

Abstract
Studies suggest that cell-cell interactions may regulate apoptosis, and in particular, the calcium-dependent cell adhesion molecule N-cadherin has been shown to be capable of modulating this process. Rat granulosa cells (GCs) are known to express N-cadherin whereas cAMP is known to induce apoptosis in human and rat GCs. Based on these observations, we hypothesized that N-cadherin regulates human GC apoptosis via a cAMP-dependent mechanism. N-cadherin expression was evaluated in ovarian follicles and corpora lutea utilizing immunohistochemical techniques and in luteinized GCs in culture using immunoblotting, flow cytometric analysis, immunohistochemistry, and indirect immunofluorescence techniques utilizing anti-N-cadherin antibodies directed against both the extracellular and cytoplasmic domains of the molecule. Apoptosis was assessed by TUNEL and DNA fragmentation analysis and confirmed by flow cytometric cell cycle analysis and electron microscopy. The rate of GC apoptosis was found to be two- to three-fold lower among aggregated cells, as compared with single cells. N-cadherin was found to be expressed by aggregating GCs in vitro and GCs cultured in the presence of either N-cadherin function disrupting antibodies or peptides exhibiting enhanced rates of apoptosis. GCs in situ stained intensely for N-cadherin in preantral and normal growing preovulatory follicles as well as early corpora lutea. N-cadherin was weak in atretic follicles and regressing corpora lutea. Exposure of GCs to cAMP increased apoptosis while decreasing N-cadherin protein expression in a dose-dependent manner. Cell culture under serum-free conditions increased apoptosis and decreased N-cadherin expression, in part through cleavage of the extracellular domain of the molecule. The metalloproteinase inhibitor 1-10-phenanthroline inhibited the cleavage of the extracellular domain of N-cadherin and concomitantly inhibited the serum-deprivation-induced apoptosis of aggregated GCs. Collectively, these observations suggest that down-regulation of N-cadherin or the absence of a functional extracellular domain of the molecule prevents cell aggregation and is associated with GC apoptosis. In addition, cAMP induces apoptosis in a dose-dependent manner, and this process is dependent, at least in part, on regulation of the N-cadherin molecule at the surface of the cells. We conclude that N-cadherin-mediated GC signaling plays a central role in follicular and luteal cell survival.
Follicular atresia and luteolysis are important events ensuring ovarian cyclicity and are achieved by a selective degeneration of follicular and luteal cells. Follicular integrity is provided by the establishment of gap junctions and cell-cell adhesion between granulosa cells (GCs). 1 Recent observations suggest that degeneration of follicular and luteal cells is mediated via programmed cell death. 2-4 Typical morphological and biochemical events characterizing apoptosis have been observed in primary human and rat GCs obtained from either preantral or preovulatory follicles. These include chromatin condensation and fragmentation of DNA. 4-6 Because follicular degeneration or corpus luteum regression are associated with loss of cell-cell adhesion sites, 2 it can be hypothesized that cell adhesion molecules (CAMs) are implicated in GC survival and death. Cadherins are a rapidly expanding family of calcium-dependent CAMs. 7 The classical cadherins (CADs) are integral membrane glycoproteins that generally promote cell adhesion through homophilic interactions. Cadherins have been shown to regulate epithelial, endothelial, neural, and cancer cell adhesion, with different CADs expressed on different cell types (reviewed in Ref. 7 ). The structures of the CADs are generally similar. CADs are composed of five extracellular domains (EC1 to EC5), a single hydrophobic domain (TM) that traverses the plasma membrane (PM), and two cytoplasmic domains (CP1 and CP2). The calcium-binding motifs DXNDN, DXD, and LDRE are interspersed throughout the extracellular domains. The first extracellular domain (EC1) contains the classical cadherin adhesion recognition (CAR) sequence HAV (His-Ala-Val). Linear synthetic peptides containing the CAR sequence (ie, FHLRAHAVDINGNQV and LRAHAVDING) and antibodies directed against the CAR sequence have been shown to inhibit CAD-dependent processes. 7-10 More recently, Gour and Blaschuk have developed higher-affinity cyclic peptides containing the CAR sequence (ie, N-Ac-CHAVC-NH2 and N-Ac-CHGVC-NH2) that have been characterized to be even more potent inhibitors of CAD-dependent processes (PCT patent WO 98/02452). Additionally, it has been demonstrated that both rat and mouse GCs express the N-cadherin molecule. 11-14 However, little information is available on the expression and role of N-cadherin on the regulation of apoptosis in human GCs.
The present study was aimed at determining whether N-cadherin, expressed in human GCs, is involved in the mechanism of apoptosis. Moreover, given the previously described role of cAMP in promoting apoptosis in human and rat GCs in vitro, 3,5,6 we investigated whether N-cadherin-mediated cell-cell adhesion counteracts the apoptosis-inducing action exerted by this cyclic nucleotide. We herein show not only that this molecule is expressed by human GCs and mediated cell-cell adhesion but also that its expression is regulated through follicle maturation and corpus luteum (CL) formation. Additionally, cAMP was shown to decrease N-cadherin in a dose-dependent manner. Moreover, we demonstrate that cell-cell aggregation promotes survival of GCs and that loss of N-cadherin from the cell surface induces apoptosis in these cells, supporting a major role of this adhesion molecule in the GC life cycle. Cleavage of the extracellular domain of N-cadherin by metalloproteinases appears to be a critical step in this process.
Materials and Methods
Reagents, Antibodies, and Peptides
All reagents were of analytical grade and were purchased from Sigma Chemical Co. (St. Louis, MO), unless otherwise stated. The monoclonal anti-N-cadherin monoclonal antibody (MAb) (A-CAM, clone GC4), which recognizes the extracellular domain of N-cadherin, was purchased from Sigma. The monoclonal anti-N-cadherin MAb (13A9), which specifically recognizes the cytoplasmic domain of N-cadherin but not P-, E-, or M-cadherin, was a gift from Dr. K. Knudsen (Lankenau Medical Research Center, Wynnewood, PA). 15
The two cyclic peptides N-Ac-CHAVC-NH2 and N-Ac-CHGVC-NH2 were synthesized using standard solid-phase peptide synthetic techniques. The peptides were assembled on a methylbenhydrylamine resin, and acetylation of the amino-terminal was performed by reacting the peptide resins with a solution of acetic anhydride in dichloromethane in the presence of diisopropylamine. The peptide was then side-chain deprotected and cleaved from the resin at 0°C with liquid HF in the presence of anisole as a carbocation scavenger. The crude linear peptides were purified by reverse-phase high-performance liquid chromatography (HPLC), and then the linear peptides were cyclized using I2 in methanol. 16 The cyclic peptide was purified by HPLC and characterized by analytical HPLC and by mass spectral analysis (PCT patent WO 98/02452). The peptides were supplied by Dr. Gour (McGill University).
Cell Culture and Treatments
Human GCs were isolated from 25 patients, aged from 25 to 43 years old, undergoing in vitro fertilization/embryo transfer. These cells had been exposed in vivo to a follicular recruitment regimen including a gonadotropin-releasing hormone (GnRH) agonist (Lupron) for pituitary suppression and to purified follicle-stimulating hormone (FSH; Metrodin or Fertinex, Serono, Randolph, MA) for follicular stimulation. Moreover, all patients had received a single dose of purified human chorionic gonadotropin (hCG) 36 hours before follicular aspiration. The follicular fluid was collected and centrifuged. The sedimented cells were resuspended in Ca2+, Mg2+-free Hanks’ balanced salt solution (Gibco BRL, Life Technologies, Grand Island, NY) overlaid on Ficoll-Hypaque (Organon Teknika, Durham, NC) and centrifuged at 400 × g for 30 minutes. The cells were collected from the interphase. The isolated human GCs were suspended, washed twice with Ca2+, Mg2+-free Hanks’ salt solution and cultured in Hams F-12:DMEM (1:1, v/v; Gibco, Grand Island, NY) media supplemented with 10% fetal bovine serum (FBS), penicillin (10 U/ml), streptomycin (0.05 mg/ml), and fungizone (0.25 mg/ml). Cultured cells were treated with combinations of 8-bromo-cAMP from 0.1 to 1.5 mmol/L, and 1-10 phenanthroline at 3 mmol/L in serum-free Hams F-12/DMEM (1:1, v/v) medium with 0.1% bovine serum albumin (BSA) and 10 mmol/L HEPES. Contamination with monocytes, identified by the anti-CD14 MAb (Becton Dickinson, Lincoln Park, NJ), was <2%. Each experiment was performed at least three times with different cell preparations to ensure consistency of the findings.
Collection and Processing of Ovarian Tissue
Ovarian cortical biopsies measuring approximately 1 mm thick were collected from four parous, healthy women, 25 to 34 years of age, undergoing elective laparoscopic tubal ligation. This work was approved by the Institutional Review Board. Participation was voluntary, and each patient gave informed consent. The ovarian tissue was transferred in sterile bicarbonate-buffered Waymouth media (Hyclone Laboratories, Logan, UT) containing 0.5% BSA. The tissue, while submerged in media, was examined under a stereomicroscope (Nikon) and loosely dissected with two sterile 27-gauge tuberculin needles. The ovarian tissue was minced and placed in a sterile capped test tube with 5 ml of enzyme/media solution that was prepared in bicarbonate-buffered Waymouth media supplemented with 0.5% BSA, 0.1% crude collagenase (collagenase type XI, 1660 U/mg solid), and 0.01% DNAse (DNAse 1, 440 Kunitz U/mg solid). Digestion was performed for 3 hours at 37°C. The solution of ovarian tissue and enzyme media was diluted 1:5 with sterile Waymouth media. The digested tissue was then scanned under the stereomicroscope. Preantral follicles were identified, isolated, recovered with hand-drawn sterilized glass micropipets, and transferred to glass slides precoated with poly-l-lysine for attachment and further processing for indirect immunofluorescence.
Electron Microscopy
Routine Embedding in Epoxy Resin for Conventional Transmission Electron Microscopy
Cells grown on human plasma fibronectin-coated (25 μg/ml; Sigma) plastic coverslips were fixed in ice-cold fixative (2% paraformaldehyde/0.5% glutaraldehyde) in 0.1 mol/L cacodylate buffer, pH 7.4, for 10 minutes. After washing in cacodylate (three times), coverslips were post-fixed in 1% OsO4 in the same buffer for 10 minutes and washed. Cells were then washed three times with 0.05 mol/L maleate buffer (pH 5.2) followed by 1% uranyl acetate in the same buffer (pH 6.0). The cells were dehydrated in a series of graded alcohols (70%, 95%, and 100%) three times for 10 minutes in each step. Sections were infiltrated with resin and allowed to polymerize at 60°C in special slide molds (Electron Microscopy Sciences (EMS), Fort Washington, PA). For immunoelectron microscopy, LR White resin was used for better antigen preservation. For this, cells were fixed and dehydrated as above, without post-contrasting with OsO4 or uranyl acetate. Resin infiltration was followed by polymerization at 55°C for 3 days.
Ultrathin Sectioning
Ultrathin sections (100 nm) were cut with a diamond knife (Diatome, Fort Washington, PA) on a Leica (Chicago, IL) ultramicrotome. LR White sections were collected on formvar-coated 150-mesh nickel grids (Electron Microscopy Sciences). After immunostaining, sections were post-contrasted in 2% uranyl acetate/0.5% aqueous lead citrate. Resin sections were collected on uncoated 150-mesh copper grids (EMS) post-contrasted with 20% uranyl acetate (Amersham, Arlington Height, IL)/0.5% lead citrate, and viewed on a Hitachi H-600 transmission electron microscope (Nissey Sangyo, Gaithersburg, MD) at 75 kV.
Immunoelectron Microscopy
Ultrathin tissue sections in LR White collected on grids were first rinsed on drops of water followed by blocking in 5% serum. Grids were then floated on drops of the primary antibody (GC4 at 1:100 dilution) for 60 minutes at room temperature, and antibody was rinsed off in wash buffer (PBS containing 0.5% BSA, 0.1% fish gelatin (Amersham, Belgium), 0.05% Tween 20, and 0.5M NaCl) to reduce background staining. Grids were then floated for 30 minutes on drops of the secondary, gold-conjugated antibody (1:25 dilution of goat-anti-mouse IgG-Au10, Amersham). Sections were rinsed and post-fixed in 2% glutaraldehyde in PBS for 2 minutes followed by distilled water and contrasting.
Indirect Immunofluorescence
Cells grown on coverslips were washed twice in prewarmed (37°C) DMEM and twice in prewarmed PBS containing 1.5 mmol/L Ca2+ and fixed in 100% methanol (−20°C for 5 minutes). Cells were incubated in 10% normal goat serum (30 minutes at room temperature) and then with the primary antibodies (2 hours at room temperature). A fluorescein-conjugated goat anti-mouse secondary antibody (Jackson Immunoresearch Laboratories, West Grove, PA) was used at 1:200 (30 minutes at room temperature). Coverslips were mounted on glass slides with Fluoromount G (Fisher Scientific, Malvern, PA) containing 1,4-diazabicyclo(2,2,2) octane (DABCO; Polysciences, Warrington, PA) to stabilize fluorescence and photographed with a Nikon microscope.
Paraffin-embedded tissue sections of 5 μm were deparaffinized at 70°C and treated with prewarmed pepsin (0.65 mg/ml in PBS) at 40°C for 5 minutes. Sections were incubated in 0.1% sodium borohydride/PBS for 1 hour at room temperature to reduce tissue endogenous fluorescence. Sections were then blocked with 25% goat serum/4% BSA in PBS. Primary antibodies were diluted in the same buffer and employed for indirect immunofluorescent studies as described above. Sections were mounted with media as above.
Immunohistochemistry
Paraffin-embedded human ovarian tissue sections (5 μm thick) were deparaffinized at 70°C in xylene, rehydrated through a graded ethanol series, and rinsed in PBS (pH 7.4). Sections were treated with 0.1% hydrogen peroxide for 30 minutes at 20°C (to inactivate endogenous peroxidase activity) and washed in PBS. Sections were then digested with prewarmed pepsin (0.65 mg/ml in PBS) at 40°C for 5 minutes, preincubated with 5% blocking serum (normal goat serum, Vector Laboratories, Burlingame, CA), and then incubated with the primary antibody (10 μg/ml) for 1 hour at room temperature. Localization of the primary antibody was performed by incubation of the sections with a biotinylated anti-mouse IgG antibody, and then biotin was detected using an avidin-biotin-peroxidase kit (Vector Laboratories) with diaminobenzidine as the chromogen. Control sections were processed in an identical manner by substitution of the primary antibody with a purified mouse IgG fraction. For the archival ovarian tissue specimens used in this study, dating of the phase of the endometrial cycle was performed on endometrial tissue specimens from the same patients, according to the criteria of Noyes et al. 17
In Situ Detection of Apoptosis
Terminal Deoxynucleotidyl Transferase (TdT)-Mediated dUTP-Biotin Nick End Labeling (TUNEL) Assay
Apoptosis was detected by in situ 3′-end labelling of DNA fragments in vitro. DNA fragments were labeled and detected by use of the reagents and procedures provided in the ApopTag in situ apoptosis detection kit (Oncor, Gaithersburg, MD). Briefly, GCs were fixed in 10% buffered formalin and washed twice in PBS. The cells were then incubated in a humidified chamber at 37°C for 1 hour in the presence of TdT and digoxigenin-11 dUTP and dATP. The cells were washed with buffer and incubated with anti-digoxigenin-fluorescein antibody for 30 minutes at room temperature. The cells were then washed with buffer and observed under epifluorescence and bright-field optics. The nuclear structures of individual cells were stained with propidium iodide.
Electron Microscopy
To induce apoptosis in vitro, cells were incubated in serum-free media for 24 hours. Cells undergoing apoptosis were identified by electron microscopic examination of epoxy-embedded tissue.
DNA Fragmentation Analysis
Detached and attached cells harvested from each set of cultures were pooled and washed in PBS, and approximately 3 × 10 6 cells were collected by centrifugation at 1000 × g for 10 minutes. The pellet of cells was lysed in 30 μl of lysis buffer containing 10 mmol/L TrisHCL, 10 mmol/L EDTA, and 0.5% Triton X-100 for 20 minutes on ice and then treated with RNAse (20 mg/ml) and proteinase K (20 mg/ml), 1 μl, at 37°C for 1 hour, respectively. Then 20 μl of the treated sample with 3 μl of loading buffer was directly added to the wells of a 2% agarose gel and electrophoresed in Tris/boric acid/EDTA (TBE) electrophoresis buffer at 50 V for 4 hours, and the DNA was visualized with ethidium bromide under ultraviolet light.
Flow Cytometry (Cell Cycle Analysis)
Trypsin-generated cell suspensions (including floating cells; minimum of 10 6 cells) were fixed in 70% ethanol for at least 16 hours, treated with RNAse A (Sigma; 500 μg/ml for 30 minutes at room temperature), stained with propidium iodide (PI; 20 μg/ml), and analyzed using an EPICS XL flow cytometer (Coulter Corp., Hialeah, FL). Data were analyzed using a Cellfit program. Cells that contained less than 2n DNA content in the cell cycle analysis profile were considered to be apoptotic. 18
Double (TUNEL) Assay and Indirect Immunofluorescence
To assess the expression of N-cadherin in GCs undergoing apoptosis in vivo and in vitro, we performed double-fluorescent staining for apoptosis (TUNEL) and N-cadherin expression. Briefly, indirect immunofluorescence for N-cadherin was performed and followed by TUNEL assay, as described above.
We first examined the expression of apoptosis in conjunction with the distribution of N-cadherin in isolated GCs, in isolated preantral follicles, and in ovarian tissue sections by using antibodies directed against both the extracellular (GC4) and cytoplasmic (13A9) domain of the molecule. Approximately 200 cells were counted and evaluated for DNA fragmentation in each treatment group. For each treatment group, the mean percentage of apoptotic nuclei ± SE were calculated. For the in vitro treatment groups the percentage of aggregated GCs was determined by counting the number of cells that were single or engaged in aggregates. The mean percentage of aggregated GCs ± SE for each treatment was also calculated.
Immunoblotting
GCs in monolayer cultures were placed on ice and washed twice with ice-cold PBS containing 2 mmol/L phenylmethylsulfonyl fluoride (PMSF). Cells were harvested in 1 ml of 5 mmol/L EDTA/PBS containing 2 mmol/L PMSF at 4°C by scraping off the plates with a cell scraper (Marsh, Rochester, NY) and were centrifuged for 5 minutes at 16,000 × g in a microfuge at 4°C. The cell pellets were lysed with 0.1 ml of PBS lysis buffer containing 0.01 mmol/L Tris acetate (pH 8.0), 0.5% Nonidet P-40, and 0.5 mmol/L Ca2+ (TNC) as well as protease inhibitors (2 mmol/L PMSF, 2 μg/ml antipain, 10 μg/ml benzamidine, 2 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin A, and 1 μg/ml chymostatin). After this procedure, pellets were dissolved through a 28.5-gauge needle (Becton Dickinson, NJ) and incubated on ice for 20 minutes with occasional vortexing. The cell extracts were centrifuged at 16,000 × g in the microfuge at 4°C for 30 minutes. Supernatants were snap-frozen in liquid N2 and kept in −80°C until use. Protein concentrations in the cell extracts were evaluated according to Bradford. 19 Samples (50 μg of protein) were heated at 100°C for 5 minutes and underwent sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), according to Laemmli, 20 in 7% gels under nonreducing conditions. The proteins were electrophoretically transferred to nitrocellulose paper (BA-S NC; Schleicher and Schuell, Keene, NH) using the procedure of Towbin et al. 21 The blots were washed twice in TBS (20 mmol/L Tris, 500 mmol/L NaCl, pH 7.5) for 10 minutes and then incubated in 5% nonfat dry milk (Carnation) TBS for 1 hour. The blots were incubated in the presence of monoclonal N-cadherin antibody (13A9; 10 μg/ml) for 2 hours, washed with TBST (50 mmol/L Tris, pH 7.5, 171 mmol/L NaCl, 0.05% (v/v) Tween 20) for 5 minutes three times, and incubated with horseradish-peroxidase-labeled anti-mouse antibody (Promega, Madison, WI) in 1:2000 dilution for 1 hour. N-cadherin was visualized using chemiluminescence according to the manufacturer’s instructions (ECL Kit, Kirkegaard and Perry, Bethesda, MD).
Flow Cytometric Analysis of Human Luteinizing GCs
Flow cytometry was performed as previously described. 22 The isolated human GCs were washed in PBS with 0.1% BSA (Sigma), sedimented by centrifugation, and incubated with the anti-N-cadherin (GC4 clone, diluted at 1:100) or the purified mouse IgG3 for 1 hour at 4°C. After washing with PBS, the cell pellet was incubated with FITC-congugated goat anti-mouse Ig, for 30 minutes at 4°C in the dark. After washing in PBS, the cells were analyzed using an EPICS XL flow cytometer (Coulter Corp.).
Cell Aggregation Assessed by Time-Lapse Videomicroscopy
Human GCs were cultured in serum-containing medium. For these studies, the Lab-Tek slide was sealed with a mixture of Vaseline and paraffin (20:1; Micro Video Instruments, Arrow, MA) to maintain pH of the medium. The Lab-Tek slide was then placed in a 37°C, humified plexiglass microscope culture chamber (Nikon Corp., Tokyo, Japan). A field containing several GC agreggates was selected and observed under phase contrast. Sequential images were collected at 1-hour intervals over a 24-hour period. To ensure an accurate assessment of cell contact, only GCs with five or fewer cell contacts were examined. A cell contact was considered lost when the cells were completely detached. The number of initial cell contacts that remained intact at each time interval was then counted, and a percentage calculated. At least 70 cell contacts were examined.
Cell-Cell Adhesion Inhibition Experiments
Human GCs were isolated as described above and plated in Lab-Tek slides coated with human plasma fibronectin (25 μg/ml; Sigma) in serum-free medium (DMEM/F12, 1:1, v/v, 15 mmol/L HEPES), for time-lapse videomicroscopy as described above. At time 0, a total of 5 × 10 5 cells per Lab-Tek slide were incubated in the presence of anti-N-cadherin antibody (GC4), 23,24 N-Ac-CHAVC-NH2, or N-Ac-CHGVC-NH2 at a final concentration of 1 mg/ml. For the experiment with the anti-N-cadherin antibody the cells were preincubated with the antibody for 30 minutes before plating. As control, for the GC4 antibody experiments, we used purified mouse IgG3. The cultures were terminated 24 hours from the initiation of the experiment. The cells were washed with PBS (three times) at room temperature, fixed with 10% buffered formalin, and washed again twice in PBS, and a TUNEL assay for detection of apoptosis was assessed as described above.
Statistical Analysis
All experiments were conducted in duplicates and repeated at least three times. The percentage data were analyzed by either one-way analysis of variance followed by Student-Newman-Keuls multiple range test or Student’s t-test. Only P < 0.05 was considered significant.
Results
Establishment of Cell-Cell Contacts Prevents Apoptosis in Granulosa Cells
GC culture for 24 hours under serum-free conditions increased apoptosis as assessed by TUNEL. Cell-cell contact significantly decreased apoptosis as assessed by TUNEL; single GCs were nearly twice as likely to be apoptotic than aggregated GCs (P < 0.05; Figure 1C ▶ ). Cell contact with just one GC resulted in a reduced rate of apoptosis compared with single GCs (P < 0.05; Figure 1C ▶ ).
Figure 1.

Granulosa cell aggregation and apoptosis. A and B: Double immunostaining for nuclei (A) and apoptotic nuclei (B) was performed as described in Materials and Methods. Note the paucity of apoptotic cells within an aggregate (arrow) whereas single cells clearly show positive staining for apoptosis (arrowheads). C: Quantitative analysis showing the marked decrease (*P < 0.05) in apoptotic nuclei when cells were in small (two- to four-cell) aggregates or large (five or more cell) aggregates.
To better define the role of aggregation in modulating GC differentiation, we used electron microscopy to analyze cell morphology in GCs cultured in the presence of serum or under serum-free conditions. Cells cultured for 24 hours spread out as single or aggregated cells. Both single and aggregated cells displayed well developed mitochondria and numerous round osmiophilic lipid-containing secretory droplets, characteristic of steroidogenic cells (Figure 2, A–C) ▶ . The matrix of the nuclei appeared physiological, with normal chromatin condensation (Figure 2, B and C) ▶ . However, many of the single cells cultured in serum-free conditions displayed a dramatic increase in chromatin condensation in the nucleus. The cytoplasm contained damaged mitochondria with ruptured internal membranes, damaged lipid droplets, and autophagic vesicles (Figure 2, A, E, and D) ▶ . In contrast, most of the aggregated cells (even under serum-free condition for 24 hours) did not display such apoptotic features and appeared to have normal ultrastructural characteristics (Figure 2, A–C) ▶ .
Figure 2.
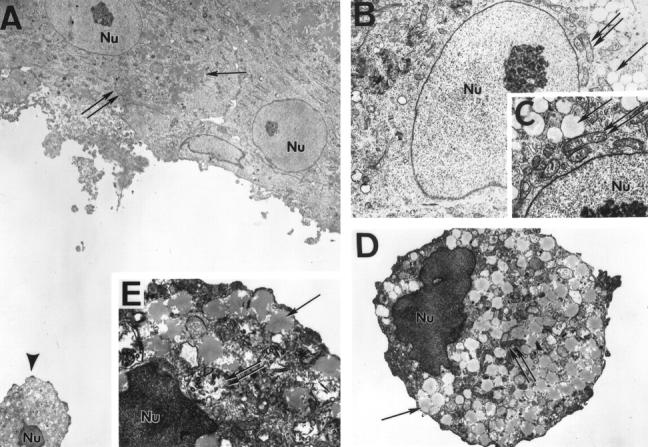
Electron microscopy of normal and apoptotic granulosa cells. A: Cells exhibiting well defined cell-cell contacts show no signs of apoptosis as judged by their ultrastructural characteristics. As shown at higher magnification in B, the nuclei (double arrow, Nu) and cell organelles are intact. Of note are the osmiophilic lipid droplets (arrow) and the intact internal membranes of the numerous mitochondria (C, double arrow). However, single cells (A, arrowhead), shown in detail in D and E, have a pathological ultrastructure: chromatin is condensed in the nuclei and the internal structure of mitochondria is damaged or non-existent (E, double arrows). Lipid droplets have a jagged membrane (E, arrow). Magnification, ×2000 (A), ×8000 (B and D), and ×12,000 (C and E).
N-Cadherin Is Expressed by GCs in Vitro
Using indirect immunofluorescence, we examined the distribution of N-cadherin in isolated GCs. By utilizing a MAb against the extracellular domain of N-cadherin (GC4), the molecule was localized to the surface membranes of single and aggregated GCs and was concentrated at the GC:GC junctional interface in aggregating GCs (Figure 3B) ▶ . Immunoelectron microscopy showed clusters of N-cadherin localized to cell surface microvilli in contact with adjacent cells (Figure 3, F and G) ▶ . Its expression was similar to that shown for E-cadherin in human trophoblast cells. 25 Controls using normal mouse IgG were negative (data not shown). Flow cytometric analysis of N-cadherin expression showed that 75 ± 8% of the cells were positive for N-cadherin (Figure 3, C and D) ▶ . Immunoblotting, performed to characterize the electrophoretic mobility of immunoreactive N-cadherin in human GCs, revealed it to be similar to that expressed in mouse brain (135 kd; Figure 3E ▶ ).
Figure 3.

N-cadherin expression in isolated human granulosa cells. A and B: Phase contrast (A) and indirect immunofluorescence (B) for N-cadherin of human GCs at 24 hours of culture. Note the distribution of N-cadherin at the points of cell-cell contact. C and D: Flow cytometric analysis of human GCs immunostained with control mouse IgG (C) and GC4 antibody (D). Note the marked shift in immunofluorescence when the specific antibody to N-cadherin was used. E: Immunoblotting of human GC membrane extract for N-cadherin (lane 1) showing a distinct band at 135 kd, which coincides with the electrophoretic mobility of the control extract from mouse brain (lane 2). F and G: Immunoelectron microscopy for N-cadherin utilizing the GC4 antibody against the extracytoplasmic domain of the molecule shows the localization of the protein in the region of villous-like protrusions on the surface of the cells (arrows). Magnification, ×68,000 (F and G); bar, 50 μm (A and B:).
Blocking N-Cadherin-Mediated Cell-Cell Adhesion Inhibits Aggregation and Promotes Apoptosis in GCs
Incubation of GCs with the blocking antibody GC4 directed against the human N-cadherin extracellular domain 23,24 inhibited GC aggregation by 25 ± 2.5% of controls when used at a concentration of 1 mg/ml (P < 0.05; Figure 4, C, D, and G ▶ ). Incubation of GCs with the synthetic cyclic peptide N-Ac-CHAVC-NH2 also inhibited GC aggregation. There was a 57 ± 2.4% aggregation of the control value of aggregation in the presence of 1 mg/ml N-Ac-CHAVC-NH2 peptide (HAV; P < 0.05; Figure 4, E–G ▶ ). The nonfunctional control peptide N-Ac-CHGVC-NH2 had no effect on GC adhesion. In the presence of the N-cadherin blocking antibody or the cyclic synthetic peptide N-Ac-CHAVC-NH2, ∼25% to 34% of the GCs formed at least a single GC contact, and these cultures had an increased number of single cells (Figure 4G) ▶ . GCs that established cell contacts in the presence of the GC4 blocking antibody or the synthetic cyclic peptide N-Ac-CHAVC-NH2 had a higher rate of apoptosis than the aggregates of control cultures (P < 0.05; Figure 4H ▶ ). Moreover, neither the antibody nor the cyclic peptide influenced the rate of apoptosis in single GCs (Figure 4H) ▶ . As a control we used the cyclic synthetic peptide N-Ac-CHGVC-NH2 or purified mouse IgG3, at a concentration of 1 mg/ml. These had no effect on GC aggregation or apoptosis.
Figure 4.

Effect of anti-N-cadherin antibody (GC4) and functional cyclic peptide N-Ac-CHAVC-NH2 (HAV) on apoptosis in single and aggregating human granulosa cells in culture. A, C, and E and in higher magnification B, D, and F: Control, anti-N-cadherin-treated (+GC4) and N-Ac-CHAVC-NH2 treated (+HAV) GCs in culture for 24 hours shows the marked interruption of aggregation of the cells and their change in configuration to a more stellate cell shape. Note the significant inhibition (*P < 0.05) of aggregation by both treatments (G). H: Over 50% of single cells exhibited apoptosis as determined by the TUNEL assay, and there was no effect of either the anti-N-cadherin antibody (GC4) or the N-Ac-CHAVC-NH2 peptide (HAV). In contrast, note the lower baseline proportion of apoptotic cells within aggregates and the induction of apoptosis by the anti-N-cadherin antibody (GC4) and the N-Ac-CHAVC-NH2 peptide (HAV) to levels comparable to single cells. (*P < 0.05).
N-Cadherin Is Expressed by Human GCs in Vivoand Is Regulated during Follicular Maturation and Corpus Luteum Formation
Using indirect immunofluorescence in isolated preantral follicles and immunohistochemistry on archival human ovarian tissue specimens, we found that N-cadherin expression on GCs is developmentally regulated. N-cadherin was detected in GCs of primordial and primary follicles (Figure 5, A–C) ▶ . Antral follicles displayed a much stronger N-cadherin staining, which was present throughout all layers of GCs (Figure 6A) ▶ . In antral atretic follicles there were fewer GC layers and the cell-cell contacts between granulosa cells appeared more disrupted (Figure 6, D–F) ▶ . These cells displayed more granular chromatin. In these atretic follicles, N-cadherin staining remained strong in a single GC layer in proximity to the follicular basement membrane and possibly theca cells, whereas there was no N-cadherin staining in the more apical cell layers (Figure 6, D–F) ▶ . Luteal cells were strongly positive for N-cadherin in the early luteal and mid-luteal phase (Figure 6B) ▶ , whereas there was only weak N-cadherin staining in luteal cells during late luteal phase (Figure 6C) ▶ . These studies demonstrated the presence of N-cadherin in human GCs in follicles at different stages of maturation.
Figure 5.

Immunolocalization of N-cadherin in isolated preantral human follicles. Human preantral follicles were isolated from ovarian biopsy specimens and immunostained for N-cadherin. Under low power (A) multiple preantral follicles of varying sizes (arrows) can be seen surrounded by N-cadherin-positive cells. Under high magnification (B and C) it can be clearly seen that the oocytes are negative for N-cadherin whereas remaining/adherent GCs stain intensely for N-cadherin (arrowhead). Magnifications, 100× (A and B), 600× (C).
Figure 6.
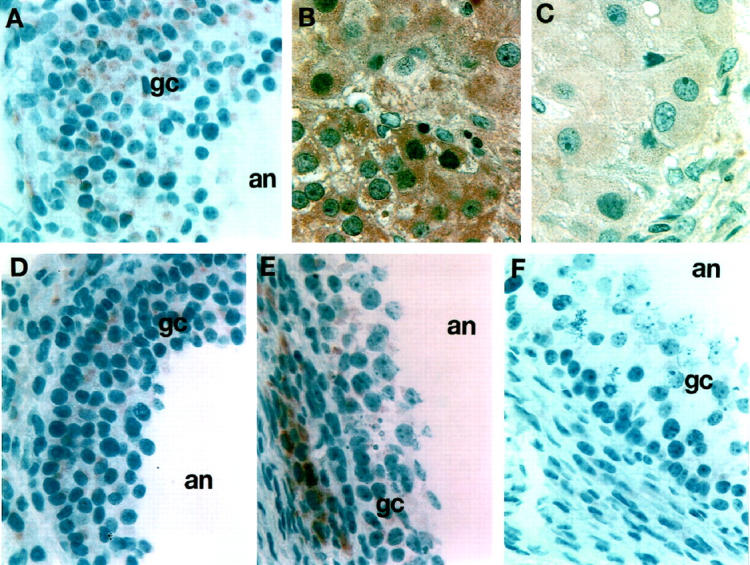
Immunohistochemical detection of N-cadherin in adult human ovary in situ. A: Normal antral follicle with multiple layers of GCs (gc) shows N-cadherin staining through all layers extending to the follicular antrum (an). B and C: Extensive staining for N-cadherin is observed in luteal cells and is more marked in the early/mid luteal phase (B) and is much weaker in a corpus luteum obtained from the late luteal phase (C). D to F: N-cadherin immunostaining of atretic ovarian follicles (at progressive stages early to late D through F), which clearly show limited numbers of GCs that appear to be losing their cell-cell junctions and their cellular integrity. Note the limited staining for N-cadherin of the granulosa cells, which is either almost absent or localized to cells closest to the basement membrane. Magnification, ×300.
cAMP-Induced Apoptosis Is Associated with Down-Regulation of N-Cadherin Expression in Granulosa Cells
To investigate the effect of cAMP on N-cadherin expression, immunoblotting revealed that cAMP decreased N-cadherin protein in a dose-dependent manner (Figure 7A) ▶ . This was also confirmed by flow cytometric analysis (Figure 7, B–D) ▶ . Incubation of GCs with 1 mmol/L cAMP for 24 hours resulted in a 10-fold decrease in N-cadherin fluorescence intensity (P < 0.05) and a 3- to 4-fold decrease in the percentage of N-cadherin-positive cells (P < 0.05; Figure 7 B–D ▶ ). By performing double-fluorescent staining for apoptosis (TUNEL) and N-cadherin in GCs exposed to 1 mmol/L 8-bromo-cyclic AMP for 24 hours we found a temporal association between the cAMP-induced down-regulation of N-cadherin and the induction of apoptosis in these cells (Figure 7, E and F) ▶ . The effect of cAMP on GC apoptosis was also confirmed by cell cycle flow cytometric analysis of DNA content (Figure 8, A–D) ▶ .
Figure 7.

Cyclic AMP regulation of N-cadherin expression in human GCs. A: Immunoblotting for N-cadherin shows a dose response to increasing concentrations of 8-bromo-cyclic AMP and reduction in N-cadherin protein in GCs in culture. Note that at the low concentration of 0.1 mmol/L, there is no effect on N-cadherin expression. B: Dose response of N-cadherin-positive cells to increasing concentrations of 8-bromo-cyclic AMP as assessed by flow cytometric analysis. Note that significant reduction in the N-cadherin-positive cells was achieved with a concentration equal to or over 0.5 mmol/L (*P < 0.05). Note also that low concentrations of cAMP (0.1 to 0.2 mmol/L) had no effect on N-cadherin expression. C and D: Example of the flow cytometric analysis of one representative experiment showing the clear shift to the left when cells were treated for 24 hours with 1 mmol/L 8-bromo-cyclic AMP (+cAMP). E and F: Double immunocytochemistry for apoptosis (E) and N-cadherin (F) in GCs in culture after exposure of the cells to 1 mmol/L 8-bromo-cyclic AMP for 24 hours. Note the large aggregate at the center of the figure indicating extensive number of apoptotic nuclei (E) and the staining with N-cadherin (F) only of the cells in the periphery of the aggregate, which show no signs of apoptosis (arrow).
Figure 8.

Serum deprivation and cAMP effect in human GC apoptosis. Cell cycle flow cytometric analysis of DNA content shows an increase in apoptosis (region E) under serum-free conditions (C, SF) and an additional increase after the addition of 1 mmol/L cAMP (D, SF+cA), as compared with control cultures (B, serum). (*P < 0.05).
N-Cadherin Extracellular Domain Is Present Only in Non-Apoptotic GCs
By performing double-fluorescent staining for N-cadherin expression and apoptosis (TUNEL), we attempted to correlate apoptosis in GCs with the expression of N-cadherin. This was carried out in cultured GCs as well as in isolated ovarian follicles and human ovarian tissue sections. Antibodies recognizing either the extracellular or cytoplasmic domains of the molecule were used. The cells expressing the extracellular domain of N-cadherin were not apoptotic and absence of expression correlated with apoptosis (Figure 9, C and D) ▶ . In preantral follicles almost all surrounding GCs were positive for N-cadherin (GC4) (Figure 5, A–C) ▶ , and we were unable to demonstrate any apoptosis in these cells (data not shown). In addition, co-localization of apoptotic cells not stained for N-cadherin (GC4) in situ corroborate our in vitro observations (Figure 9, I–K) ▶ . In contrast, by using an antibody recognizing the cytoplasmic domain of N-cadherin (13A9), we could identify numerous apoptotic GCs that were positive for N-cadherin (Figure 9, E and F) ▶ . This suggested a strong correlation between cleavage of the extracellular domain of N-cadherin and activation of apoptosis.
Figure 9.

Immunohistochemical localization of N-cadherin in GCs induced to become apoptotic after serum deprivation for 24 hours. A, B, G, and H: Control phase (A and G) and indirect immunofluorescence (B and H) showing extensive N-cadherin immunolocalization at cell-cell borders immediately after serum removal from the culture. C to F: Double staining to detect apoptotic nuclei (C and E) and with anti-N-cadherin antibody against the extracellular domain (D) or the cytoplasmic domain of N-cadherin (F). Note that when the antibody against the extracellular domain to N-cadherin was used, the apoptotic cells (C, arrows) do not stain for N-cadherin although a clearly non-apoptotic cell at the center of the field (gc) is markedly positive for N-cadherin (D, open arrows). In contrast, utilizing the N-cadherin antibody directed against the cytoplasmic domain of the molecule indicates that apoptotic cells (E, arrows) continue to stain for N-cadherin (F, arrowhead), suggesting that cleavage of the extracellular domain of the molecule participates in the events leading to apoptosis of the cells. I, J, and K: Phase (I) and double staining to detect apoptotic nuclei (J) and N-cadherin antibody against the extracellular domain (K) in an atretic follicle in situ. Note that all are apoptotic cells (J, arrows) and do not stain for N-cadherin (K, arrows).
Inhibition of Cleavage of the Extracellular Domain of N-Cadherin Prevents Apoptosis in Granulosa Cells
To better define the relationship between the cleavage of the extracellular domain of N-cadherin and activation of apoptosis, we incubated GCs in serum-free culture media for 24 hours in the presence of 1–10 phenanthroline, a metalloproteinase inhibitor that blocks the cleavage of the extracellular domain of N-cadherin. 26 We found that 1–10 phenanthroline inhibited the serum-deprivation-induced decrease of N-cadherin as assessed by immunoblotting (Figure 10, A and B) ▶ and flow cytometric analysis (Figure 10, D–G ▶ ; P < 0.05). When the antibody against the cytoplasmic domain was used (13A9), a 40-kd cleavage fragment representing the cytoplasmic tail of the molecule was evident (Figure 10A) ▶ . In addition, another fragment of ∼90 kd was also evident. The significance of this larger N-cadherin fragment is unclear as it has not been described before (see Discussion). In addition, 1–10 phenanthroline decreased the serum-deprivation-induced apoptosis of aggregated human GCs by 50% of control as assessed by TUNEL (P < 0.05; Figure 10C ▶ ). The rate of apoptosis in single cells was unaffected by treatment of cells with 1–10 phenanthroline (Figure 10C) ▶ . DNA fragmentation electrophoretic analysis clearly demonstrated that serum deprivation induced this late event of apoptosis (Figure 11 ▶ , lane 2), whereas 1–10 phenanthroline inhibited it (Figure 11 ▶ , lane 3). In a parallel experiment and in agreement with the results presented in Figures 4H, 7, E and F, and 8 ▶ ▶ ▶ , treatment of the cells in culture with the HAV peptide or cAMP further confirmed that these treatments induce apoptosis by demonstrating DNA fragmentation (Figure 11 ▶ , lanes 1 and 5).
Figure 10.

Effect of 1–10 phenanthroline on N-cadherin expression and apoptosis on GCs in culture. Immunoblotting for N-cadherin by utilizing the 13A9 antibody against the cytoplasmic domain of N-cadherin (A) or the GC4 antibody against the extracellular domain of N-cadherin (B). Note the reduction of the 135-kd N-cadherin protein in GCs in the absence of serum (A and B, SF) and the appearance of lower molecular weight fragments when the cytoplasmic domain antibody was used (A). In contrast, 1–10 phenanthroline reverses this effect (A and B). C: Note that the metalloproteinase inhibitor had no effect on the percentage of apoptotic single cells but significantly decreased (*P < 0.05) the percentage of apoptotic nuclei in cellular aggregates. D to F: Flow cytometric analysis for N-cadherin of one representative experiment showing the clear shift to the right when cells were treated for 24 hour with 3 mmol/L 1–10 phenanthroline (NS+Phen). G: Quantitative analysis clearly indicates a significant increase of immunodetectable N-cadherin in the presence of 1–10 phenanthroline (*P < 0.05).
Figure 11.
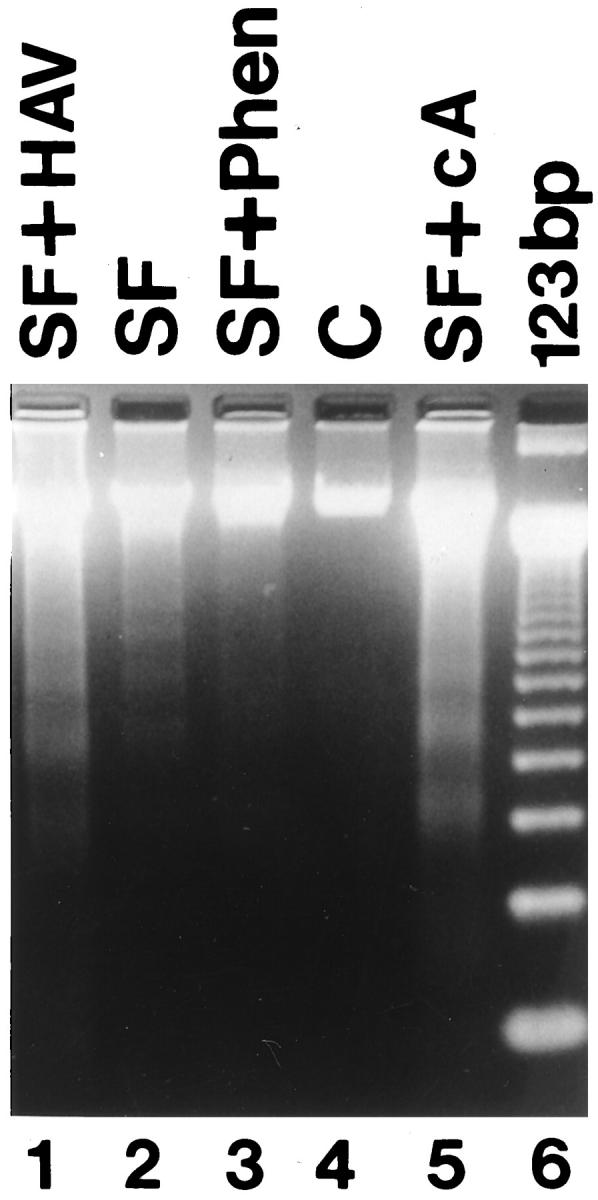
Electrophoretic analysis of DNA fragmentation. Adherent and detached GCs from flasks were pooled and resolved by electrophoresis on 2% agarose gel as described in Materials and Methods. The appearance of the banding pattern after serum removal (SF) indicates that DNA fragmentation characteristic of apoptosis occurred. Note that DNA fragmentation was further increased in the presence of cAMP (SF+cA) or in the presence of the blocking N-cadherin cyclic peptide (SF+HAV). Note also that the presence of 1–10 phenanthroline (SF+Phen) reversed the effect on DNA fragmentation. C: Serum, DNA markers (123 bp).
Discussion
This study demonstrates that human GC aggregation promotes cell survival and that N-cadherin expression by these cells mediates homophilic-homotypic cell adhesion. These observations along with the pattern of the in vivo expression of N-cadherin in the human ovary and the promotion of apoptosis when the N-cadherin-mediated aggregation was blocked in vitro suggest a role for this molecule in GC programmed cell death. In addition, our data extend these conclusions by providing two possible mechanisms by which apoptosis may be controlled in these cells: 1) through a cAMP-mediated down-regulation of N-cadherin and 2) through the enzymatic cleavage of the extracellular domain of the molecule. Therefore, the regulation of expression and processing of N-cadherin by human GCs may play a critical role in normal follicular development and the maintenance of the corpus luteum.
There is strong evidence in the literature supporting the hypothesis that both follicular atresia and corpus luteum regression are mediated by apoptosis. 2,4 In the present study, we found that cell-cell contacts provide a potent stimulus promoting GC survival under apoptosis-inducing conditions, such as growth factor deprivation. In particular, the data indicate that under serum-depleted conditions, aggregated GCs were two times less likely to be apoptotic compared with single cells. Moreover, GCs engaging in aggregate formation displayed a well developed apparatus of micro-organelles, including secretory lipid droplets, mitochondria, and surface microvilli, whereas many single cells displayed ultrastructural features of cells undergoing apoptosis.
Using an antibody (13A9) directed against the highly conserved cytoplasmic region of human N-cadherin, a protein approximately 135 kd in size was detected in GCs. The electrophoretic mobility of immunoreactive N-cadherin in humans is similar to that expressed in rat (120 to 140 kd), 11 and this is consistent with a high degree of conservation at both the amino acid and gene levels reported for the cadherin molecules, which is as high as 90% for the carboxyl-terminal portion of the protein. 27-29 In addition, our studies demonstrated that N-cadherin was localized to the junctional interface of aggregating GCs both by immunofluorescence and by immunoelectron microscopy.
If N-cadherin-mediated GC aggregation plays an important role in promoting cell survival, then it would be anticipated that: 1) disruption of N-cadherin-mediated cell-cell adhesion would promote apoptosis, 2) factors known to induce GC apoptosis might affect N-cadherin expression, and 3) prevention of N-cadherin cleavage from the cell surface would prevent apoptosis. We were able to demonstrate that N-cadherin fulfilled all of these anticipated events.
In the first of a series of functional studies, we investigated the effect of inhibition of N-cadherin-mediated cell aggregation utilizing an antibody or a blocking peptide. Our data clearly show that the use of these molecules not only inhibited GC aggregation but also increased apoptosis in these cells, thus strongly suggesting a direct link between N-cadherin-mediated GC adhesion and cell survival. Nevertheless, it should also be noted that, despite the presence of a blocking antibody or the N-Ac-CHAVC-NH2 peptide, 25% to 34% of GCs formed at least a single cell-cell contact, suggesting that other cell adhesion molecules may mediate adhesion between human GCs. 30 We found a higher degree of disruption of human GCs in the N-Ac-CHAVC-NH2 cyclic-peptide-treated cultures compared with the blocking antibody (GC4). This may be due to the smaller size of the peptide, allowing it to penetrate better in the intercellular spaces and disrupt cell-cell adhesion. Of note is that our data showed a higher inhibition of aggregation in human GCs compared with what has been demonstrated in rat GCs. 11 This may be due to species differences or, alternatively, to our use of a different function-blocking peptide. N-Ac-CHAVC-NH2 (HAV) was designed as a cadherin adhesion recognition (CAR) sequence homologue, which could act as an inhibitor of cell adhesion. To serve as effective CAR sequence homologues, peptides must be able to bind to the cadherin molecule with high affinity. Cyclization of peptides enhances the affinity of these molecules to their respective ligands and have proven more effective in the study of function of cell adhesion molecules. The information necessary for cadherin-mediated adhesion resides with the HAV motif. Differential antagonistic activities of differently constrained HAV-containing peptides can be explained by the relative orientations of the His and Val side chains. Consequently, to retain biological activity, cyclic constructs must influence the backbone conformation without compromising the crucial side chain interaction with the ligand molecule. When this is accomplished, incorporation of the HAV sequence into a cyclic structure restricts the conformations available to the peptide backbone and leads to dramatic increases in potency when compared with the linear analogues. This is the case with N-Ac-CHAVC-NH2.
We then focused our attention on the expression of N-cadherin in vivo. It was felt that correlation of our in vitro data with the in vivo situation was critical to determine the relevance of our studies to the normal physiology of follicular development and corpus luteum formation and regression. Our data strongly suggest that the expression of N-cadherin is regulated in human GCs in vivo during follicular maturation and corpus luteum formation. Preantral follicles isolated from human ovaries 31,32 provided us with the opportunity to pinpoint the ontogeny of N-cadherin protein in single, well defined follicular stages. N-cadherin protein expression was found in almost all GCs in primordial, primary, and secondary follicles. These data were confirmed by immunohistochemical studies performed on archival ovarian tissue. The molecule was expressed throughout all stages of early follicular development. Moreover, although in healthy antral follicles N-cadherin protein was detected in all GC layers, in atretic antral follicles we observed only a single cell layer expressing the molecule. This was in cells of the compact layer adjacent to the basement membrane. In contrast, the more apical layers, which exhibited features of cellular degeneration, displayed no N-cadherin immunoreactivity. We also found strong staining for N-cadherin in almost all luteal cells in early luteal phase corpora lutea with weak or absent N-cadherin protein staining in the late luteal phase. All of the in situ observations suggest a direct correlation between the presence of the N-cadherin molecule and absence of features characteristic of cellular apoptosis. Our finding that apoptosis does not occur in preantral follicles isolated from human ovarian biopsies parallels the observations in the rat ovary, where apoptosis does not occur in primordial and primary follicles, 33 and is also in agreement with recent findings utilizing archival human ovarian tissue. 2 Taken together, the in situ observations suggest that follicular cell apoptosis is initiated when follicles begin to enter the antral stage of development. 2 What initiates departure from the resting pool and apoptosis in some but not all of these follicles is, at present, unresolved.
Given both the functional in vitro data on the role of N-cadherin in GC apoptosis and the correlation of follicular atresia and corpus luteum regression with the absence of N-cadherin, it was felt that understanding the regulation of expression and processing of the molecule was critical. We initiated the studies by evaluating the role of cAMP on N-cadherin expression. The rationale behind these studies was based on observations that cAMP may have both pro- and anti-apoptotic action(s) on GCs. As assessed by immunoblotting and flow cytometric analysis, we found that low concentrations of cAMP (0.1 to 0.2 mmol/L) had no effect on N-cadherin protein expression, whereas higher concentrations of cAMP (up to 1.5 mmol/L) decreased N-cadherin protein in a dose-dependent manner. Thus, in our experimental system, cAMP proved to be predominantly pro-apoptotic. We suggest that N-cadherin may be intimately involved in the early cascade of events that mediate cAMP-induced apoptosis in human GCs. That intracellular cAMP can induce apoptosis is a well established phenomenon in a variety of tissue culture models, including GCs. 5,6,34,35 This appears somewhat paradoxical, as cAMP, as second messenger of both FSH and LH/hCG action on GCs, may have anti-apoptotic effects. 5,6 It has been suggested that this difference is likely due to the significantly higher intracellular concentration of cAMP achieved through in vitro exposure of the cells to exogenous cAMP analogues compared with endogenous cAMP generated after exposure of GCs to the gonadotropin. 5,6 That high concentrations of cAMP can induce apoptosis has been previously shown in human GCs after exposure of the cells to forskolin and, even more dramatically, to 8-bromo-cAMP. 6 These observations are in line with data obtained in the rat GCs. 5 The pro-apoptotic action of cAMP does not appear to be a pharmacological but rather a physiological phenomenon, given that the effects were observed using higher but, nevertheless, physiological concentrations of the cAMP analogues. Alternatively, an explanation for these paradoxical observations may be that gonadotropins exert their protective effect through a different intracellular pathway and that cellular fate is determined by a quantitative balance between pro- and anti-apoptotic intracellular signals. Recent findings support such a hypothesis as FSH was shown to activate protein phosphorylation through tyrosine kinase activation. 35 In addition, signaling pathways involving protein kinase A and intracellular Ca2+ have also been implicated in apoptosis of other cell systems. 36,37 Experimental evidence suggests that high levels of intracellular cAMP may induce the release of calcium ions activating calcium-dependent endonucleases responsible for DNA fragmentation. 38 Of interest is that despite the induction of apoptosis in the above referenced studies using GCs, steroidogenesis did not decline. 6 This would suggest that apoptotic cells can still be actively involved in steroid production until total cell collapse has occurred. This conclusion is further strengthened by recent findings that, in apoptotic cells, steroidogenic organelles, including lipid droplets, mitochondria, and smooth endoplasmic reticulum, remain intact during bleb formation and nuclear fragmentation. 3 Moreover, it has been shown that these organelles are clustered more densely in the perinuclear region during the early phases of apoptosis, and it was suggested that coupling between the organelles carrying the steroidogenic enzymes may become more efficient. 3 Our findings provide additional insight into the molecular mechanism(s) associated with cAMP-induced apoptosis in GCs; specifically, our data demonstrate that cAMP regulation of expression of N-cadherin by GCs is critical in GC-GC adhesion and cell survival.
An alternative pathway for the modulation of cell surface expression and function of N-cadherin is its processing through cleavage of the extracellular domain of the molecule. This led us to hypothesize that one mechanism of regulation of N-cadherin function in GCs could be its proteolytically mediated turnover at the cell surface. Metalloproteinases (MMPs) are able to cleave the extracellular domain of cadherins. Specifically, metalloproteinase activity at the cell surface has been shown to cleave the N-cadherin molecule and to release a soluble 97-kd amino-terminal fragment in the culture media. 26 Of particular interest is that MMP-2 and MMP-9 have been identified in human luteinized GCs. 39 MMP activity is regulated by tissue inhibitors of metalloproteinases (TIMPs). 41 A TIMP-like protein has been identified in the human follicular fluid with increasing activity during follicular maturation, 39,40 and TIMP-1 mRNA has also been identified in preovulatory human GCs. 41 TIMP-1 and TIMP-2 have also been proposed to play a regulatory role in ovarian connective tissue remodeling. 39 In the present studies, our data demonstrate that, under apoptosis-inducing conditions (serum free), N-cadherin is cleaved and its cleavage is inhibited by 1–10 phenanthroline, an MMP inhibitor. Immunoblot studies utilizing an antibody against the cytoplasmic domain of N-cadherin showed the presence of a 135-kd product in the presence of serum and a 40-kd fragment, representing the cytoplasmic domain. A third fragment of approximately 90 kd was identified in the absence of serum, which could represent the intracellular domain and a portion of the extracellular domain. This suggests that in GCs an alternative cleavage site (and thus inactivation) of the N-cadherin molecule may exist. When an antibody against the extracellular domain of the molecule was used for immunoblotting, a decrease in the detectable membrane-associated N-cadherin was observed. This finding is also in line with cleavage of the extracellular domain of the molecule under apoptosis-inducing conditions. It should be kept in mind that the soluble, cleaved fragment may be functionally active in culture and contribute to further inhibition of the binding of N-cadherin molecules between GCs still possessing intact molecules on their cell surface. In support of our findings is the observation that VE-cadherin was shown recently to be involved in endothelial cell survival and, specifically, that shedding of the molecule from the cell surface promoted apoptosis. 42 Thus, in the present studies, we showed that GC apoptosis was associated with the cleavage of the extracellular domain of the N-cadherin molecule and that prevention of this molecular event was specifically capable of preventing apoptosis as assessed by a number of independent experimental methods.
Taken together, the present data provide strong evidence that N-cadherin-mediated cell-cell adhesion is critical for the survival of GCs. The signaling cascade following the homotypic/homophilic interactions between N-cadherin molecules on aggregating GCs remains to be elucidated. Despite the variability of the extracellular domain between different cadherins, all members of this family possess a highly conserved cytoplasmic domain that functions as a binding site for catenins, which mediate binding to the cytoskeleton. It is possible that occupation of the cell recognition site on the extracellular domain is followed by activation of signal transduction pathways ultimately leading to up-regulation of survival factors. In support of this hypothesis is the interaction of N-cadherin with catenins that was recently shown to promote cell viability. 43 Alternatively, it is possible that occupation of the extracellular domain of N-cadherin induces conformational changes in the cytoskeleton and cell shape that might provide nonspecific signals promoting cell survival. Cleavage of the molecule by metalloproteinases may result in the withdrawal of these nonspecific supporting mechanisms, leading the cell to apoptosis. In any case, regulation of N-cadherin expression by extracellular proteolysis provides for a number of theoretical possibilities for modulation of cell adhesive interactions participating in the survival of human GCs. If the expression and/or the activity of an N-cadherin cleaving proteinase can be controlled locally, this could provide an additional mechanism for the fine-tuning of the GC position, cell-cell contact, and survival of the cell and hence the follicle or corpus luteum.
In summary, the present studies have shown that N-cadherin is expressed by human GCs and that this molecule mediates cell-cell adhesion between GCs. We observed a strong correlation between N-cadherin expression by granulosa or luteal cells and follicular survival in isolated follicles and archival tissue sections. In fact, we found strong expression of the molecule by GCs in follicles of the resting pool, of growing antral follicles, and of healthy corpora lutea. In contrast, the molecule was lost in degenerating GCs of atretic follicles and in luteal cells of the late luteal phase. Furthermore, we demonstrated that cell-cell adhesion is critical to the survival of GCs and that N-cadherin-mediated cell-cell adhesion is a critical mediator of survival signals and inhibits apoptosis in these cells. Our data provide two possible mechanisms by which apoptosis may be triggered in GCs, namely, through cAMP-mediated pathways that also induce down-regulation of N-cadherin and through the enzymatic cleavage of the extracellular domain of the molecule. Taken together, our experimental observations strongly support the concept that this adhesion molecule may be intimately involved in determining the fate of follicles and of the corpus luteum. Specifically, we propose that N-cadherin-mediated GC-GC adhesion initiates a signal transduction cascade that can modulate, at least in part, apoptosis of follicular and luteal cells.
Acknowledgments
We thank Celeste Ferreira-Cornwell for fruitful discussions, Samantha Bunso and Jennifer Bucci for their expert isolation of the GCs used in these studies, Sacha Montas and Eugenia Argyris for their excellent technical assistance, and Valerie Baldwin for her help in the preparation of this manuscript.
Footnotes
Address reprint requests to Dr. Christos Coutifaris, Division of Human Reproduction, Department of Obstetrics and Gynecology, University of Pennsylvania Medical Center, 3400 Spruce Street, 106 Dulles Building, Philadelphia, PA 19104. E-mail: ccoutifaris@obgyn.upenn.edu.
Supported by NIH grant HD-31903 (C. Coutifaris), by the Alexander Onassis Foundation (A. Makrigiannakis), and by the Medical Research Council of Canada (O. Blaschuk).
Presented in part at the 80th Meeting of the Endocrine Society (New Orleans, LA, June, 1998).
References
- 1.Amsterdam A, Plehn DD, Suh BS: Structure-function relationships during differentiation of normal and oncogene-transformed granulosa cells. Biol Reprod 1992, 46:513-522 [DOI] [PubMed] [Google Scholar]
- 2.Yuan W, Judice L: Programmed cell death in human ovary is a function of follicle and corpus luteum status. J Clin Endocrinol Metab 1997, 82:3148-3155 [DOI] [PubMed] [Google Scholar]
- 3.Keren-Tal J, Suh BS, Dantes A, Lindner S, Oren M, Amsterdam A: Involvement of p53 expression in cAMP-mediated apoptosis in immortalized granulosa cells. Exp Cell Res 1995, 218:283-295 [DOI] [PubMed] [Google Scholar]
- 4.Tilly JL, Kowalski Kl, Johnson AL, Hsueh AJW: Involvement of apoptosis in ovarian follicular atresia and postovulatory regression. Endocrinology 1991, 129:2799-2801 [DOI] [PubMed] [Google Scholar]
- 5.Aharoni D, Dantes A, Obey M, Amsterdam A: cAMP-mediated signals as determinants for apoptosis in primary granulosa cells. Exp Cell Res 1995, 218:271-282 [DOI] [PubMed] [Google Scholar]
- 6.Breckwoldt M, Selaraj N, Aharoni D, Barash A, Segal I, Insler V, Amsterdam A: Expression of Ad4-BP/cytochrome 450 side chain cleavage enzyme and induction of cell death in long term cultures of human granulosa cells. Mol Hum Reprod 1996, 6:391-400 [DOI] [PubMed] [Google Scholar]
- 7.Munro SB, Blaschuck OW: The structure, function and regulation of cadherins. Brodt P eds. Cell Adhesion and Invasion in Cancer Metastasis. 1996, :pp 17-34 TX, RG Landes, Austin [Google Scholar]
- 8.Alexander JS, Blaschuk OW, Haselton FR: An N-cadherin-like protein contributes to solute barrier maintenance in cultured endothelium. J Cell Physiol 1993, 156:610-618 [DOI] [PubMed] [Google Scholar]
- 9.Blaschuk OW, Pouliot Y, Holland PC: Identification of a conserved region common to cadherins and influenza strain A hemagglutinins. J Mol Biol 1990, 211:679-682 [DOI] [PubMed] [Google Scholar]
- 10.Blaschuk OW, Sullivan R, David S, Pouliot Y: Identification of a cadherin cell adhesion recognition sequence. Dev Biol 1990, 139:227-229 [DOI] [PubMed] [Google Scholar]
- 11.Peluso JJ, Pappalardo A, Trolice MP: N-cadherin-mediated cell contact inhibits granulosa cell apoptosis in a progesterone-independent manner. Endocrinology 1996, 137:1196-1203 [DOI] [PubMed] [Google Scholar]
- 12.MacCalman CD, Farookhi R, Blaschuk OW: Estradiol regulates E-cadherin mRNA levels in the surface epithelium of the mouse ovary. Clin Exp Metastasis 1994, 12:276-282 [DOI] [PubMed] [Google Scholar]
- 13.Blaschuk OW, Farookhi R: Estradiol stimulates cadherin expression in rat granulosa cells. Dev Biol 1989, 136:564-567 [DOI] [PubMed] [Google Scholar]
- 14.Farrookhi R, Blaschuk OW: Cadherins and ovarian follicular development. Gibori G eds. Signaling Mechanism and Gene Expression in the Ovary. 1991, :pp 254-260 Springer-Verlag, New York [Google Scholar]
- 15.Knudsen KA, Soler AP, Johnson KR, Wheelock MJ: Interaction of α-actinin with the cadherin cell-cell adhesion complex via α-catenin. J Cell Biol 1995, 130:67-77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kamber B: Cystine peptides from (S-acetamidomethyl-cysteine)-peptides through oxidation with iodine: synthesis of cyclo-l-cystine. Helvetica Chim Acta 1971, 54:927-930 [DOI] [PubMed] [Google Scholar]
- 17.Noyes RW, Hertig AT, Rock J: Dating the endometrial biopsy. Fertil Steril 1950, 1:3-7 [DOI] [PubMed] [Google Scholar]
- 18.McGahon AJ, Martin SJ, Bissonette RP, Mahboubi A, Shi Y, Mogil RJ, Nishioka WK, Green DR: The end of the (cell) line: methods for the study of apoptosis in vitro. Methods Cell Biol 1995, 46:153-185 [DOI] [PubMed] [Google Scholar]
- 19.Bradford MM: A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976, 72:248-254 [DOI] [PubMed] [Google Scholar]
- 20.Laemmli UK: Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227:680-685 [DOI] [PubMed] [Google Scholar]
- 21.Towbin H, Stachelin T, Gordon J: Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA 1979, 76:4350-4354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fujiwara H, Fukuoka M, Yasuda K, Ueda M, Imai K, Goto Y, Suginami H, Kanzaki H, Maeda M, Mori T: Cytokines stimulate dipeptidyl peptidase-IV expression on human leutenizing granulosa cells. J Clin Endocrinol Metab 1994, 79:1007-1011 [DOI] [PubMed] [Google Scholar]
- 23.Volk T, Geiger B: A 135-kd membrane protein of intercellular adherens junctions. EMBO J 1984, 3:2249-2260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Volk T, Volberg T, Sabanay I, Geiger B: Cleavage of A-CAM by endogenous proteinases in cultured lens and developing chick embryos. Dev Biol 1990, 139:314-326 [DOI] [PubMed] [Google Scholar]
- 25.Coutifaris C, Kao L, Sehdev H, Chin U, Babaola G, Blaschuk O, Strauss J, III: E-cadherin expression during the differentiation of human trophoblasts. Development 1991, 113:767-777 [DOI] [PubMed] [Google Scholar]
- 26.Paradies NE, Grunwald GB: Purification and separation of NCAD90, a soluble endogenous form of N-cadherin, which is generated by proteolysis during retinal development and retains adhesive and neurite-promoting function. J Neurosci Res 1993, 36:33-45 [DOI] [PubMed] [Google Scholar]
- 27.Nose A, Nagafuchi A, Taceichi M: Isolation of placental cadherin cDNA: identification of a novel gene family of cell-cell adhesion molecules. EMBO J 1987, 6:3655-3661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ringwald M, Schuh R, Vestweber D, Eistetter H, Lottspeich F, Engel J, Dolz R, Jahnig F, Eppen J, Mayer S, Muller C, Kemler R: The structure of cell adhesion molecule uvomorulin: insights into the mollecular basis of Ca2+-dependent cell adhesion. EMBO J 1987, 6:3647-3653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bussemakers MJG, Bokhoven A, Van Mees SGM, Kelmer R, Schlaken JA: Molecular cloning and characterization of human E-cadherin cDNA. Mol Biol 1993, 17:123-128 [DOI] [PubMed] [Google Scholar]
- 30.Mayerhoffer A, Lahr G, Froihlich U, Zienecker R, Sterzik K, Gratzl M: Expression and alternative splicing of the neural cell adhesion molecule NCAM in human granulosa cells during leuteinization. FEBS Lett 1994, 346:207-212 [DOI] [PubMed] [Google Scholar]
- 31.Oktay K, Nugent D, Newton H, Salha O, Chatterjee P, Gosden RG: Isolation and characterization of primordial follicles from fresh and cryopreserved human ovarian tissue. Fertil Steril 1997, 67:481-486 [DOI] [PubMed] [Google Scholar]
- 32.Barnhart K, De Mola R, Garside B, Heyner S, Schultz R, Eppig J, Kopf G, Coutifaris C: Morphometric analysis of isolated human preantral follicles and oocytes. 1995. Seattle, WA, Oct Presented at the American Society for Reproductive Medicine 51st Annual Meeting
- 33.Hsueh AJ, Eisenhauer K, Chun SY, Hsu SY, Billig H: Gonadal cell apoptosis. Recent Prog Horm Res 1996, 51:433-455 [PubMed] [Google Scholar]
- 34.Duprez E, Gjersten BT, Bernard O, Lanotte M, Doskeland SO: Antiapoptotic effect of heterozygously expressed mutant RI (Ala336 → Asp) subunit of cAMP kinase I in a rat leukemia cell line. J Biol Chem 1993, 268:8332-8340 [PubMed] [Google Scholar]
- 35.Aharoni D, Dantes A, Amsterdam A: Cross-talk between adenylate cyclase activation and tyrosine phosphorylation leads to modulation of the actin cytoskeleton and to acute progesterone secretion in ovarian granulosa cells. Endocrinology 1993, 133:1426-1436 [DOI] [PubMed] [Google Scholar]
- 36.Mcconkey DJ, Jonadal M, Orrenius S: Cellular signaling in thymocyte apoptosis. Semin Immunol 1992, 4:371-377 [PubMed] [Google Scholar]
- 37.McConkey DJ, Orrenius S, Jonadal M: Agents that elevate cAMP stimulate DNA fragmentation in thymocytes. J Immunol 1990, 145:1227-1230 [PubMed] [Google Scholar]
- 38.Schwartzman RA, Cidlowski JA: Apoptosis: the biochemistry and molecular biology of programmed cell death. Endocr Rev 1993, 14:133-151 [DOI] [PubMed] [Google Scholar]
- 39.Stamouli A, O’Sullivan MJB, Frankel S, Thomas EJ, Richardson MC: Suppression of matrix metalloproteinase production by hCG in cultures of human leuteinized granulosa cells as a model for gonadotropin-induced luteal rescue. J Reprod Fertil 1996, 105:235-239 [DOI] [PubMed] [Google Scholar]
- 40.Curry TE, Sanders SL, Pedigo NG, Estes RS, Wilson EA, Vernon MW: Identification and characterization of metalloproteinase inhibitor of activity in human ovarian follicular fluid. Endocrinology 1988, 123:1611-1618 [DOI] [PubMed] [Google Scholar]
- 41.Rapp G, Freudenstein J, Klaudiny J, Mucha J, Wempe Zimmer M, Scheit KH: Characterization of three abundant mRNAs from human ovarian granulosa cells. DNA Cell Biol 1990, 40:1170-1178 [DOI] [PubMed] [Google Scholar]
- 42.Herren B, Levkau B, Raines E, Ross R: Cleavage of β-catenin and plakoglobin and shedding of VE-cadherin during endothelial apoptosis: evidence for a role for caspases and metalloproteinases. Mol Biol Cell 1998, 9:1589-1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hermiston ML, Gordon JJ: In vivo analysis of cadherin function in mouse intestinal epithelium: essential roles in adhesion, maintenance of differentiation, and regulation of programmed cell death. J Cell Biol 1995, 129:489-506 [DOI] [PMC free article] [PubMed] [Google Scholar]