) ▶ .
Figure 5.

Western blot analysis for the detection of p53 in lung extracts. p53 was undetectable in control lungs treated with saline (NaCl) or with KGF and was strongly expressed in hyperoxia-exposed lungs. KGF treatment before or concomitantly to oxygen exposure decreased significantly the expression of p53. The film was exposed for 30 seconds. Quantification of three different samples for each condition was performed by scanning densitometry. Values represent the mean ± SD of pixel density (*P < 0.05 compared with NaCl-treated group).
Figure 6.
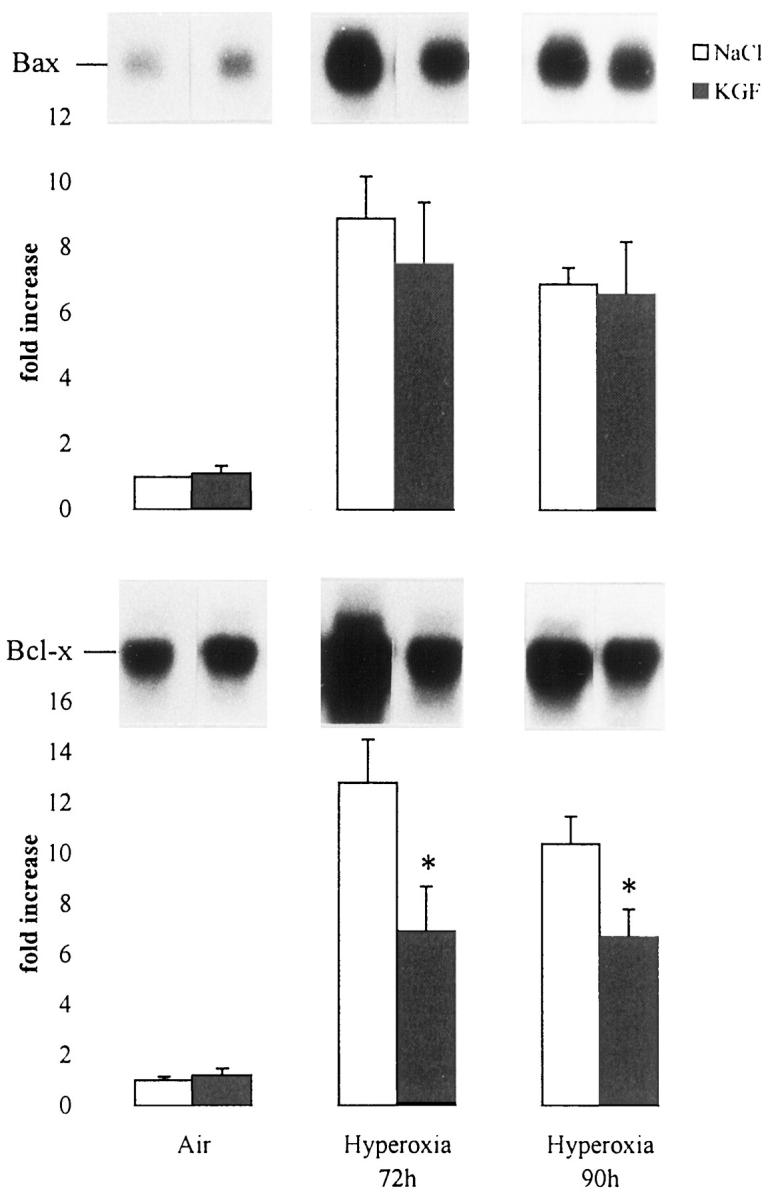
Effect of rhKGF (day −2, day −1) treatment on bax and bcl-x in murine lung during hyperoxia. Mice were exposed to air or to hyperoxia for the indicated times. Total lung RNA was isolated, and 10 μg was electrophoresed, transferred onto nitrocellulose membrane, and hybridized with 32P-labeled RNA probes. Quantification of the samples was performed with a Molecular Dynamics PhosphorImager and adjusted to the quantity of 18 S rRNA detected with methylene blue staining. Values represent means ± SD of three animals and are expressed as fold increases over air-breathing animals set as 1. KGF treatment did not change the basal level of bax and bcl-x in air-breathing animals, whereas it decreased significantly hyperoxia-induced bcl-x (P < 0.05). This effect was not significant with bax.
Expression of SPA and SPB mRNAs
Some studies have shown that KGF administration increases the synthesis of surfactant proteins in vitro and in vivo. 9,27 It has been postulated that one of the KGF actions might be related to the increase in surfactant production. We found a twofold increase in SPA and SPB mRNA of the whole-lung extract from KGF-treated mice compared with saline-treated mice. This difference was similar in air-breathing mice or oxygen-exposed mice (not shown)
KGF Restores Fibrinolytic Activity and Decreases Fibrin Deposition within the Lung
As we demonstrated that fibrin deposition contributes to the pathogenesis of hyperoxic lung injury, we analyzed the fibrinolytic activity of the BAL by zymography and fibrin deposition by immunostaining. KGF restored the fibrinolytic activity, which was markedly inhibited during hyperoxia (Figure 7) ▶ . Fibrin deposition was diminished in KGF-treated mice (18.5 ± 7.8 arbitrary units) compared with NaCl-treated mice (28.9 ± 6.9; P < 0.0001). To know whether the regulation of plasminogen activator and inhibitor within the lungs was changed by KGF administration, 14 the expression of uPA, tPA, and PAI-1 mRNAs was assessed by RNAse protection analysis. Plasminogen activator (uPA and tPA) mRNAs were significantly increased by hyperoxia, and this was not modified by KGF treatment (Figure 8) ▶ . PAI-1 mRNA was expressed in air- breathing mice receiving saline, and no difference was seen in mice receiving KGF. Lungs from mice exposed for 90 hours to hyperoxia and treated with saline showed a 50-fold increased PAI-1 mRNA content, whereas lungs treated with KGF showed only a 24-fold increase in PAI-1 mRNA content (P < 0.05). We also examined the amount of PAI-1 protein present in hyperoxic lungs. Saline-treated mice show an eightfold greater amount of PAI-1 than KGF-treated mice (P < 0.05; Figure 9 ▶ ). These data clearly indicate that KGF administration can also influence the fibrinolytic balance during the course of hyperoxic injury.
Figure 7.

Zymographic analysis of PA activities in BAL. A total of 20 μg of protein recovered in BAL from individual mice was loaded onto each lane. Mice were exposed to hyperoxia for 72 hours and treated with KGF (lanes 1 to 3) or with NaCl (lanes 4 to 6). Lanes 7 to 8, air-breathing mice. The migration of tPA and uPA was determined with purified standard proteins electrophoresed in adjacent lanes. Note the marked induction of tPA-mediated activity in KGF-treated mice compared with NaCl-treated mice. The photograph was taken after 48 hours of incubation at 37°C.
Figure 8.

Quantification of uPA, tPA, and PAI-1 in murine lungs. Mice were exposed to air or to hyperoxia for the indicated time and treated with saline (NaCl) or rhKGF on days 0 and 1, and 5 μg of total lung RNA was hybridized to 32P-labeled UTP cRNA probes and RNAse-resistant hybrids analyzed after separation in urea/polyacrylamide gels. Quantification of the samples was performed as described. Values represent means ± SD of three animals and are expressed as fold increase compared with NaCl controls set as 1. uPA mRNA level increased two to threefold in NaCl- or in KGF-treated mice, tPA mRNA increased eight- to ninefold, whereas PAI-1 mRNA increase was significantly less in KGF-treated mice exposed to hyperoxia. *P < 0.05 versus NaCl-treated mice exposed to hyperoxia.
Figure 9.

Western blot analysis for the detection of PAI-1 in lung tissue extracts during hyperoxic injury. PAI-1 was almost undetectable in the lung of an air-exposed animal (not shown). KGF treatment before or concomitantly to oxygen exposure decreased significantly the expression of PAI-1. The film was exposed for 30 seconds. Quantification of three different samples for each condition was performed by scanning densitometry. Values represent the mean ± SD of pixel density (*P < 0.05 compared with NaCl-treated group).
Discussion
Administration of KGF has been reported to increase the resistance of the lung and other organs to variety of injuries, including hyperoxia, chemotherapy, irradiation, and administration of acid. 3-5,7,28 In general, KGF is efficient when administered before but not after injury, and it is generally assumed that KGF protects lungs more efficiently when administered intratracheally instead of i.v. 4 Recently, it has been shown that alveolar lesions during oxygen toxicity could be attenuated also by giving KGF i.v. 28 These observations have some importance for understanding of the KGF mode of action. In this study, we administered KGF i.v. and during oxygen exposure.
KGF is known to increase proliferation in many epithelia, including the bronchial and alveolar epithelium. 2 A proliferative response was obvious in air-breathing animals injected i.v. with KGF, indicating that this route of administration can reach the alveolar epithelium. During oxygen administration, the turnover of alveolar cells was markedly reduced, and KGF administration was not able to restore the turnover rate to levels similar to those seen in air-breathing mice. Despite this low proliferation rate, KGF was still able to attenuate the alveolar damage. This observation indicates that the protective effect within the lung is not necessarily associated with its capacity to sustain proliferation.
Exposure to oxygen is known to result in the death of alveolar epithelial and endothelial cells. 1 Precise evaluation of different cellular compartment damage is, however, difficult. By an evaluation of the cellular volume using semiquantitative EM, oxygen toxicity appears as a loss of endothelial cell volume and as an interstitial edema, but not as a loss of epithelial volume as already described. 29 Indeed, epithelium damage is evident by plasma membrane disruption and condensation of nuclear chromatin, but semiquantitative analysis of cell volume does not take into account these changes. Therefore, we established an injury score of type I cells by another semiquantitative approach. This score was significantly different between the saline- and the KGF-treated group on day 0 and day 1, suggesting that KGF exerts a clear protective effect on epithelial cells.
KGF is believed to be highly specific for epithelial cells, 6,30 and as expected, KGF increased the epithelial cell volume, especially when given to air-breathing mice and, to a more limited extent, when given to oxygen-breathing mice. Surprisingly, administration of KGF has also a protective effect on endothelium by preventing its destruction induced by oxygen as well as the resulting increase in interstitial volume. Until now, no precise evaluation by EM has been made on different lung constituents on KGF treatment. These observations indicate that in vivo the cytoprotective effect of KGF is not limited to its first target, the epithelium, but concerns the whole alveolar septum. One is tempted to speculate that KGF can exert a paracrine effect on endothelium via the epithelium.
The oxygen-induced lung injury presents features of both necrosis and apoptosis. 11 Internucleosomal DNA degradation, a modification specific to apoptosis, is evident during oxygen-induced injury. However, it has not been possible so far to attenuate oxygen-induced injury by anti-apoptotic strategies. 11 Surprisingly, we were unable to demonstrate an effect of KGF on the extent of internucleosomal degradation, suggesting that KGF does not influence oxygen-induced apoptosis. Expression of p53, a factor associated with the regulation of the cell cycle and cell death, is up-regulated during hyperoxia, and KGF administration clearly decreased its expression, suggesting that this down-regulation might prevent alveolar cell death. However, this mechanism seems unlikely in the oxygen-induced alveolar damage, as p53 −/− mice do not manifest an increased resistance to oxygen. 11 Our results are similar to a report showing that KGF treatment decreased the amount of p53 in an epithelial type II cell line, whereas it does not inhibit DNA laddering induced by hyperoxia. 8 Thus, if KGF can attenuate programmed cell death in some cell lines, there is no strong evidence that this is its mode of action in the oxygen-induced alveolar injury.
Among the other effects of KGF that might play a role in the resistance to hyperoxic lung injury is the increased production of some surfactant proteins. 9 Surfactant inhibition is believed to be an important mechanism of respiratory distress in several acute lung pathologies, 31 and therefore surfactant replacement has become a widely applied therapeutic regimen. Currently, there is still a debate as to the dose of surfactant that should be used, but it is generally accepted that it should be given at the highest possible dosage. 32 In our conditions, KGF increased moderately SPA and SPB mRNA levels within the lung, suggesting that this effect might indeed contribute to the improvement of the alveolar function.
Oxygen-induced alveolar injury is associated with an antifibrinolytic state favoring fibrin deposition, which is believed to contribute to the impairment of the alveolar function and gas exchange. 14 Fibrin is also known to impair the function of surfactant. 33 As PA and PA inhibitor are produced by epithelial cells, we postulated that KGF might modulate alveolar fibrinolysis. There are reports showing that KGF increases the PA activity in cultured epithelial cells. 10,34 In vivo, KGF did not change the level of uPA or tPA mRNA in air-breathing or in oxygen-exposed mice. It would be unlikely that we would see any effect of KGF on tPA, as tPA is mainly produced by endothelial cells that do not bear KGF receptors. PAI-1 is produced by endothelial and epithelial cell lines 35,36 and in vivo by epithelial type II cells. 14 KGF administration down-regulates PAI-1 expression during hyperoxia and by this way prevents the disturbance of fibrinolytic activity within the alveolar space. As we previously showed that PAI-1-deficient mice survive longer than their +/+ wild-type controls, 14 one of the major effects of KGF, independent of cell proliferation, might be due to an increase of fibrinolysis.
Oxygen is a widely used therapy for several respiratory insufficiencies, but breathing a high oxygen concentration carries a risk to worsen the alveolar injury. It is thus of importance for clinical use that KGF cannot only prevent but also attenuate alveolar damage when given during oxygen therapy.
Acknowledgments
We thank Amgen, Inc. (Thousand Oaks, CA) for providing the human recombinant KGF, D. Belin for helping in preparing the cRNA probes, M. Redard for immunohistochemistry preparations, P. Henchoz for electron microscopy preparations, J. C. Rumbeli for his photographic work, and G. Brighouse for reading the manuscript.
Footnotes
Address reprint requests to Dr. Constance Barazzone, Departments of Pediatrics and Pathology, Centre Médical Universitaire, 1211 Geneva, Switzerland. E-mail: constance.barazzone@medecine.unige.ch.
C. Barazzone and P.F. Piguet are supported by grant 32–47284.96 from the Fonds National Suisse de la Recherche Scientifique.
References
- 1.Crapo JD, Barry BE, Foscue A, Shelburne J: Structural and biochemical changes in rat lung during exposure to lethal and adaptative doses of oxygen. Am Rev Respir Dis 1980, 122:123-143 [DOI] [PubMed] [Google Scholar]
- 2.Ulich TR, Yi ES, Longmuir K, Yin S, R. B, Morris CF, Housley RM, Pierce GF: Keratinocyte growth factor is a growth factor for type II pneumocytes in vivo. J Clin Invest 1994, 93:1298–1236 [DOI] [PMC free article] [PubMed]
- 3.Yano T, Deterding RR, Scott Simonet W, Shannon J, Mason R: Keratinocyte growth factor reduces lung damage due to acid instillation in rats. Am J Respir Cell Mol Biol 1996, 15:433-442 [DOI] [PubMed] [Google Scholar]
- 4.Panos RJ, Bak PM, Scott Simonet W, Rubin JS, Smith LJ: Intratracheal instillation of keratinocyte growth factor decreases hyperoxia-induced mortality in rats. J Clin Invest 1995, 96:2026-2033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yi ES, Williams ST, Lee H, Malicki D, Chin EM, Yin S, Tarpley J, Ulich T: Keratinocyte growth factor ameliorates radiation- and bleomycin-induced lung injury. Am J Pathol 1996, 149:1963-1970 [PMC free article] [PubMed] [Google Scholar]
- 6.Rubin JS, Osada H, Finch PW, Taylor WG, Rudikoff S, Aaronson SA: Purification and characterization of a newly identified growth factor specific for epithelial cells. Proc Natl Acad Sci USA 1989, 86:802-806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takeoka M, Ward WF, Pollack H, Kamp DW, Panos RJ: KGF facilitates repair of radiation-induced DNA damage in alveolar epithelial cells. Am J Physiol 1997, 272:L1174-L1180 [DOI] [PubMed] [Google Scholar]
- 8.Buckley S, Barsky L, Driscoll B, Weinberg K, Anderson KD, Warburton D: Apoptosis and DNA damage in type 2 alveolar epithelial cells cultured from hyperoxic rats. Am J Physiol 1998, 74:L714-L720 [DOI] [PubMed] [Google Scholar]
- 9.Xu X, McCormick-Shannon K, Voelker DR, Mason RJ: KGF increases SP-A and SP-D mRNA levels and secretion in cultured rat alveolar type II cells. Am J Respir Cell Mol Biol 1998, 18:168-178 [DOI] [PubMed] [Google Scholar]
- 10.Tsuboi R, Sato C, Kurita Y, Rubin JS, Ogawa H: Keratinocyte growth factor (FGF-7) stimulates migration and plasminogen activator activity of normal human keratinocytes. J Invest Dermatol 1993, 101:49-53 [DOI] [PubMed] [Google Scholar]
- 11.Barazzone C, Horowitz S, Donati YR, Rodriguez I, Piguet PF: Oxygen toxicity in mouse lung: pathways to cell death. Am J Respir Cell Mol Biol 1998, 19:577-581 [DOI] [PubMed] [Google Scholar]
- 12.O’Reilly MA, Staversky RJ, Stripp BR, Finkelstein JN: Exposure to hyperoxia induces p53 expression in mouse lung epithelium. Am J Respir Cell Mol Biol 1998, 18:43-50 [DOI] [PubMed] [Google Scholar]
- 13.McGrath S: Induction of p21 WAF/CIP1 during hyperoxia. Am J Respir Cell Mol Biol 1998, 18:179-187 [DOI] [PubMed] [Google Scholar]
- 14.Barazzone C, Belin D, Piguet PF, Vassalli JD, Sappino AP: Plasminogen activator inhibitor-1 in acute hyperoxic mouse lung injury. J Clin Invest 1996, 98:2666-2673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barazzone C, Tacchini Cottier F, Vesin C, Rochat AF, Piguet PF: Hyperoxia induces platelet activation and lung sequestration: an event dependent on tumor necrosis factor-α and CD11a. Am J Respir Cell Mol Biol 1996, 15:107-114 [DOI] [PubMed] [Google Scholar]
- 16.Bachofen M, Weibel ER: Alterations of the gas exchange apparatus in adult respiratory insufficiency associated with septicemia. Am Rev Respir Dis 1977, 116:589-615 [DOI] [PubMed] [Google Scholar]
- 17.Fracica PJ, Knapp MJ, Crapo JD: Patterns of progression and markers of lung injury in rodents and subhuman primates exposed to hyperoxia. Exp Lung Res 1988, 14:869-885 [DOI] [PubMed] [Google Scholar]
- 18.Weibel ER: Morphometry: stereological and practical methods. Gil J eds. Models of Lung Disease, Microscopy, and Structural Methods. 1990, :pp 199-252 Dekker, New York [Google Scholar]
- 19.Piguet PF, Vesin C: Pulmonary platelet trapping induced by bleomycin: correlation with fibrosis and involvement of the β-2 integrins. Int J Exp Pathol 1994, 70:321-328 [PMC free article] [PubMed] [Google Scholar]
- 20.Sappino AP, Huarte J, Vassalli JD, Belin D: Sites of synthesis of urokinase and tissue-type plasminogen activators in the murine kidney. J Clin Invest 1991, 87:962-970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sappino AP, Madani R, Huarte J, Belin D, Kiss JZ, Wohlwend AL, Vassalli JD: Extracellular proteolysis in the adult murine brain. J Clin Invest 1993, 92:679-685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Debrock S, Declerk PJ: Neutralization of plasminogen activator inhibitor-1 inhibitory properties: identification of two different mechanisms. Biochem Biophys Acta 1997, 1337:257-266 [DOI] [PubMed] [Google Scholar]
- 23.Bradford MM: A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976, 72:248-254 [DOI] [PubMed] [Google Scholar]
- 24.Vassalli JD, Dayer JM, Wohlwend A, Belin D: Concomitant secretion of prourokinase and of plasminogen activator-specific inhibitor by stimulated macrophages. J Exp Med 1984, 159:1653-1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cazals VB, Mouhieddine B, Maitre B, Le Bouc Y, Chadelat K, Brody JS, Clement A: Insulin-like growth factors, their binding proteins, and transforming growth factor β-1 in oxidant-arrested lung alveolar epithelial cells. J Biol Chem 1994, 209:14111-14117 [PubMed] [Google Scholar]
- 26.Mantell LL, Kazzaz JA, Xu J, Palaia TA, Piedboeuf B, Hall S, Rhodes GC, Niu G, Fein AF, Horowitz S: Unscheduled apoptosis during acute hyperoxia. Cell Death Differ 1997, 4:600-607 [DOI] [PubMed] [Google Scholar]
- 27.Panos RJ, Bak PM: Keratinocyte growth factor (KGF) increases whole lung SP-A, B and C transcript levels. Am J Respir Crit Care Med 1996, 153:A642 [Google Scholar]
- 28.Guo J, Yi ES, Havill A, Sarosi I, Whitcomb L, Yin S, Middleton S, Piguet PF, Ulich TR: Intravenous keratinocyte growth factor protects against experimental pulmonary injury. Am J Physiol 1998, 275:L800-L805 [DOI] [PubMed] [Google Scholar]
- 29.Folz RJ, Piantadosi CA, Crapo JA: Oxygen toxicity. Crystal RG West JB Barnes PJ Weibel ER eds. The Lung, 1997, vol 2.:pp 2713-2722 Lippincott-Raven, Philadelphia [Google Scholar]
- 30.Miki T, Fleming TP, Bottaro DP, Rubin JS, Aaronson SA: Expression cDNA cloning of KGF receptor by creation of a transforming autocrine loop. Science 1991, 251:72-75 [DOI] [PubMed] [Google Scholar]
- 31.Lachmann B, Eijking EP, So KL, Gommers D: In vivo evaluation of the inhibition capacity of human plasma on exogenous surfactant function. Intensive Care Med 1994, 20:6-11 [DOI] [PubMed] [Google Scholar]
- 32.Gregory TJ, Longmore WJ, Moxley MA: Surfactant chemical composition and biophysical activity in acute respiratory distress syndrome. J Clin Invest 1991, 88:1976-1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seeger W, Stoehr G, Wolf HD, Neuhof H: Alteration of surfactant function due to protein leakage: special interaction with fibrin monomer. J Appl Physiol 1985, 58:326-338 [DOI] [PubMed] [Google Scholar]
- 34.Putnins EE, Firth JD, Uitto VJ: Keratinocyte growth factor stimulation of gelatinase (matrix metalloproteinase-9) and plasminogen activator in histiotypic epithelial cell culture. J Invest Dermatol 1995, 104:989-994 [DOI] [PubMed] [Google Scholar]
- 35.Loskutoff DJ, Edgington TS: Synthesis of a fibrinolytic activator and inhibitor by endothelial cells. Proc Natl Acad Sci USA 1977, 74:3903-3907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gross T, Simon R, Kelley C, Sitrin R: Rat alveolar epithelial cells concomitantly express plasminogen activator inhibitor-1 and urokinase. Am J Physiol 1991, 260:L286-L295 [DOI] [PubMed] [Google Scholar]