

Abstract
Astrocytic tumors occasionally arise in the central nervous system following radiotherapy. It is not clear if these gliomas represent a unique molecular genetic subset. We identified nine cases in which an astrocytoma arose within ports of previous radiation therapy, with total doses ranging from 2400 to 5500 cGy. Irradiated primary lesions included craniopharyngioma, pituitary adenoma, Hodgkin’s lymphoma, ependymoma, pineal neoplasm, rhabdomyosarcoma, and three cases of lymphoblastic malignancies. Patients ranged from 9 to 60 years of age and developed secondary tumors 5 to 23 years after radiotherapy. The 9 postradiation neoplasms presented as either anaplastic astrocytoma (3 cases) or glioblastoma multiforme (6 cases). Two of the latter contained malignant mesenchymal components. We performed DNA sequence analysis, differential polymerase chain reaction (PCR), and quantitative PCR on DNA from formalin-fixed, paraffin-embedded tumors to evaluate possible alterations of p53, PTEN, K-ras, EGFR, MTAP, and p16 (MTS1/CDKN2) genes. By quantitative PCR, we found EGFR gene amplification in 2 of 8 tumors. One of these demonstrated strong immunoreactivity for EGFR. Quantitative PCR showed chromosome 9p deletions including p16 tumor suppressor gene (2 of 7 tumors) and MTAP gene (3 of 7). Five of 9 tumors demonstrated diffuse nuclear immunoreactivity for p53 protein. Sequencing of the p53 gene in these 9 cases revealed a mutation in only one of these cases, a G-to-A substitution in codon 285 (exon 8). Somewhat unexpectedly, no mutations were identified in PTEN, a commonly altered tumor suppressor gene in de novo glioblastoma multiformes. Unlike some radiation-induced tumors, no activating point mutations of the K-ras proto-oncogene or base pair deletions of tumor suppressor genes were noted. These radiation-induced tumors are distinctive in their high histological grade at clinical presentation. The spectrum of molecular genetic alterations appears to be similar to that described in spontaneous high grade astrocytomas, especially those of the de novo type.
Radiation therapy is often administered as an adjuvant in the treatment of incompletely resected tumors of the central nervous system (CNS) and pituitary gland. The brain is also exposed to radiation during the treatment of head and neck tumors and of hematological malignancies involving the central nervous system. A significant but rare long-term complication of CNS radiotherapy is a secondary, presumably radiation-induced, neoplasm. 1-3 Epidemiological studies suggest an increased risk of developing gliomas for patients exposed to ionizing radiation, but little is known about the molecular genetic events underlying such tumorigenesis. 4-6 If unique, the underlying genetic alterations introduced by ionizing radiation could serve as a diagnostic molecular fingerprint for these lesions. For example, an unusual 3-bp homozygous deletion in exon 7 of the p53 gene has been described in a radiation-induced glioblastoma multiforme (GBM). 7 Activating K-ras mutations have also been described in radiation-induced tumors, including a primitive neuroectodermal tumor of the cerebral hemispheres. 8,9 Alternatively, genetic alterations introduced by radiation could resemble those identified in spontaneous astrocytic neoplasms, including EGFR amplification, p16 (MTS1/CDKN2) deletions, PTEN mutations, and p53 mutations. 10-16 Comparable alterations in spontaneous and radiation-induced tumors may suggest similar pathogenetic intermediate mechanisms. To address these issues, we have evaluated potential genetic alterations in a series of nine radiation-induced astrocytic neoplasms.
Materials and Methods
We searched the Johns Hopkins Oncology Center Information System for astrocytic tumors that arose following radiation therapy involving the CNS and recovered six cases. Three cases were received in consultation. In all cases, clinical information was reviewed to ensure that tumors arose within ports of prior radiation and that a sufficient latency period existed between radiation treatment and tumor development. The shortest latency period in this series of tumors was 5 years. Hematoxylin and eosin (H&E) slides were reviewed for classification and grading. All of the included astrocytic neoplasms differed histologically from the primary irradiated neoplasm. None of the astrocytic tumors studied was a recurrence and none had been treated with chemotherapy or radiotherapy.
Immunohistochemistry
For immunohistochemical studies, paraffin-embedded specimens were sectioned at 4 μm, deparaffinized, and treated for antigen enhancement by either limited protein digestion or steaming (20 minutes at 80°C). Slides were then incubated at room temperature with antibodies to EGFR (monoclonal, 1:15; Zymed, South San Francisco, CA) or p53 (monoclonal, 1:250; Dako Co., Carpenteria, CA). Antibody was detected using the avidin-biotin complex method and diaminobenzidine as the chromogen. p53 staining of nuclei was graded in the region of greatest staining for each tumor as 3+ (>50% of nuclei), 2+ (>20% and <50%), 1+ (>0 and <20%), or absent. Intensity of cytoplasmic and cell membrane immunoreactivity for EGFR was graded as absent, mild, moderate, or strong.
DNA Isolation
For each paraffin-embedded block containing tumor, corresponding H&E slides were reviewed to ensure that DNA was isolated from tissue containing at least 90% tumor. Ten unstained histological sections containing neoplastic tissue, each 10 μm thick, were cut from paraffin blocks, deparaffinized in xylene, octane, or Histoclear, and scraped into microcentrifuge tubes containing 300 μl of a mixture consisting of 3 mmol/L Tris (pH 7.5), 0.2 mmol/L EDTA, 1 mg/ml proteinase K, 0.5% Tween 20, and 0.05% sodium dodecyl sulfate (SDS). Samples were incubated at 58°C for 5 hours, and fresh proteinase K/Tween 20/SDS was added for a minimum of 12 hours. Following protein digestion, samples were incubated at 95°C for 10 minutes. Samples were assessed for purity and DNA concentration spectrophotometrically at 260 and 280 nm.
PCR/Sequencing of Exons 5–8 of p53
Purified genomic DNA (100 ng) was PCR amplified in a 50-μl reaction containing 50 mmol/L KCl, 10 mmol/L Tris (pH 8.3), 1.5 mmol/L MgCl2, 1.0 μl 10 mmol/L dNTPs (2.5 mmol/L each dATP, dCTP, dGTP, and dTTP), 1.0 U Taq polymerase (Sigma, St. Louis, MO), and 0.4 μmol/L forward and reverse primers. Primers were designed to flank each exon and include the intron-exon junctions. One of the primers in each pair was 5′-biotinylated to allow for purification of single-stranded template for sequencing using a streptavidin-coated magnetic bead protocol. 17 DNA was denatured at 94°C for 10 minutes. Thermocycler conditions were set for 30 cycles at 94°C for 30 seconds each, an empirically determined, optimal annealing temperature for each primer pair for 30 seconds and at 72°C for 30 seconds. A final extension step was 72°C for 7 minutes. The annealing temperatures and primer pairs for each exon were as follows: exon 5, 58°C for primer set Sarker 5U + Koga 5L-Bio, or 62°C for primer set 5U-Bio + 5L; exon 6, 66°C for Ohgaki 6U + 6L-Bio or 64°C for Ohgaki 6U-Bio + Sakar 6L; exon 7, 66°C for Vogel 7U + Sakar 7L-Bio or 62°C for 7U + 7L; and exon 8, 60°C for both 8U + 8L-Bio or 8U-Bio + Sakar 8L. 18-21 Sequencing was performed using Sequenase v. 2.0 and dideoxy-chain termination methods incorporating 35S-dATP, as previously described. 17 Internal primers were used for sequencing reactions and were annealed at 60°C. Labeling mix (dGTP) was diluted 1:10 and buffer supplemented with manganese was used in all reaction mixes. Sequencing was duplicated in the forward direction and confirmed in the reverse direction for each exon.
PCR/Sequencing of PTEN
One microliter of the DNA isolates were amplified in 50-μl reactions containing the following: 20 pmol/L forward and reverse primers (see Table 1 ▶ for primer sequences and annealing temperatures), 200 μmol/L each dNTP, 1.5 mmol/L MgCl2, 50 mmol/L KCl, 10 mmol/L Tris-HCl (pH 8.3), 0.001% gelatin, 5% DMSO, and 1.25 units Amplitaq Gold (Perkin-Elmer, Foster City, CA). Reactions were carried out in a Perkin-Elmer 9600 thermocycler using the following conditions: 95°C hold for 9 minutes, followed by 50 cycles of 95°C for 30 seconds, 55°C or 48°C for 30 seconds, 72°C for 1 minute, followed by a 10-minute hold at 72°C. Forty microliters of each PCR product were treated with 40 U exonuclease I and 8 U shrimp alkaline phosphatase (PCR Product Pre-Sequencing kit, Amersham, Arlington, IL) by incubating at 37°C for 15 minutes, then at 80°C for 15 minutes. Approximately 100 ng of the treated PCR products were then sequenced using 2 pmol/L of forward or reverse primer and Thermosequenase kit (Amersham). Cycle sequencing conditions were 95°C for 20 seconds, 58°C for 30 seconds, and 72°C for 1 minute for 30 cycles. Reactions were quenched, denatured at 95°C for 2 minutes, and electrophoresed through a 6% polyacrylamide gel with 15% formamide and 7 mol/L urea. Gels were dried for 1 hour and exposed to film overnight.
Table 1.
EGFR Primers
| Exon | Primer name | Primer sequence | PCR product size | TM* |
|---|---|---|---|---|
| 1 | 1S | gccaccagcagcttctgcc | 187 | 55 |
| 1AS | ctaagagagtgacagaaagg | |||
| 2 | 2S1 | accttttattactccagcta | 191 | 48 |
| 2AS1 | ttcctgtatacgccttcaag | |||
| 2S2 | caaacattattgctatggga | 169 | 48 | |
| 2AS2 | atctttttctgtggcttaga | |||
| 3 | 3S | gctcattttgttaatgg | 135 | 55 |
| 3AS | tagaagatatttgcaagc | |||
| 4 | 4S | ataaagattcaggcaatgtt | 184 | 48 |
| 4AS | atcgggtttaagttatacaa | |||
| 5 | 5S1 | agtttgtatgcaggcaatgtt | 240 | 55 |
| 5AS1 | atcattacaccagttcgtccc | |||
| 5S2 | gcagcaattcactgtaaagc | 179 | 55 | |
| 5AS2 | cagatccaggaagaggaaagg | |||
| 6 | 6S1 | ttttcaatttggcttctctt | 143 | 48 |
| 6AS1 | catcttgtgaaacaacagtgcc | |||
| 6S2 | cctgttaaagaatcatctgg | 137 | 48 | |
| 6AS | ctgttccaatacatggaagg | |||
| 7 | 7S1 | ttcctgtgaaataatactgg | 179 | 55 |
| 7AS1 | gaactctactttgatatcacc | |||
| 7S2 | ttcatgtactttgagttccc | 131 | 55 | |
| 7AS2 | ccttattttggatatttctccc | |||
| 8 | 8A1 | tgcaaatgtttaacataggtga | 170 | 55 |
| 8AS1 | ttccttgtcattatctgcacg | |||
| 8S2 | caagaaatcgatagcatttgc | 181 | 55 | |
| 8AS2 | atacatacaagtcaacaaccccc | |||
| 9 | 9S1 | taagatgagtcatatttgtggg | 179 | 55 |
| 9AS1 | cagagtcagtggtgtcaga | |||
| 9S2 | tagaggagccgtcaaatcca | 194 | 48 | |
| 9AS2 | catggtgnnttatccctctt |
*TM, annealing temperature (°C).
PCR/Seqencing of K-ras
A 190-bp segment of the K-ras gene was amplified using primers K-ras-F (5′-ACTGAATATAAACTTGTGGTAGTTGGAG) and K-ras-R (5′-TCATGAAAATGGTCAGAGAAACC). PCR reactions containing 1× Gene Amp PCR buffer (Perkin-Elmer), 400 μmol/L dNTPs, 0.8 μmol/L K-ras-F, 0.8 μmol/L K-ras-R, and 1.5 U Taq polymerase (Life Technologies, Gaithersburg, MD) were amplified using 32 cycles of 30 seconds at 94°C, 30 seconds at 56°C, and 45 seconds at 72°C, followed by a 1-minute extension at 72°C. The resulting PCR products were evaluated on 2% ethidium bromide-stained agarose gels. Confirmed PCR products were purified for direct sequencing by treatment with 10.0 U Exonuclease I and 2.0 U Shrimp Alkaline Phosphatase (Amersham) at 37°C for 15 minutes, followed by a brief incubation at 80°C for 15 minutes. The purified PCR products were directly sequenced using the ThermoSequenase radiolabeled terminator cycle sequencing kit (Amersham) according to manufacturer’s instructions. Products of the cycle sequencing reactions were resolved on 6% acrylamide-8 mol/L urea denaturing gels. Gels were fixed in a solution of 5% methanol and acetic acid, dried, and subjected to radiography.
Differential PCR of EGFR
PCR amplifications (Perkin-Elmer apparatus) were carried out in a final volume of 25 μl as previously described. 22 Each reaction mixture contained 0.15–0.20 μg of template, 10 mmol/L Tris-Cl (pH 8.3), 50 mmol/L KCI, 1.5 mmol/L MgCl2, 0.001% gelatin (w/v), 200 μmol/L dNTPs, 0.05 μmol/L oligonucleotide primers for reference and target gene, and 0.0625 U AmpliTaq DNA polymerase (Perkin-Elmer, Norwalk, CT). PCR cycles were carried out as follows: 92°C for 2 minutes; cycles 1–5 at 97°C for 30 seconds, 53°C for 1 minute, and 72°C for 1 minute; cycles 6–25 or 6–30 at 95°C for 1 minute, 53°C for 1 minute, and 72°C for 1 minute; and a final extension after the last cycle at 72°C for 10 minutes. After PCR, the entire reaction product was electrophoresed on a 2% agarose gel and stained with ethidium bromide. Fragments from X174-HaeIII digested DNA (Promega, Madison, WI) were used as markers to document amplification of appropriately sized fragments. Gels were photographed using the Gel-doc 1000 photodocumentation system (Bio-Rad, Philadelphia, PA) and resulting images were quantitatively analyzed using Molecular Analyst software. This method of differential PCR used with appropriate controls has demonstrated 100% concordance with slot blot analysis for EGFR amplification in a series of astrocytic neoplasms. 22
Primers for EGFR used in this study (sense, AGCCATGCCCGCATTAGCTC; antisense, AAAGGAATGCAACTTCCCAA) amplify a 110-bp genomic fragment corresponding to bases 3901–4010 of the cDNA sequence. The reference primers for the detection of increased EGFR gene dosage amplify a 70-bp genomic fragment from the β-globin gene (sense, TGACTCCTGAGGAGAAGTCTGC [cDNA bases 164–186]; antisense, TCACCACCAACTTCATCACGT [cDNA bases 212–233]), and a 79-bp fragment of the cystic fibrosis gene (sense, GGCACCATTAAAGAAATATCATCTT [cDNA bases 1630–1655]; antisense, GTTGGCATGCTTTGATGACGCTTC [cDNA bases 1685–1708]), respectively.
Quantitive PCR Analysis of p16 and MTAP
Quantitative PCR assays were performed using primers from p16 exon 1 and MTAP exon 8 with an MTAP pseudogene as reference, as described previously. 23 MTAP is located approximately 100 kb telomeric to p16 on chromosome 9p and encodes methylthioadenosine phosphorylase. Amplification was carried out in a total volume of 50 μl containing 100 ng of genomic DNA, 1× PCR buffer (10 mmol/L Tris-HCl (pH 8.3), 50 mmol/L KCl, 1.5 mmol/L MgCl2, 0.01% gelatin), 200 μmol/L of each dNTP including 80 μmol/L digoxigenin-labeled dUTP (Boehringer Mannheim, Indianapolis, IN), 150 ng each of sense and anti-sense primers, and 2.5 units of Taq DNA polymerase (AMITaq Gold; Perkin-Elmer, Foster City, CA). Following incubation at 94°C for 5 minutes, 26 cycles were performed consisting of 94°C denaturation (1 minute), 64°C annealing (1 minute), and 72°C extension (1 minute).
Aliquots of the PCR products were resolved on 2% agarose gels (Gibco, Grand Island, NY) for qualitative purposes. The remaining PCR products were subjected to purification through QiaQuick Spin Columns (Qiagen Inc., Chatsworth, CA) to remove unincorporated primers. After binding the PCR products to a 96-well enzyme-linked immunosorbent assay plate coated with streptavidin, the incorporated digoxigenin-dUTP in the PCR products was detected with enzyme-linked immunosorbent assay. To construct calibration curves, PCR assays were carried out in parallel using standard DNA templates that contained mixtures of p16-negative, MTAP-negative Jurkat leukemic cells and normal human diploid fibroblast DNA (WI-38) in varying proportions. The optical density reading of each sample was used to determine the status of p16 exon 1 or MTAP exon 8 by calculating the relative gene dosage (Dp16 and DMTAP) as follows:
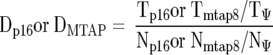 |
where Tp16, Tmtap8, and TΨ represent the tumor optical density readings of p16 exon 1, MTAP exon 8, and the MTAP pseudogene, respectively, and Np16, Nmtap8, and NΨ represent the normal fibroblast optical density readings of p16 exon 1, MTAP exon 8, and the MTAP pseudogene, respectively. 23 Dp16 and DMTAP values were plotted as standard curves for determination of relative gene doses in primary tumor samples. Homozygous deletion of p16 or MTAP genes was defined as a relative gene dosage less than 30%.
Results
Radiation-induced astrocytomas included in this study occurred in nine patients ranging in age from 9 to 60 years old (see Table 2 ▶ ). These tumors arose within ports of external beam radiation therapy ranging in dose from 2400 to 5500 cGy and occurred between 5 and 25 years after radiotherapy (Figure 1) ▶ . Irradiated primary tumors included Hodgkin’s disease, pituitary adenoma, rhabdomyosarcoma, craniopharyngioma, and CNS involvement by lymphoblastic malignancies (three cases). All astrocytic tumors studied were primary lesions (ie, none were recurrences). None of the astrocytomas included was irradiated before this study. All of the radiation-induced tumors in this series were high grade at clinical presentation, including six glioblastomas and three anaplastic astrocytomas. Two of the glioblastomas had malignant mesenchymal components but did not satisfy criteria for the diagnosis of gliosarcoma. The molecular genetic alterations identified in each tumor are listed in Table 3 ▶ .
Table 2.
Clinical Characteristics of Radiation-Induced Astrocytomas
| No. | Age/sex | Primary diagnosis | Chemotherapy | Radiation dose | Interval (years) | Secondary neoplasm |
|---|---|---|---|---|---|---|
| 307 | 13 /M | ALL | yes | 2400 cGy | 8 | GBM |
| 308 | 34 /M | Pineal tumor | no | 4570 cGy | 23 | GBM |
| 309 | 31 /F | Hodgkin’s disease | yes | 4500 cGy | 8 | AA |
| 310 | 19 /F | Lymphoblastic lymphoma | yes | 3000 cGy | 5 | GBM |
| 311 | 28 /F | Ependymoma | no | 5400 cGy | 7 | AA |
| 312 | 60 /M | Pituitary adenoma | no | 4500 cGy | 15 | GBM |
| 313 | 9 /M | Rhabdomyosarcoma | yes | 5400 cGy | 7 | GBM |
| 314 | 18 /F | Craniopharyngioma | no | 5500 cGy | 11 | GBM |
| 363 | 20 /M | ALL | yes | 3600 cGy | 7 | AA |
ALL, acute lymphoblastic leukemia; cGy, centiGray; AA, anaplastic astrocytoma; GBM, glioblastoma multiforme.
Figure 1.

Gadolinium-enhanced magnetic resonance images of a patient who was treated with 4500 cGy of radiation therapy for a residual pituitary adenoma (A, arrow) following incomplete surgical resection. Isodose curves of radiotherapy (shown in red, with percentages of maximal dose in white) demonstrate the isocenter of radiation therapy at the residual adenoma. Fifteen years after surgery and radiotherapy, the patient developed a glioblastoma multiforme involving the left and right hemispheres, crossing at the corpus callosum (B). The new lesion was radiographically distinct from the residual adenoma (B, arrow) and occurred within the port of previous radiation therapy.
Table 3.
Molecular Genetic Alterations in Radiation-Induced Astrocytomas
| No. | Dx. | p53 IHC | p53 gene | K-ras | EGFR gene | p16 gene (p16/ΨMTAP) | MTAP gene (MTAP/ΨMTAP) | PTEN gene |
|---|---|---|---|---|---|---|---|---|
| 307 | GBM | negative | WT | WT | nl. | nl. (0.58) | deletion (0.02) | WT |
| 308 | GBM | +++ | WT | WT | nl. | deletion (0.26) | deletion (0.18) | WT |
| 309 | AA | negative | WT | WT | amplified | nl. (0.71) | nl. (0.92) | WT |
| 310 | GBM | negative | WT | WT | nl. | nl. (0.66) | nl. (0.96) | WT |
| 311 | AA | +++ | WT | WT | nl. | deletion (0.15) | deletion (0.15) | WT |
| 312 | GBM | negative | WT | WT | amplified | nl. (0.84) | nl. (1.00) | WT |
| 313 | GBM | +++ | WT | WT | nl. | NA | NA | WT |
| 314 | GBM | + | G → A | WT | NA | NA | NA | WT |
| codon 285 | ||||||||
| 363 | AA | + | WT | NA | nl. | nl. (0.41) | nl. (0.97) | WT |
Dx., diagnosis; AA, anaplastic astrocytoma; GBM, glioblastoma multiforme; IHC, immunohistochemical staining; WT, wild-type; NA, not available; nl., normal.
p53 scores: +++, strong; +, weak diffuse nuclear staining.
p53
Complete genomic DNA sequence analysis of exons 5–8 of the p53 gene revealed a single point mutation in one of nine tumors sequenced. This tumor was a glioblastoma multiforme that occurred in the suprasellar region of an 18-year-old female who had been radiated 11 years earlier following incomplete resection of a craniopharyngioma. No associated low grade astrocytoma was noted histologically. The point mutation was a G-to-A transition within codon 285 (exon 8) that results in an amino acid substitution from glutamic acid to lysine. Sequencing in the reverse direction confirmed the mutation. Each of the other eight tumors contained wild-type p53 sequence within exons 5–8.
Three of nine tumors had strong (3+) diffuse nuclear immunoreactivity for p53 protein, while two had weak (1+) diffuse nuclear staining. Four tumors had no immunoreactivity for p53. The point mutation in the p53 gene was present in a tumor that had distinct but weak (1+) nuclear immunoreactivity for p53 protein.
EGFR Amplification
Multiplex PCR of eight tumor specimens revealed preferential amplification of the EGFR fragment compared to both β-globin and cystic fibrosis reference in two tumors, T309 and T312 (Figure 2) ▶ . A breast tumor cell line (MDA-468; ATCC), a glioblastoma cell line (SK-MG3), and an epidermoid carcinoma cell line (A-431; ATCC), each containing known EGFR amplification, showed significantly increased bands of amplified EGFR gene compared to references and served as positive controls. 22 In six of the astrocytic tumors, no evidence of increased EGFR band intensity was seen over the reference level.
Figure 2.

Multiplex PCR reaction products of EGFR gene and reference genes (top, β-globin; bottom, cystic fibrosis) resolved by polyacrylamide gel electrophoresis and stained with ethidium bromide. Preferential PCR amplification of the EGFR gene fragment compared to reference gene was seen in tumors T309 and T312. Cell lines SK-MG3, MDA-468, and A-431 are known to contain EGFR amplification and served as positive controls.
One of the two tumors (T312) that demonstrated EGFR amplification by differential PCR also contained strong cytoplasmic and cell membrane immunoreactivity for antibody directed at the extracellular domain of the EGFR protein (Figure 3) ▶ . In the other tumor with EGFR gene amplification (T309), immunostaining was mild. Mild staining was also noted in three of the tumors with no evidence of gene amplification. In four tumors, immunoreactivity was not detected.
Figure 3.
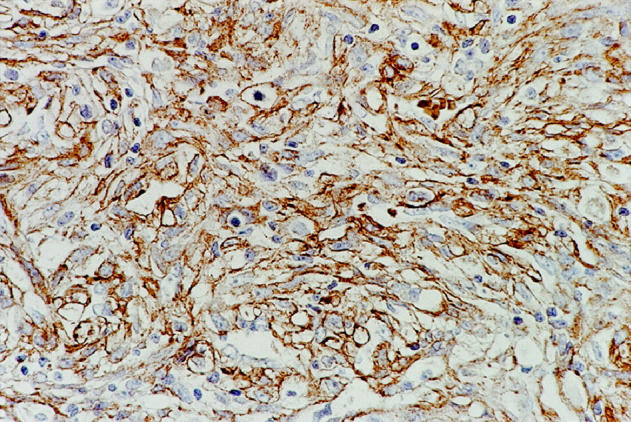
Strong cell membrane and cytoplasmic immunoreactivity for EGFR protein was seen in one of the two tumors that contained amplified EGFR gene by differential PCR (T309).
p16 and MTAP
Seven of the nine radiation-induced astrocytomas provided DNA suitable for deletion analyses of both the p16 and MTAP genes. Relative gene dosages (Dp16 or DMTAP) of p16 and MTAP for each tumor are listed in Table 3 ▶ . Two of the tumors (T308 and T311) had homozygous deletions that included both exon 1 of p16 and exon 8 of MTAP. These genes are located approximately 100 kb apart on chromosome 9p21, suggesting that these two tumors had large deletions of this region of the chromosome. One tumor (T307) had a deletion that encompassed the MTAP gene but not p16.
PTEN
All nine tumors contained wild-type sequence of exons 1–9 of the PTEN gene. No mutations or deletions were noted.
K-ras Proto-oncogene
Wild-type sequence in the region of K-ras proto-oncogene containing codons 12 and 13 was present in all eight tumors tested. No activating point mutations were identified.
Discussion
Epidemiological evidence suggests a relationship between exposure to ionizing radiation and the development of glial neoplasms. For example, patients who receive radiation therapy (mean dose, 4500 cGy) following the surgical resection of pituitary adenomas have a relative risk of 7.9 for the development of subsequent gliomas. 5 Even low doses of radiation given to children for the treatment of tinea capitis have been associated with an increased risk of developing brain tumors. 4 Despite this relationship, no specific radiographic or histopathological features distinguish between radiation-induced and spontaneous astrocytic neoplasms. To determine whether these tumors represent a unique molecular subset of gliomas, we assessed potential genetic alterations of infiltrating astrocytomas that fulfilled clinical definitions of radiation-induced tumors. These astrocytomas occurred within ports of previous external beam radiation, differed histologically from the primary (irradiated) neoplasm, and arose after a latency period of at least 5 years in this series.
With a few exceptions, the spectrum of genetic alterations that we found in radiation-induced gliomas were similar to those described in spontaneous high grade astrocytic tumors (ie, glioblastoma multiforme) of the primary or de novo type. 24,25 Specifically, both p16 deletions and EGFR amplifications were more common in the de novo pathway of GBM formation, in which tumors arise without a well-defined low-grade precursor lesion. 26 We detected EGFR amplifications in two of eight tumors and p16 deletions in two of seven tumors. Although the number of neoplasms analyzed was small, we found a slightly lower percentage of EGFR and p16 alterations in radiation-induced tumors than have been reported for de novo GBMs. In larger series of astrocytic neoplasms, EGFR amplifications have been noted in approximately 10% of anaplastic astrocytomas and 40% of glioblastomas, 10,11 whereas p16 homozygous deletions have been detected in 40–60% of malignant gliomas. 14,23 Homozygous p16 deletions are more common in GBMs with EGFR amplification than in those without EGFR amplification. 27 One study found that 71% of GBMs with EGFR amplification also had homozygous p16 deletions. 27 Within the present series, p16 deletions and EGFR amplifications did not occur in the same neoplasms. This finding may suggest some molecular genetic differences between radiation-induced and spontaneous astrocytomas, but analysis of a larger series will be required to address this more completely.
In the two tumors with p16 deletions, the MTAP gene, located 100 kb telomeric to p16 and encoding methylthioadenosine phosphorylase, was codeleted, indicating a sizable deletion involving chromosome 9p. In one tumor, the MTAP gene was deleted but the p16 gene was not. This genotype has been noted in other spontaneous astrocytomas and may suggest that another tumor suppressor gene is present on chromosome 9 telomeric to p16. 23
Only one of the nine tumors had a p53 mutation detected by direct sequence analysis. p53 mutations have been noted in both progression and primary lesions, but occur with higher frequency in the former. They are thought to represent an early genetic alteration in the progression pathway of GBMs and are therefore common in low grade astrocytomas as well as in high grade tumors that have evolved from a lower grade. 16,25 The tumor in the present series that contained the p53 point mutation was a GBM at clinical presentation, with no histological evidence of an associated lower grade precursor lesion. In keeping with previous studies that have concluded that p53 mutations and EGFR amplification are statistically exclusive events, we did not detect any p53 mutations in tumors that had EGFR amplification.
We did not detect any mutations of the PTEN tumor suppressor gene in the nine tumors that were sequenced. Mutations in PTEN are relatively common in infiltrating astrocytic neoplasms, and have been described in both the de novo and progression pathway of GBMs. They are more common in de novo GBMs and occur in approximately 25–30% of these high grade tumors. 12,28,29 Although the absence of PTEN mutations is these radiation-induced tumors is somewhat surprising, the number of cases analyzed in our series was small.
Unlike some experimental, radiation-induced malignancies, the radiation-induced astrocytic tumors in this series did not harbor activating K-ras mutations in codons 12 or 13. These mutations are a relatively common tumorigenic event in spontaneous neoplasms of the colon, pancreas, lung, and other organs, but are not common in primary brain tumors. 30 The only report of a K-ras mutation in a primary brain tumor is that of an activating point mutation in codon 12 in a pediatric primitive neuroectodermal tumor (PNET) that occurred 9 years after radiation therapy for acute lymphoblastic leukemia involving the CNS. 8 Two other radiation-induced PNETs in the same report did not contain K-ras mutations. Our data do not support a role for activating point mutations in K-ras in the development of radiation-induced astrocytic tumors.
The astrocytic tumors that arose following radiation therapy were all high grade at clinical presentation, and contained primarily molecular genetic alterations similar to those reported for the de novo pathway of GBMs. In this regard, radiation-induced tumors are distinct from infiltrating astrocytic neoplasms that arise spontaneously. In general, de novo GBMs occur in patients in their sixth or seventh decades, whereas GBMs that progress from low-grade precursors occur in patients in their third and fourth decades. 31,32 These radiation-induced gliomas occurred in patients of all ages, but particularly in patients less than 30 years old (66%) who had received radiation therapy as children. Although the low number of patients in this study precludes a statistical analysis, a recent review of the literature concluded that radiation-induced gliomas are nearly all high grade at clinical presentation and that they occur in a younger patient population than would be expected for de novo high grade gliomas. 3 In addition, almost all radiation-induced gliomas are astrocytic in their differentiation (ie, not oligodendroglial or ependymal).
Another unique feature of these tumors is their anatomical distribution. Although radiation-induced astrocytic tumors occur most often in the temporal and frontal lobes, they also occur at sites that are not typical for spontaneous infiltrating gliomas. For example, spontaneous high grade astrocytomas of the suprasellar region and cerebellum are rare, yet they have been noted in our series and in other reports of radiation-induced gliomas. 3 It has also been suggested that radiation-induced gliomas may be more likely to be multifocal. 33 This feature is unlikely to be specific, because multifocal spontaneous high grade gliomas have been well-documented, and it is unclear if multifocal lesions are more frequent following radiation therapy. All of the lesions in the present series were solitary.
In this set of astrocytomas, we did not find any specific or unique mutations, such as base pair deletions in tumor suppressor genes, that might help distinguish radiation-induced tumors from spontaneous ones. Base pair deletions result from the misrepair of double-stranded DNA breaks, a type of DNA damage that occurs following high-dose ionizing radiation. 34,35 Other types of DNA injury do not result in double-strand breaks, and single-strand breaks are repaired without the introduction of base pair deletions. Because of this, base pair deletions could potentially represent a marker for DNA damage caused by radiation. One such homozygous 3-bp deletion in exon 7 of the p53 gene was recently described in a radiation-induced glioblastoma. 7 In our DNA sequence analyses of nine radiation-induced astrocytic tumors, which included exons 1–9 of the PTEN gene, exons 5–8 of the p53 gene, and a short segment of the K-ras gene, we found no evidence of base pair deletions that might be specific for radiation-associated DNA damage. On the basis of our results, it appears that base pair deletions are not a frequent or consistent finding in radiation-induced astrocytic tumors, at least in the genes that we have analyzed.
Only a small percentage of patients who receive CNS radiation therapy will develop subsequent gliomas. One study suggests that the cumulative risk for developing a second brain tumor after radiation therapy for pituitary adenoma is 1.9% over the course of 20 years, and of these only a subset will be gliomas. 5 Outside of the well-known but rare genetic syndromes of radiation sensitivity (eg, ataxia telangiectasia, hereditary retinoblastoma, xeroderma pigmentosa, and others), little is known regarding the susceptibility of individuals within the general population to radiation-induced tumorigenesis. 36 In vitro studies of radiation-induced chromatid damage in peripheral blood lymphocytes have demonstrated that relative genetic instability may be a risk factor for the development of spontaneous malignancies, including gliomas. 37 Similar studies may be helpful in assessing susceptibility to radiation-induced neoplasia. 38
Footnotes
Address reprint requests to Daniel J. Brat, M.D., Ph.D., Department of Pathology, Carnegie 484, Johns Hopkins Hospital, Baltimore, MD 21287. E-mail: dbrat@pds.path.jhu.edu.
Supported in part by National Institutes of Health grants CA 64928, CA 64976, and CA 55728.
D. Connolly’s current address: Department of Pathology, University of Michigan, Ann Arbor, MI.
References
- 1.Harrison MJ, Wolfe DE, Lau T, Mitnick RJ, Sachdev VP: Radiation-induced meningiomas: experience at the Mount Sinai Hospital and review of the literature. J Neurosurg 1991, 75:564-574 [DOI] [PubMed] [Google Scholar]
- 2.Kaschten B, Flandroy P, Reznik M, Hainaut H, Stevenaert A: Radiation-induced gliosarcoma: case report and review of the literature. J Neurosurg 1995, 83:154-162 [DOI] [PubMed] [Google Scholar]
- 3.Simmons NE, Laws ER, Jr: Glioma occurrence after sellar irradiation: case report and review. Neurosurgery 1998, 42:172-178 [DOI] [PubMed] [Google Scholar]
- 4.Ron E, Modan B, Boice JD, Jr., Alfandary E, Stovall M, Chetrit A, Katz L: Tumors of the brain and nervous system after radiotherapy in childhood. N Engl J Med 1988, 319:1033-1039 [DOI] [PubMed] [Google Scholar]
- 5.Brada M, Ford D, Ashley S, Bliss JM, Crowley S, Mason M, Rajan B, Traish D: Risk of second brain tumour after conservative surgery and radiotherapy for pituitary adenoma. Br Med J 1992, 304:1343-1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rimm IJ, Li FC, Tarbell NJ, Winston KR, Sallan SE: Brain tumors after cranial irradiation for childhood acute lymphoblastic leukemia: a 13-year experience from the Dana-Farber Cancer Institute and The Children’s Hospital. Cancer 1987, 59:1506-1508 [DOI] [PubMed] [Google Scholar]
- 7.Tada M, Sawamura Y, Abe H, Iggo R: Homozygous p53 gene mutation in a radiation-induced glioblastoma 10 years after treatment for an intracranial germ cell tumor: case report. Neurosurgery 1997, 40:393-396 [DOI] [PubMed] [Google Scholar]
- 8.Brüstle O, Ohgaki H, Schmitt HP, Walter GF, Ostertag H, Kleihues P: Primitive neuroectodermal tumors after prophylactic central nervous system irradiation in children: association with an activated K-ras gene. Cancer 1992, 69:2385-2392 [DOI] [PubMed] [Google Scholar]
- 9.Guerrero I, Villasante A, Corces V, Pellicer A: Activation of a c-K-ras oncogene by somatic mutation in mouse lymphomas induced by γ radiation. Science 1984, 225:1159-1162 [DOI] [PubMed] [Google Scholar]
- 10.Eckstrand AJ, James CD, Cavenee WK, Seliger B, Petterrson RF, Collins VP: Genes for epidermal growth factor receptor, transforming growth factor α, and epidermal growth factor and their expression in human gliomas in vivo. Cancer Res 1991, 51:2164-2172 [PubMed] [Google Scholar]
- 11.James CD, Carlbloom E, Dumanski JP, Hansen MF, Nordenskjöld M, Collins VP, Cavenee W: Clonal genomic alterations in glioma malignancy stages. Cancer Res 1988, 48:5546-5555 [PubMed] [Google Scholar]
- 12.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R: PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostrate cancer. Science 1997, 275:1943-1946 [DOI] [PubMed] [Google Scholar]
- 13.Jen J, Harper W, Bigner SH, Bigner DD, Papadopoulos N, Markowitz S, Wilson JKV, Kinzler KW, Vogelstein B: Deletion of p16 and p15 genes in brain tumors. Cancer Res 1994, 54:6353-6358 [PubMed] [Google Scholar]
- 14.Giani C, Finocchiaro G: Mutation rate of the CDKN2 gene in malignant gliomas. Cancer Res 1994, 55:6338-6339 [PubMed] [Google Scholar]
- 15.Sidransky D, Mikkelsen T, Schwecheimer K, Rosenblum ML, Cavenee W, Vogelstein B: Clonal expansion of p53 mutant cells is associated with brain tumor progession. Nature 1992, 355:846-847 [DOI] [PubMed] [Google Scholar]
- 16.Louis DN, Gusella JF: A tiger behind many doors: multiple genetic pathways to malignant glioma. Trends Genet 1995, 11:412-415 [DOI] [PubMed] [Google Scholar]
- 17.Schwengel DA, Nouri N, Meyers DA, Levitt RC: Linkage mapping of the human thromboxane A2 receptor (TBXA2R) to chromosome 19p13.3 using transcribed 3′ untranslated DNA sequence polymorphisms. Genomics 1993, 18:212-215 [DOI] [PubMed] [Google Scholar]
- 18.Sakar FH, Kupsky WJ, Li Y-W, Sreepathi P: Analysis of p53 gene mutations in human gliomas by polymerase chain reaction-based single strand conformation polymorphisms and DNA sequencing. Diagn Mol Pathol 1994, 3:2-8 [DOI] [PubMed] [Google Scholar]
- 19.Koga H, Zhang S, Kumanishi T, Washiyama K, Ichikawa T, Tanaka R, Mukawa J: Analysis of p53 gene mutations in low- and high-grade astrocytomas by polymerase chain reaction-assisted single strand conformation polymorphism and immunohistochemistry. Acta Neuropathol 1994, 87:225-232 [DOI] [PubMed] [Google Scholar]
- 20.Oghaki H, Eibl R, Wiestler O, Yasargil M, Newcomb E, Kleihues P: p53 mutations in nonastrocytic human brain tumors. Cancer Res 1991, 51:6202-6205 [PubMed] [Google Scholar]
- 21.Kovach J, McGovern R, Cassady J, Swanson S, Wold L, Vogelstein B, Somer S: Direct sequencing from touch preparations of human carcinomas: analysis of p53 mutations in breast carcinomas. J Natl Cancer Inst 1991, 83:1004-1009 [DOI] [PubMed] [Google Scholar]
- 22.Hunter SB, Abbott A, Varma VA, Olson JJ, Barnett DW, James CD: Reliability of differential PCR for the detection of EGFR and MDM2 gene amplification in DNA extracted from FFPE glioma tissue. J Neuropathol Exp Neurol 1995, 54:57-64 [DOI] [PubMed] [Google Scholar]
- 23.Perry A, Nobori T, Ru N, Anderl K, Borell TJ, Mohapatra G, Feuerstein BG, Jenkins RB, Carson DA: Detection of p16 gene deletions in gliomas: A comparison of fluorescence in situ hybridization (FISH) versus quantitative PCR. J Neuropathol Exp Neurol 1996, 56:999-1008 [DOI] [PubMed] [Google Scholar]
- 24.von Deimling A, von Ammon K, Schoenfeld D, Wieslter OD, Seizinger BR, Louis DN: Subsets of glioblastoma multiforme defined by molecular genetic anaylsis. Brain Pathol 1993, 3:19-26 [DOI] [PubMed] [Google Scholar]
- 25.Watanabe K, Tachibana O, Sato K, Yonekawa Y, Kleihues P, Ohgaki H: Overexpression of the EGF receptor and p53 mutations are mutually exclusive in the evolution of primary and secondary glioblastomas. Brain Pathol 1996, 6:217-224 [DOI] [PubMed] [Google Scholar]
- 26.Biernat W, Tohma Y, Yonekawa Y, Kleihues P, Ohgaki H: Alterations of cell cycle regulatory genes in primary (de novo) and secondary glioblastomas. Acta Neuropathol 1994, 94:303-309 [DOI] [PubMed] [Google Scholar]
- 27.Hayahi Y, Ueki K, Waha A, Wiestler OD, Louis DN, von Deimling A: Association of EGFR gene amplification and CDKN2 (p16/MTS1) gene deletion in glioblastoma multiforme. Brain Pathol 1997, 7:871-875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rasheed BK, Stenzel TT, McLendon RE, Parsons R, Friedman AH, Bigner DD, Bigner SH: PTEN gene mutations are seen in high-grade but not in low-grade gliomas. Cancer Res 1997, 57:4187-4190 [PubMed] [Google Scholar]
- 29.Tohma Y, Gratas C, Biernat W, Peraud A, Fukuda M, Yonekawa Y, Kleihues P, Ohgaki H: PTEN (MMAC1) mutations are frequent in primary glioblastomas (de novo) but not in secondary glioblastomas. J Neuropathol Exp Neurol 1998, 57:684-689 [DOI] [PubMed] [Google Scholar]
- 30.Bos JL: Ras oncogenes in human cancer: a review. Cancer Res 1990, 49:4682-4689 [PubMed] [Google Scholar]
- 31.Burger PC, Scheithauer BW, Vogel FS: Surgical Pathology of the Nervous System and its Coverings. 3rd edition. New York, Churchill Livingstone, 1991
- 32.Burger PC, Scheithauer BW: Tumors of the Central Nervous System. Atlas of Tumor Pathology, 3rd Series, Fascicle 10. 1994, Armed Forces Institute of Pathology Washington, DC
- 33.Fontana M, Stanton C, Pompili A, Amadori S, Mandelli F, Meloni G, Riccio A, Rubinstein LJ: Late multifocal gliomas in adolescents previously treated for acute lymphoblastic leukemia. Cancer 1987, 60:1510-1518 [DOI] [PubMed] [Google Scholar]
- 34.Breimer LH: Ionizing radiation-induced mutagenesis. Br J Cancer 1988, 57:6-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Withers HR: Biological basis of radiation therapy for cancer. Lancet 1992, 339:156-159 [DOI] [PubMed] [Google Scholar]
- 36.Sankaranarayanan K, Chakraborty R: Cancer pre-disposition, radiosensitivity and the risk of radiation-induced cancers. Part I: Background. Radiat Res 1995, 143:121-143 [PubMed] [Google Scholar]
- 37.Bondy ML, Kyritsis AP, Gu J, de Andrade M, Cunningham J, Levin VA, Bruner JM, Wei Q: Mutagen sensitivity and risk of gliomas: a case-control anaylsis. Cancer Res 1996, 56:1484-1486 [PubMed] [Google Scholar]
- 38.Busch D: Genetic susceptibility to radiation and chemotherapy injury: diagnosis and treatment. Int J Radiat Oncol Biol Phys 1994, 30:997-1002 [DOI] [PubMed] [Google Scholar]