

Abstract
The regulatory role of interferon-γ receptor (IFN-γR)- and tumor necrosis factor receptor (TNFR)-mediated immune reactions for the activation of cerebral endothelial cells, microglia, and astrocytes was evaluated in a model of murine Toxoplasma encephalitis (TE). Brain endothelial cells of wild-type mice reacted in response to Toxoplasma infection with a strong up-regulation of the vascular cell adhesion molecule, the intercellular adhesion molecule (ICAM)-1, and major histocompatibility complex (MHC) class I and II antigens. A similar response was seen in mice genetically deficient for either TNFR1, TNFR2, or both TNFRs, whereas IFN-γR-deficient (IFN-γR0/0) mice were found to be defective in the up-regulation of these molecules. However, recruitment of leukocytes to the brain and their intracerebral movement were not impaired in IFN-γR0/0 mice. In addition, microglia of Toxoplasma gondii-infected IFN-γR0/0 mice failed to induce expression of ICAM-1, leukocyte function-associated antigen (LFA)-1, and MHC class I and II antigens, whereas wild-type and TNFR-deficient mice up-regulated these molecules. Moreover, TNF-α mRNA production of F4/80+ microglia/macrophages was impaired in IFN-γR0/0 mice, but not in TNFR-deficient mutants. However, induction of interleukin (IL)-1β, IL-10, IL-12p40, and IL-15 mRNA was independent of IFN-γR and TNFR signaling. In conclusion, IFN-γR, but not TNFR signaling, is the major pathway for the activation of endothelial cells and microglia in murine TE. These findings differ from observations in other inflammatory central nervous system disorders, indicating specific regulatory mechanisms in this parasitic cerebral infection.
It has evolved that resident brain cells actively participate in intracerebral immune reactions and that they are rapidly activated under a variety of pathological conditions. Among various brain cell populations, endothelial cells, microglia, and astrocytes have gained particular attention, and it has been suggested that these cells play an important role in intracerebral immune reactions. Endothelial cells, astrocytes, and microglia are major components of the blood-brain barrier (BBB), 1 which controls entry of immune cells to the brain. In addition, microglia, the resident macrophage population of the central nervous system (CNS), and astrocytes are ubiquitously distributed throughout the brain.
Previous studies have revealed that oral infection of mice with Toxoplasma gondii induces an encephalitis characterized by a strikingly strong activation of brain parenchymal cells. In particular, there is a prominent induction of major histocompatibility complex (MHC) class I and II antigens as well as of the intercellular adhesion molecule (ICAM)-1 and the vascular cell adhesion molecule (VCAM) on endothelial cells. 2,3 In addition, microglia are rapidly activated throughout the brain as seen by the strong induction of MHC class I and II antigens and ICAM-1 and an up-regulation of LCA, F4/80, LFA-1, and CD43 molecules. 2,3 Furthermore, astrocytes are activated and form hypertrophic cells with long, elongated processes and exhibit an enhanced glial fibrillary acidic protein (GFAP) expression. Whereas astrocytes show a prominent induction of MHC class I molecules, they differ from microglia and endothelium by the lack of a significant induction of MHC class II antigens as well as ICAM-1, which are expressed only on single astrocytic processes in full-blown Toxoplasma encephalitis (TE). 2
The induction of these various cell surface molecules on endothelial cells, microglia, and astrocytes can be observed in a number of autoimmune and infectious diseases of the CNS, including multiple sclerosis, experimental allergic encephalomyelitis (EAE), and various bacterial, viral, and parasitic infections. 2,4-15 However, it is still largely unknown which factors regulate the activation of endothelial cells, microglia, and astrocytes in vivo. In vitro studies using isolated neonatal human, rat, or mouse endothelial cells, microglia, and astrocytes have yielded contradictory results. For endothelial cells, either interferon (IFN)-γ or tumor necrosis factor (TNF)-α, or both cytokines have been described as the main inducer of MHC class I and II antigens as well as ICAM-1 and VCAM. 16-21 In addition, in vitro data on the relative role of IFN-γ and TNF-α for the activation of microglia and astrocytes are conflicting. Whereas some authors identified IFN-γ as the major activator of microglia and astrocytes, others attributed microglial and astrocytic cell activation to TNF-α or to the synergistic action of both cytokines. 22-27 However, it is still unresolved whether IFN-γ, which binds to the IFN-γ receptor (IFN-γR), and/or TNF-α, which binds to the TNFR1 in its soluble form 28 and to the TNFR2 in its membranous form, 29 are crucial for the activation of cerebral endothelium, microglia, and astrocytes in vivo.
Immunity to T. gondii is T cell mediated. 30,31 Both CD4+ and CD8+ T-cell-derived IFN-γ and TNF-α, which is also produced by macrophages/microglia in the brain and a few astrocytes, 32 are critical regulators of the intracerebral immune response to T. gondii. Both IFN-γ and TNF-α are indispensable for an effective anti-parasitic intracerebral immune response, 33,34 and they may exert their protection in part through the activation of brain parenchymal cells. A functional disturbance of this cytokine-mediated interaction of immune cells with brain parenchymal cells may also contribute to the reactivation of latent cerebral toxoplasmosis in AIDS patients, who are at high risk for an opportunistic TE. Therefore, the analysis of IFN-γ- and TNF-α-mediated regulation of the immunological activity of resident brain cell populations is also of clinical interest.
To analyze in detail the in vivo role of IFN-γ and TNF-α in the regulation of endothelial cells, microglia, and astrocytes in TE, we took advantage of mice genetically deficient for the IFN-γR (IFN-γR0/0), 35 TNFR1 (p55, TNFR10/0), 36 TNFR2 (p75, TNFR20/0), 37 or both TNFR1 and TNFR2 (TNFR1/20/0). Here, we show that IFN-γ, but not TNF-α, is the major in vivo activator of endothelial and microglial cells in murine TE.
Materials and Methods
Animals
The following female mice at the age of 8 to 10 weeks were used for the experiments: 129/Sv IFN-γR0/0 mice 35 and the corresponding 129/Sv wild-type controls (IFN-γR+/+) and 129/Sv × C57BL/6 TNFR10/0, TNFR20/0, and double-deficient TNFR1/20/0 mice 36,37 and their corresponding 129/Sv × C57BL/6 wild-type mice (TNFR1/2+/+). Breeding pairs of IFN-γR0/0 and IFN-γR+/+ 129/Sv mice were obtained from B&K Universal (Bicester, UK). All animals were kept in an isolation facility before and throughout the studies.
T. gondii Infection
Parasites were harvested from the brains of mice chronically infected with a low-virulent strain of T. gondii (DX strain). Brain tissue of these animals was dispersed in 0.1 mmol/L PBS (pH 7.4). The final concentration of the infectious agents was adjusted to a dose of 5 cysts/0.5 ml, which was administered orally to the experimental animals by gavage.
Experimental Procedures and Tissue Processing
Uninfected and T. gondii-infected (day 10 after infection (p.i.)) mice of all strains were studied. At the respective dates, animals were perfused intracardially with 0.9% saline in deep Metofane (Janssen, Neuss, Germany) anesthesia.
For immunohistochemistry on cryostat sections and reverse transcription polymerase chain reaction (RT-PCR) analysis, brains were dissected. Blocks of tissue were mounted on thick filter paper with Tissue-Tek O.T.C. Compound (Miles Scientific, Naperville, IL), snap-frozen in isopentane (Fluka, Neu-Ulm, Germany) precooled on dry ice, and stored at −80°C.
For the isolation of CNS-derived leukocytes from the normal and T. gondii-infected brain, brain tissue of the various experimental groups was passed through a 100-mesh stainless steel sieve, followed by collagenase/DNAse (Boehringer-Ingelheim Bioproducts, Heidelberg, Germany; Sigma, Deisenhofen, Germany) digestion and Percoll (Pharmacia, Freiburg, Germany) gradient separation as described previously. 32 Either isolated cells were analyzed by flow cytometry or F4/80+ macrophages/microglial cells were further purified using the high-gradient magnetic activated cell separation system (MACS, Milteny Biotec, Bergisch-Gladbach, Germany).
For the isolation of F4/80+ macrophages/microglia, leukocytes obtained from the brain were incubated with rat anti-mouse F4/80, followed by goat anti-rat IgG F(ab′)2 coupled with paramagnetic beads (Milteny Biotec) and fluorescein isothiocyanate (FITC)-labeled goat anti-rat IgG F(ab′)2 (Southern Biotechnology Associates-Biozol, Freising, Germany). Labeled cells were passed twice over separation columns. The purity of positively labeled cells was analyzed by flow cytometry and exceeded 97% in every case. Immediately after magnetic separation, mRNA was extracted from the purified cells.
Monoclonal and Polyclonal Antibodies
The following rat anti-mouse monoclonal antibody-producing hybridomas were obtained from the American Culture Collection (Rockville, MD) and kept under standard hybridoma conditions: anti-CD4 (clone G.K.1.5., rat IgG2b), anti-CD8 (clone 2.43, rat IgG2b), anti-CD45 (LCA, clone M1/9.3.4.HL.2, rat IgG2a), F4/80 (clone F4/80, rat IgG2b), anti-CD54 (ICAM-1, clone YN1/1.7.4, rat IgG2b), anti-VCAM (clone M/K-2.7, rat IgG1), anti-MHC class I (H-2, clone M1/42.3.9.8.HLK, IgG2a), anti-MHC class II (I-Ab,d,q, clone M5/114.15.2, rat IgG2b), anti-LFA-1α (CD11a/CD18, clone FD441.8, IgG2b).
Rat anti-mouse TNF-α (clone MP6-XT22) and FITC-conjugated rat anti-mouse CD8 were obtained from PharMingen (Hamburg, Germany). A polyclonal rabbit anti-T. gondii antiserum was purchased from Biogenex (Duiven, The Netherlands). Peroxidase-conjugated goat anti-rabbit IgG F(ab′)2 fragments, Texas-Red-conjugated goat anti-rabbit IgG F(ab′)2 fragments, biotinylated mouse serum-preadsorbed mouse anti-rat IgG F(ab′)2 fragments were from Dianova (Hamburg, Germany). Rabbit anti-cow GFAP and peroxidase-linked streptavidin-biotin complex were obtained from Dakopatts (Hamburg, Germany). Peroxidase-conjugated sheep anti-rat IgG F(ab′)2 fragments were from Amersham-Buchler (Braunschweig, Germany). Mouse serum preadsorbed phycoerythrin (PE)-conjugated goat anti-rat IgG and avidin PE/Cy5 were obtained from Southern Biotechnology Associates-Biozol.
Immunohistochemistry
Immunohistochemistry was performed on 10-μm cryostat sections as described previously. 3 In brief, for the detection of CD45 (LCA), CD4, CD8, LFA-1, and T. gondii, an indirect protocol using peroxidase-conjugated sheep anti-rat IgG F(ab′)2 fragments or peroxidase-conjugated goat anti-rabbit IgG F(ab′)2 fragments, respectively, as secondary antibody was used. The avidin-biotin complex technique was used for demonstration of MHC class I and II antigens, CD54 (ICAM-1), VCAM, F4/80, and TNF-α. Peroxidase reaction products were visualized using 3,3′-diaminobenzidine (Sigma) and H2O2 as co-substrate. Sections were in part lightly counterstained with hemalum (Merck, Darmstadt, Germany).
Simultaneous staining of GFAP and TNF-α on cryostat sections was performed with a double-labeling immunofluorescence technique. The incubation steps were 1) rat anti-mouse TNF-α, 2) biotinylated mouse anti-rat IgG F(ab′)2 fragments, 3) FITC-conjugated avidin, 4) rabbit anti-cow GFAP antiserum, and 5) Texas-Red-conjugated goat anti-rabbit IgG F(ab′)2 fragments.
To control for nonspecific reactions, incubations with either irrelevant species-specific IgG antibodies instead of the primary antibody or with omission of the primary antibody were performed.
Flow Cytometry Analysis of Brain-Derived Leukocytes
Brain-derived leukocytes were analyzed by triple- or double-immunofluorescence staining followed by flow cytometry as described previously. 32 CD4+ T cells were identified by staining with rat anti-mouse CD4 followed by goat anti-rat PE. After blocking of free binding sites of goat anti-rat PE with rat IgG, CD8+ T cells were detected with rat anti-mouse FITC-labeled CD8. Macrophages and microglial cells were identified by staining with FITC-conjugated F4/80 and LCA-biotin followed by avidin-PE/Cy5. As described and illustrated recently, 32 macrophages are F4/80+LCAhigh, whereas murine microglia are F4/80+LCAlow. To analyze activation of macrophages and microglia, brain-derived leukocytes were stained with rat anti-mouse anti-MHC class I, anti-MHC class II, and anti-ICAM-1, respectively, followed by goat anti-rat PE before incubation with F4/80-FITC, LCA-biotin, and avidin-PE/Cy5.
Control staining included incubation of brain-derived leukocytes with unlabeled or fluorochrome-labeled control antibodies.
Flow cytometry was performed on a FACScan (Becton-Dickinson, Heidelberg, Germany), and the data were analyzed with the Cell Quest Software (Becton-Dickinson).
Detection of Cytokine mRNA by RT-PCR
IFN-γ, TNF-α, and hydroxyphosphoribosyltransferase (HPRT) mRNA transcripts were analyzed in brain tissue homogenates according to a protocol described in detail before. 38 In addition, IL-1β, IL-10, IL-12p40, IL-15, TNF-α, and HPRT mRNA transcripts were assessed in brain-derived F4/80+ macrophages/microglia selectively isolated from cerebral leukocytes by MACS. Primer sequences and oligonucleotide probes were identical to those described previously. 32 Primer and probe sequences for Il-12p40 and IL-15 were as follows: IL-12p40, 5′-GTGAAGCACCAAATTACTCCGG-3′ (sense), 5′-GCTTCATCTGCAAGTTCTTGGG-3′ (antisense), and 5′-CAGTGTCCTGCCAGGAGGATGT-3′ (probe); IL-15, 5′-GTTCTCTTCTTCATCCTCCC-3′ (sense), 5′-GTGTTCTTAAGGACCTCACC-3′ (antisense), and 5′-CTTGCAGTGCATCTCCTTAC-3′ (probe).
In brief, mRNA was extracted from either brain tissue or 5 × 10 5 cells of uninfected and T. gondii-infected mice of the various strains by use of a mRNA extraction kit (Pharmacia). After reverse transcription of mRNA using the Superscript RT kit (Life Technologies, Eggenstein, Germany), PCRs were carried out in a volume of 30 μl. PCR conditions were optimized for each set of primers to ensure that amplification occurred in the linear range. PCR products were subjected to electrophoresis through an agarose gel, and the DNA was transferred to a nylon membrane (Amersham). Blots were hybridized using specific oligonucleotide probes, which were 3′-end labeled with digoxigenin by use of a DIG oligonucleotide 3′-end labeling kit (Boehringer, Mannheim, Germany). A DIG luminescent kit (Boehringer) was used to visualize the hybridization products.
Quantitation of mRNA was performed with an imaging densitometer (Scanpack, Biometra, Göttingen, Germany). The relative intensity of bands for each cytokine mRNA was determined and compared with the intensity of the autoradiographic band used for the internal control, ie, HPRT. The results are expressed as the degree of increase over mRNA levels for the cytokines in cell populations of uninfected mice of the various strains, respectively.
Statistical Evaluation
The numbers of VCAM-positive cerebral blood vessel endothelial cells were determined on anti-VCAM-stained sections in three to five T. gondii-infected mice per experimental group. Five immunostained sections from various brain regions per animal were evaluated. At least 100 high-power fields were analyzed per section, which were randomly selected. The statistical significance of the differences was evaluated by using the Mann-Whitney U test. P values < 0.05 were accepted as significant.
Results
Toxoplasma Encephalitis Is Induced in 129/Sv IFN-γR+/+, 129/Sv × C57BL/6 TNFR1/2+/+, and IFN-γR- and TNFR-Mutant Mice
At day 10 p.i, T. gondii had infected the brain of mice from all experimental groups (Figure 1) ▶ . However, IFN-γR0/0, TNFR1/20/0, and TNFR10/0 mice had a significantly increased parasitic load as compared with the respective wild-type mice (data not shown), which is consistent with previous studies in toxoplasmosis of IFN-γR0/0, IFN-γ0/0, and TNFR-deficient mice. 39,40
Figure 1.

Toxoplasma encephalitis in 129/Sv IFN-γR+/+, 129/Sv × C57BL/6 TNFR1/2+/+ mice as well as in IFN-γR- and TNFR-deficient mutants. At day 10 p.i., small numbers of T. gondii cysts are present in the cortex of 129/Sv IFN-γR+/+ (a), 129/Sv × C57BL/6 TNFR1/2+/+ (c), and TNFR20/0 (e) mice. At the same time point, groups of T. gondii antigen are detectable in the brains of IFN-γR0/0 (b), TNFR10/0 (d), and TNFR1/20/0 (f) mice. Anti-T. gondii immunostaining, slight counterstaining with hemalum; magnification, ×62.5.
Differential Induction of IFN-γ and TNF-α mRNA in the Brains of T. gondii-Infected 129/Sv IFN-γR+/+, 129/Sv × C57BL/6 TNFR1/2+/+, and IFN-γR- and TNFR-Mutant Mice
In the uninfected brain of all strains of mice, cytokine levels were low. Only occasional TNF-α mRNA transcripts could be detected in single animals, but IFN-γ mRNA was consistently negative (Figure 2) ▶ . In TE, IFN-γ mRNA was equally induced in 129/Sv IFN-γR+/+ and IFN-γR0/0 mice. In contrast to IFN-γR+/+ mice, IFN-γR0/0 mice failed to up-regulate TNF-α mRNA (Figure 2) ▶ . To further dissect the role of TNF-α and TNF receptors in TE, TNFR1- and/or TNFR2-deficient mice, which have a normal IFN-γR signaling, were studied. T. gondii-infected 129/Sv × C57BL/6 TNFR mutant mice showed an equal production of IFN-γ mRNA as compared with wild-type mice. In addition, up-regulation of TNF-α mRNA was indistinguishable in TNFR mutant and wild-type mice (Figure 2) ▶ .
Figure 2.

Expression of IFN-γ and TNF-α mRNA in normal and T. gondii-infected brains of 129/Sv IFN-γR+/+ and 129/Sv × C57BL/6 TNFR1/2+/+ mice as well as IFN-γR- and TNFR-mutant strains. IFN-γ, TNF-α, and HPRT mRNA transcripts were analyzed by RT-PCR in brain tissue homogenates at the indicated days. Three animals per groups were analyzed, and representative bands are shown. In a repeat experiment, identical data were obtained.
Impaired Activation of Cerebral Blood Vessel Endothelial Cells in IFN-γR0/0, but Not in TNFR10/0, TNFR20/0, and TNFR1/20/0 mice
The brains of uninfected mice of all strains showed an immunologically down-regulated phenotype, which included the absence of MHC class I and II antigens on endothelial cells of blood vessels (Figure 3) ▶ . Only single VCAM+ and ICAM-1+ endothelial cells were observed (Figure 3) ▶ . In TE, however, blood vessel endothelial cells from wild-type mice were strongly activated (Figures 3 and 4) ▶ ▶ .
Figure 3.

Expression of VCAM, ICAM-1, and MHC class II antigens in the CNS of uninfected and T. gondii-infected 129/Sv IFN-γR+/+ and IFN-γR0/0 mice. a to c: Uninfected mice of all strains analyzed expressed low levels of VCAM (a) and ICAM-1 (b) on cerebral blood vessel endothelial cells, and MHC class II antigens (c) were negative. 129/Sv IFN-γR+/+ mice are shown; identical results were obtained from uninfected 129/Sv × C57BL/6 TNFR1/2+/+, IFN-γR,0/0 TNFR1,0/0 TNFR20/0, and TNFR1/20/0 mice. d to f: In T. gondii-infected IFN-γR+/+ mice, VCAM was up-regulated on cerebral endothelial cells (d). ICAM-1 (e) and MHC class II antigens (f) were induced on endothelial cells of blood vessels (arrows) as well as on microglia throughout the brain (arrowheads). Microglial cells show the characteristic activated phenotype with short, elongated processes. g to i: In T. gondii-infected IFN-γR0/0 mice, VCAM (g) and ICAM-1 (h) were expressed on a significantly lower number of cerebral endothelial cells as compared with 129/Sv IFN-γR+/+ mice (d and e), and MHC class II antigens were not induced (i. In i, some I-A-positive inflammatory leukocytes reside in the leptomeninges. Anti-VCAM (a, d, and g), anti-ICAM-1 (b, e, and h), and anti-I-A (c, f, and i) immunostaining, slight counterstaining with hemalum; magnification, ×62.5.
Figure 4.

Expression of VCAM, ICAM-1, and MHC class II antigens in the CNS of T. gondii-infected 129/Sv × C57BL/6 TNFR1/2+/+ as well as TNFR-deficient mice. On infection with T. gondii, wild-type as well as TNFR1,0/0 TNFR20/0, and TNFR1/20/0 mice showed an equally strong up-regulation of VCAM (a, d, g, and k), ICAM-1 (b, e, h, and l), and MHC class II antigens (c, f ,i, and m) on blood vessel endothelial cells. Microglial cells also exhibited an equally strong up-regulation of ICAM-1 (b, e, h, and l) and MHC class II antigens (c, f, i, and m). Anti-VCAM (a, d, and g), anti-ICAM-1 (b, e, and h), and anti-I-A (c, f, and i) immunostaining, slight counterstaining with hemalum; magnification, ×62.5.
To evaluate whether there were significant differences in the activation of endothelial cells between IFN-γR0/0 mice and IFN-γR+/+ as well as between TNFR1/2+/+, TNFR10/0, TNFR20/0, and TNFR1/20/0 mice, VCAM expression was quantitated (Table 1) ▶ . This parameter is suitable to selectively analyze endothelial cells, as VCAM expression in the murine brain parenchyma is restricted to blood vessel endothelial cells. 2 Uninfected mice of all strains had weakly stained VCAM+ blood vessel endothelial cells in their brains without significant differences between the various strains (Figure 3 ▶ ; Table 1 ▶ ). In TE, the number of VCAM+ blood vessel endothelial cells increased significantly in all experimental groups as compared with the respective uninfected strain (P < 0.0001). However, the number of VCAM+ cerebral endothelial cells was significantly lower in T. gondii-infected IFN-γR0/0 mice as compared with IFN-γR+/+ animals (P < 0.001). In contrast, the number of VCAM+ blood vessels did not differ between TNFR1/2+/+ mice and the various TNFR mutants.
Table 1.
Expression of VCAM on Cerebral Endothelial Cells of Uninfected and T. gondii-Infected 129/Sv, 129/Sv × C57BL/6, IFN-γR0/0, TNFR10/0, TNFR20/0, and TNFR1/20/0 Mice
| Mouse strain | VCAM+ blood vessels | |
|---|---|---|
| Uninfected | T. gondii-infected | |
| 129/Sv | 0.47 ± 0.042 | 10.46 ± 0.28* |
| IFN-γR0/0 | 0.42 ± 0.043 | 2.42 ± 0.09† |
| 129/Sv× C57BL/6 | 0.58 ± 0.045 | 11.02 ± 0.32‡ |
| TNFR10/0 | 0.33 ± 0.042 | 10.38 ± 0.29 |
| TNFR20/0 | 0.47 ± 0.044 | 10.58 ± 0.39 |
| TNFR1/20/0 | 0.43 ± 0.045 | 11.37 ± 0.41 |
The number of VCAM-positive blood vessels was determined in at least 100 high-power fields (HPF) of anti-VCAM-stained sections. The mean value of VCAM+ blood vessels per HPF ± SEM is shown. Four animals per group were analyzed.
* In all experimental groups, the number of VCAM+ blood vessels significantly increased in TE as compared with the uninfected brain (P < 0.001 for infected IFN-γR0/0 versus uninfected IFN-γR0/0 mice; P < 0.0001 for all other infected versus uninfected strains).
† The number of VCAM+ cerebral blood vessels was significantly reduced in T. gondii-infected IFN-γR0/0 mice as compared with T. gondii-infected 129/Sv IFNγR+/+, 129/Sv × C57BL/6 TNFR1/2+/+, TNFR10/0, TNFR20/0, and TNFR1/20/0 mice (P < 0.0001).
‡ There were no statistically significant differences in the number of VCAM+ cerebral blood vessels between T. gondii-infected wild-type, TNFR10/0, TNFR20/0, and TNFR1/20/0 mice (P > 0.05).
In addition, only single ICAM-1+ blood vessels were detectable in the brains of uninfected and T. gondii-infected IFN-γR0/0 animals (Figure 3) ▶ . Moreover, IFN-γR0/0 mice completely lacked induction of MHC class I and II antigens on cerebral endothelium in TE (Figure 3) ▶ . In contrast to IFN-γR0/0 mice, neither TNFR1- nor TNFR2-mediated immune responses were required for the in vivo activation of cerebral endothelial cells, and all cell surface molecules were similarly strongly up-regulated irrespective of the TNFR mutant (Figure 4) ▶ . Thus, these data point to IFN-γ as a major activator of endothelial cells in vivo, and in the absence of IFN-γ-mediated effects, only a weak, but with respect to VCAM, significant activation of cerebral blood vessel endothelial cells occurs.
Normal Recruitment and Intracerebral Cell Movement in T. gondii-infected IFN-γR0/0 and TNFR Mutant Mice
To evaluate whether the impaired up-regulation of cell adhesion molecules on cerebral endothelial cells of IFN-γR0/0 mice resulted in a reduced recruitment of inflammatory leukocytes to the T. gondii-infected brain, intracerebral leukocytes were isolated, quantitated, and analyzed by flow cytometry. In all mouse strains, TE was characterized by the recruitment of leukocytes to the brain across the blood-brain barrier (Figures 5 and 6) ▶ ▶ . Flow cytometry revealed that the inflammatory infiltrates were predominantly composed of CD4+ and CD8+ T cells as well as of macrophages (Figure 6) ▶ . With respect to the number CD4+ and CD8+ T cells, there were no significant differences between the various strains of mice. However, wild-type mice recruited more macrophages to the brain than TNFR10/0, TNFR20/0, and TNFR1/20/0 mice (Figure 6) ▶ . Once recruited to the brain, inflammatory leukocytes of all mouse strains efficiently moved into the brain parenchyma, where LCA+ inflammatory leukocytes were immunohistochemically detected (Figure 5) ▶ .
Figure 5.

Inflammatory leukocytes in the brains of T. gondii-infected 129/Sv IFN-γR+/+ and 129/Sv × C57BL/6 TNFR1/2+/+ mice as well as IFN-γR- and TNFR-deficient strains. a: LCA+ cells are part of an infiltrate in the upper layer of the frontal cortex in a 129/Sv IFN-γR+/+ mouse. Leukocytes have already moved into the brain tissue (arrows). In addition, meningeal infiltrates are present (arrowhead). b: Perivascular infiltrate in the rostral basal ganglia of an IFN-γR0/0 mouse. Some LCA+ leukocytes have invaded the adjacent brain tissue (arrows). c to e: LCA+ leukocytes are present in both perivascular cuffs as well as diffusely infiltrating the basal ganglia in a 129/Sv × C57BL/6 TNFR1/2+/+ (c), TNFR10/0 (d), and a TNFR20/0 mouse. f: Many LCA+ cells diffusely infiltrate the basal ganglia in the adjacent neighborhood of the lateral ventricle in a TNFR1/20/0 mouse. A prominent ventriculitis has also developed, and the lumen and the wall of the ventricle are heavily infiltrated by leukocytes. Anti-LCA (CD45) immunostaining, slight counterstaining with hemalum; magnification, ×62.5.
Figure 6.
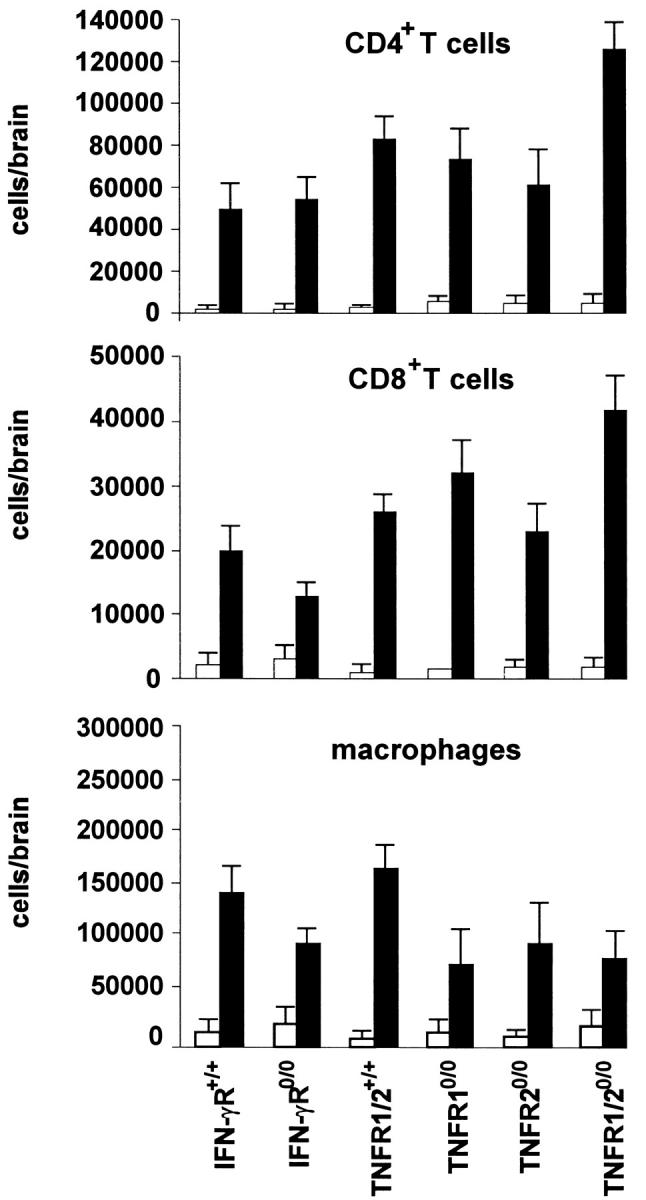
Quantitative assessment of intracerebral CD4+ and CD8+ T cells and macrophages in the normal and T. gondii-infected brain of 129/Sv IFN-γR+/+ and 129/Sv × C57BL/6 TNFR1/2+/+ mice as well as IFN-γR- and TNFR-deficient mice. Brain-resident leukocytes were isolated from the normal and T. gondii-infected brain (day 10 p.i.) by mechanical and enzymatic disruption of brain tissue followed by density gradient centrifugation. The total number of leukocytes was counted, and the absolute numbers of CD4+ and CD8+ T cells as well as macrophages per brain were calculated from the percentage of CD4+ and CD8+ cells as well as F4/80+LCAhigh macrophages in flow cytometry. Data represent the mean value ± SE from three mice of each experimental group. Similar data were obtained in a repeat experiment.
Thus, IFN-γR- and TNFR-mediated immune reactions were not necessary for the recruitment and intracerebral movement of inflammatory leukocytes.
Reduced Microglial Activation in T. gondii-Infected IFN-γR-Deficient Mice, but Not in TNFR-Deficient Strains
In all uninfected strains of mice, resting microglia expressed low levels of the F4/80 antigen and LCA, whereas MHC class I and II molecules, ICAM-1, and LFA-1 could not be detected. In 129/Sv and 129/Sv × C57BL/6 wild-type mice, microglial activation was indicated by the up-regulation of the F4/80 antigen, LCA, and LFA-1 as revealed by immunohistochemistry. Furthermore, there was a prominent, ubiquitous de novo induction of MHC class I and II antigens and ICAM-1 on microglia (Figures 3 and 4) ▶ ▶ . In contrast, microglia of IFN-γR0/0 mice was not activated as indicated by the lack of an induction of MHC class I and II antigens as well as ICAM-1 (Figure 3) ▶ . In addition, up-regulation of the F4/80 antigen, LCA, and LFA-1 was drastically reduced. In contrast, immunohistochemistry showed an equally strong activation of microglia in T. gondii-infected TNFR10/0, TNFR20/0, and TNFR1/20/0 mice (Figure 4) ▶ .
In addition to this topographical analysis, microglial cell activation was quantitatively studied by flow cytometry (Figure 7) ▶ . As shown previously, 32,41 microglia are F4/80+LCAlow and can thereby be differentiated from macrophages, which are F4/80+LCAhigh. Resting microglia of uninfected mice did not express MHC class I and II antigens, ICAM-1, and LFA-1. In TE, activated microglia and macrophages of both strains of wild-type mice expressed MHC class I and II antigens, ICAM-1, and LFA-1 (Figure 7) ▶ . Whereas TNFR10/0, TNFR20/0, and TNFR1/20/0 mice exhibited an equally strong expression of MHC class I and II antigens, ICAM-1, and LFA-1 as compared with the respective 129/Sv and 129/Sv × C57BL/6 wild-type mice, T. gondii-infected mice of IFN-γR0/0 mice did not express these molecules on their microglia and macrophages (Figure 7) ▶ .
Figure 7.

Induction of MHC class I and II antigens, LFA-1, and ICAM-1 in the T. gondii-infected brain of 129/Sv IFN-γR+/+ and 129/Sv × C57BL/6 TNFR1/2+/+ mice as well as IFN-γR- and TNFR-deficient mice. Intracerebral leukocytes were isolated from the normal and T. gondii-infected brain (day 10 p.i.) of 129/Sv IFN-γR+/+, IFN-γR0/0, 129/Sv × C57BL/6 TNFR1/2+/+, and TNFR10/0 and/or TNFR20/0 mice. To extract a sufficient number of intracerebral leukocytes, the brains of three mice per group were pooled. By flow cytometry, microglia were identified as F4/80+LCAlow, whereas macrophages were F4/80+LCAhigh. Combined staining with anti-MHC class I, anti-MHC class II, anti-LFA-1, and anti-ICAM-1 was performed to assess the activation of microglia and macrophages in TE. The mean fluorescence intensity of MHC class I, MHC class II, LFA-1, and ICAM-1 expression of microglia and macrophages was determined by flow cytometry, and data represent the fold increase of the mean fluorescence intensity in T. gondii-infected mice over uninfected mice of the respective mouse strain. In a second experiment, which included six mice per experimental group, identical data were obtained.
As illustrated in Figure 7 ▶ , induction of MHC class I and II antigens, ICAM-1, and LFA-1 on intracerebral F4/80+LCAhigh macrophages as well as on F4/80+LCAlow microglia, which is ontogenetically derived from bone marrow macrophages, was also dependent on IFN-γR, but not on TNFR, signaling.
Cytokine mRNA Production of F4/80+ Macrophages/Microglia from T. gondii-Infected Wild-Type Mice and from IFN-γR0/0, TNFR10/0, TNFR20/0, TNFR1/20/0 Mice
F4/80+ cells were further purified from cerebral leukocytes by MACS. In uninfected mice, F4/80+ cells were predominantly composed of microglia in all experimental groups (>94% microglia, Table 2 ▶ ). In TE, the majority of F4/80+ cells was also composed of microglia (73.4% to 90.5%, Table 2 ▶ ). However, macrophages recruited to the brain also contributed to this cell population.
Table 2.
Frequency of Microglia and Macrophages in the Normal and T. gondii-Infected Brain
| Mouse strain | % cells | |||
|---|---|---|---|---|
| Uninfected | Infected | |||
| Microglia | Macrophages | Microglia | Macrophages | |
| 129/Sv | 95.7 ± 0.5 | 4.4 ± 0.4 | 78.0 ± 3.0 | 22.0 ± 3.0 |
| IFN-γR0/0 | 95.4 ± 0.8 | 4.6 ± 0.8 | 90.5 ± 3.5 | 9.6 ± 3.9 |
| 129/Sv× C57BL/6 | 97.0 ± 0.4 | 3.0 ± 0.4 | 73.4 ± 3.1 | 26.6 ± 3.2 |
| TNFR10/0 | 96.4 ± 0.4 | 3.6 ± 0.4 | 89.5 ± 1.8 | 10.5 ± 1.8 |
| TNFR20/0 | 98.0 ± 0.3 | 2.0 ± 0.3 | 87.0 ± 1.6 | 13.0 ± 1.5 |
| TNFR1/20/0 | 93.8 ± 1.6 | 6.2 ± 1.7 | 88.7 ± 5.4 | 11.3 ± 5.3 |
Leukocytes were isolated from the brain by collagenase/DNAse digestion and density gradient centrifugation. Isolated cells were stained with F4/80-FITC and LCA-biotin followed by avidin-PE/Cy5. The percentage of F4/80+ LCAlow microglia and F4/80+ LCAhigh macrophages was determined by flow cytometry.
RT-PCR analysis of purified F4/80+ cells revealed occasionally weak TNF-α, IL-1β, IL-10, IL-12p40, and IL-15 mRNA transcripts in uninfected mice of all experimental groups. There were no major differences in the basal expression of these cytokines between the various non-infected strains. By day 10 p.i., 129/Sv and 129/Sv × C57BL/6 wild-type mice reacted with an increased transcription of IL-1β, IL-10, IL-12p40, and TNF-α mRNA in F4/80+ cells (Figure 8) ▶ . Due to the lack of IFN-γ-mediated immune responses, F4/80+ microglia/macrophages of 129/Sv IFN-γR0/0 mice, however, failed to up-regulate TNF-α mRNA, whereas induction of IL-1β, IL-10, and IL-12p40 was unimpaired (Figure 8) ▶ . In contrast to IFN-γR0/0 mice, IL-1β, IL-10, and TNF-α cytokine mRNA production of F4/80+ cells from 129/Sv × C57BL/6 TNFR10/0, TNFR20/0, and TNFR1/20/0 mice did not differ from their respective wild-type mice. Interestingly, IL-12p40 mRNA increased to higher levels in F4/80+ microglia/macrophages of TNFR10/0, TNFR20/0, and TNFR1/20/0 as compared with wild-type mice. Induction of IL-15 mRNA was either weak or absent in wild-type animals and in IFN-γR- and TNFR-deficient strains. Thus, major differences in IL-15 mRNA levels could not be detected between the various experimental groups at day 10 p.i.
Figure 8.

IL-1β, IL-10, IL-12p40, IL-15, and TNF-α mRNA induction in intracerebral F4/80+ cells of normal and T. gondii-infected 129/Sv IFN-γR+/+ and 129/Sv × C57BL/6 TNFR1/2+/+ as well as IFN-γR- and TNFR-deficient mice. Top: Intracerebral leukocytes were isolated from the normal and T. gondii-infected brain (day 10 p.i.), and F4/80+ microglia and macrophages were further separated by MACS. The purity of isolated F4/80+ cells was controlled by flow cytometry, and the data represent F4/80 staining before MACS (filled histograms) and after MACS (open histograms). The purity of F4/80+ cells after MACS always exceeded more than 97%. Bottom: Relative levels of cytokine mRNA expression in F4/80+ cells at day 10 p.i. Data are expressed as relative increase of cytokine mRNA levels in F4/80+ cells at day 10 p.i. over values obtained from F4/80+ cells of uninfected brains of the respective mouse strain. Data represent the value obtained from pooled cells of six to nine mice in each experimental group. A second experiment yielded identical results.
Equally Weak Activation of Astrocytes in T. gondii-Infected Wild-Type Mice and in IFN-γR0/0, TNFR10/0, TNFR20/0, and TNFR1/20/0 Mice
At day 10 p.i., astrocytes were only marginally activated as evidenced by a weak up-regulation of GFAP only in subleptomeningeal and subependymal areas of the brain. Double-fluorescence studies for GFAP and TNF-α identified small numbers of TNF-α-expressing cells in wild-type and in TNFR10/0, TNFR20/0, and TNFR1/20/0 mice, but not in IFN-γR0/0 mice. However, in none of these strains were GFAP+ astrocytes co-labeled by TNF-α (data not shown).
Discussion
This study demonstrates that IFN-γ is the major regulator of cerebral endothelium and microglia in TE. This conclusion is based on two observations. First, activation of cerebral endothelial cells and microglia was significantly impaired in IFN-γR0/0 mice. Second, in contrast to IFN-γR0/0 mice, the activation of endothelial cells and microglia was normal in mice deficient for either TNFR1, TNFR2, or both TNF receptors. To distinguish between IFN-γR- and TNFR-mediated effects on the activation of resident brain cell populations in murine TE, it was mandatory to study both IFN-γR- and TNFR-mutant mice, as IFN-γR deficiency was associated with reduced levels of TNF-α.
On cerebral endothelial cells, the coordinate, maximal up-regulation of VCAM, which is exclusively expressed on endothelial cells in the murine brain parenchyma, 2 as well as ICAM-1 and MHC class I and II antigens was critically dependent on IFN-γ, but not on TNF-α. As TNFR10/0, TNFR20/0, and TNFR1/20/0 as well as wild-type mice showed an equally strong expression of these cell surface molecules, a co-stimulatory signal provided by TNF-α is not required for IFN-γ-induced up-regulation of cell adhesion molecules and MHC antigens on cerebral endothelium in vivo. Previous studies, most of which were performed on cultivated endothelium isolated from brain microvessels, attributed the induction of cell adhesion molecules and MHC antigens to either IFN-γ or TNF-α or to a synergistic action of both cytokines 16,18,20,21,42-44 . Recently, a central role has been proposed for TNF-α in the induction of ICAM-1 in cerebral malaria, as TNF-α/β-deficient mice did not exhibit a Plasmodium berghei-induced ICAM-1 expression on cerebral endothelial cells. 45 In these animals, however, intracerebral IFN-γ levels were also reduced.
Although maximal induction of VCAM, ICAM-1, and MHC class I and II antigens was IFN-γ dependent, IFN-γR0/0 mice also showed a significant up-regulation of VCAM on infection. This finding indicates that in addition to IFN-γ other factors are also able to up-regulate these cell surface molecules, albeit in a significantly reduced manner. One candidate may be IL-1β, which can induce ICAM-1 and VCAM on cultured endothelial cells 44,46 and which is also efficiently produced in TE of IFN-γR0/0 mice. Whereas in murine TE IFN-γ-independent factors play a limited role with respect to the induction of cell adhesion molecules on cerebral endothelial cells, there is evidence that these IFN-γ-independent factors are more important in other CNS infections. This is illustrated in murine encephalitis caused by the lymphocytic choriomeningitis virus (LCMV), where a normal induction of ICAM-1 and VCAM on cerebral blood vessels was observed in IFN-γ-deficient mice. 47
A major function of cell adhesion molecules is to coordinate and regulate the recruitment of leukocytes from blood vessels to parenchymal inflammatory foci. Importantly, in our study, flow cytometry demonstrated that all subsets of leukocytes were still efficiently recruited to the brain in IFN-γR mutant mice. Therefore, the induction of low numbers of cell adhesion molecules in IFN-γR0/0 mice is sufficient to ensure the entry of leukocytes into the T. gondii-infected brain (Figures 5 and 6) ▶ ▶ . In addition, these data clearly show that there is no direct correlation between the number of ICAM-1- and VCAM-positive endothelial cells and the amount of immune cells recruited to the brain. Interestingly, in 129/Sv × C57BL/6 wild-type animals significantly more macrophages were recruited to the T. gondii-infected brain as compared with TNFR mutants, which, however, did not differ among each other. Although the mechanism of the impaired recruitment of macrophages to the brain of TNFR10/0, TNFR20/0, and TNFR1/20/0 mice remains at present unclear, this observation is in line with a recent study in murine EAE. In this latter model, TNF-α-deficient mice also had an impaired recruitment of macrophages, but not of T cells, to the brain. 48
Moreover, all TNFR-deficient mutants as well as IFN-γR0/0 mice and the respective wild-type strains had a normal movement of leukocytes from cerebral blood vessels to intraparenchymatous, T. gondii-associated inflammatory foci. This is in contrast to murine EAE in TNF-α-deficient mice, where direction of inflammatory leukocytes to the brain parenchyma was grossly impaired. 48 The divergent role of TNF-α for the direction of inflammatory leukocytes into the brain parenchyma in murine TE and EAE may be explained by the different pathogenetic mechanisms of these diseases. Whereas in TE the parasite causes local tissue destruction, thereby attracting protective leukocytes, in EAE, autoimmune T cells move to the brain parenchyma without an exogenous stimulus and initiate tissue destruction.
In TE, IFN-γ was also the major activator of microglia. In uninfected mice from all strains, microglia showed a resting phenotype. In TE, microglia were strongly, ubiquitously activated in both wild-type strains and TNFR10/0, TNFR20/0, and TNFR1/20/0 mice as evidenced by the induction of MHC class I and II antigens, ICAM-1, and LFA-1. In contrast, microglia of T. gondii-infected IFN-γR0/0 mice were not activated, even in close vicinity to T. gondii and perivascular inflammatory infiltrates. These experiments extend previous in vivo studies in which intrathecal, intravenous, or intraperitoneal application of IFN-γ resulted in microglial activation and induction of MHC antigens. 49-51 Moreover, in our study, maximal induction of MHC class I and II antigens and ICAM-1 was independent of TNF-α, and there was no synergistic effect of TNF-α with IFN-γ. Whereas the capacity of IFN-γ to activate microglia is generally accepted, the regulatory effects of TNF-α have been controversially discussed. Previous studies either attributed an activating or inhibiting effect or no effect to TNF-α regarding microglial stimulation and up-regulation of MHC antigens and ICAM-1. 22-24,52-55 A regulatory role for TNF-α in the activation of microglia was recently suggested in rat EAE, where a reduced activation of microglia was achieved by treating animals before the onset of EAE with a TNFR-IgG fusion protein directed against TNF. 56 However, as the application of a TNFR-IgG fusion protein prevents the normal systemic activation of T cells, a reduced microglial activation most probably reflects a downstream effect explained by diminished cytokine production, including IFN-γ, by T cells.
In our study, IFN-γ also regulated microglial cytokine production. To investigate the regulatory role of IFN-γ and TNF-α on microglial cytokine production, leukocytes were isolated from the brains of IFN-γR0/0, TNFR10/0, TNFR20/0, TNFR1/20/0, and TNFR1/20/0 mice as well as wild-type controls. These cells were further magnetically purified resulting in highly pure F4/80+ cells, which consisted of more than 94% microglia in the normal brain. In T. gondii-infected mice, F4/80+ cells were also mainly composed of microglia (73.4% to 90.5%), but a significant number of macrophages (9.6% to 26.6%) were co-isolated. Interestingly, RT-PCR revealed that F4/80+ cells of IFN-γR0/0 mice were unable to increase their TNF-α production. In contrast, a prominent increase in TNF-α mRNA transcripts was noticed in TNFR10/0, TNFR20/0, TNFR1/20/0, and wild-type control mice. Although we cannot differentiate by our method between TNF-α mRNA production of microglia and macrophages, the lack of TNF-α mRNA up-regulation in T. gondii-infected IFN-γR0/0 mice illustrates that microglial cells also do not increase their TNF-α mRNA production. Thus, TNF-α synthesis cannot only be induced in microglia by IFN-γ, as has been described for isolated microglia in vitro, 57 but, moreover, is strictly dependent on IFN-γ in TE, whereas the induction of TNF-α mRNA is independent of TNFR expression of microglia. Indeed, activated T lymphocytes, which are the major source of IFN-γ in TE, 32 as well as Th1-derived cytokines 58 have recently been shown to induce microglial TNF-α production, 59 and this mechanism may also be important in our model. TNF-α, however, was the only IFN-γ-dependent cytokine, as the induction of IL-1β, IL-10, IL-12p40, and IL-15 mRNA transcripts was not impaired in IFN-γR0/0 mice. Interestingly, although TNF-α promotes microglial cytokine production in vitro, 60 we obtained no evidence for a critical role of TNF-α for microglial cytokine induction in vivo, as microglia from TNFR-deficient mice and TNFR wild-type animals all showed a strong induction of TNF-α, IL-1β, IL-10, IL-12p40, and IL-15 mRNA.
In contrast to microglia and cerebral endothelium, there were no differences in the activation of astrocytes in TE between the various strains. In the present study, which was performed in the early phase of TE (10 days p.i.), activation of astrocytes was confined to perivascular, subleptomeningeal, and subependymal areas. We have previously reported that astrocyte activation is more prominent at later stages of TE, when these cells express MHC class I antigens and produce TNF-α. 2,32 In contrast, MHC class II antigens and ICAM-1 are only weakly expressed on single astrocytic processes even at maximal activation. 2,32 As both IFN-γR0/0 and TNFR1/20/0 mice succumb to toxoplasmosis within 17 days, ie, before the onset of a maximal astrocytic activation, 40 we cannot exclude that IFN-γ and/or TNF-α may be required for the induction of TNF-α, MHC class II antigens, and ICAM-1 on astrocytes in later stages of the disease.
In conclusion, the combined use of mice deficient in either IFN-γR or TNFR signaling enabled us to selectively dissect the relative role of IFN-γ and TNF-α for the activation of brain parenchymal cells in a model of murine TE. The results of this study illustrate that in vivo IFN-γR signaling, but not TNFR signaling, is crucial for the activation of cerebral blood vessel endothelial cells and microglia in this experimental CNS infection.
Acknowledgments
We thank Dr. V. Hans for statistical help and H. U. Klatt for photographic assistance.
Footnotes
Address reprint requests to Dr. Martina Deckert-Schlüter, Institut für Neuropathologie, Universitätskliniken Bonn, Sigmund-Freud-Strasse 25, D-53105 Bonn, Germany. E-mail: umt91c@ibm.rhrz.uni-bonn.de.
Supported by the Deutsche Forschungsgemeinschaft (grant De 485/6–1).
References
- 1.Lassmann H, Zimprich F, Vass K, Hickey WF: Microglial cells are a component of the perivascular glia limitans. J Neurosci Res 1991, 28:236-243 [DOI] [PubMed] [Google Scholar]
- 2.Deckert-Schlüter M, Schlüter D, Hof H, Wiestler OD, Lassmann H: Differential expression of ICAM-1, VCAM and their ligands LFA-1, Mac-1, CD43, VLA-4, and MHC class II antigens in murine Toxoplasma encephalitis: a light microscopic and ultrastructural immunohistochemical study. J Neuropathol Exp Neurol 1994, 53:457-468 [DOI] [PubMed] [Google Scholar]
- 3.Schlüter D, Löhler J, Deckert M, Hof H, Schwendemann G: Toxoplasma encephalitis of immunocompetent and athymic nude mice: immunohistochemical characterisation of Toxoplasma antigen, infiltrates and major histocompatibility complex gene products. J Neuroimmunol 1991, 31:185-198 [DOI] [PubMed] [Google Scholar]
- 4.Shrikant J, Chung IY, Ballestas ME, Benveniste EN: Regulation of intercellular adhesion molecule-1 gene expression by tumor necrosis factor-α, interleukin-β, and interferon-γ in astrocytes. J Neuroimmunol 1994, 51:209-220 [DOI] [PubMed] [Google Scholar]
- 5.Sobel RA, Mitchell ME, Fondren G: Intercellular adhesion molecule-1 (ICAM-1) in cellular immune reactions in the human central nervous system. Am J Pathol 1990, 136:1309-1316 [PMC free article] [PubMed] [Google Scholar]
- 6.Bo L, Mork S, Kong PA, Nyland H, Pardo CA, Trapp BD: Detection of MHC class II antigens on macrophages and microglia, but not on astrocytes and endothelia in active multiple sclerosis. J Neuroimmunol 1994, 51:135-146 [DOI] [PubMed] [Google Scholar]
- 7.Vass K, Lassmann H, Wekerle H, Wisniewski HM: Distribution of Ia antigen in the lesions of rat acute experimental allergic encephalomyelitis. Acta Neuropathol 1986, 70:149-160 [DOI] [PubMed] [Google Scholar]
- 8.Cannella B, Cross AH, Raine CS: Upregulation and coexpression of adhesion molecules correlate with relapsing autoimmune demyelination in the central nervous system. J Exp Med 1990, 172:1521-1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Engelhardt B, Conley FK, Butcher EC: Cell adhesion molecules on vessels during inflammation in the mouse central nervous system. J Neuroimmunol 1994, 51:199-208 [DOI] [PubMed] [Google Scholar]
- 10.Massa PT, Dörries R, ter Meulen V: Viral particles induce Ia antigen expression on astrocytes. Nature 1986, 320:543-546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Massa PT, Dörries R, Wege H, ter Meulen V: Analysis and pathogenetic significance of class II MHC (Ia) antigen induction on astrocytes during JHM coronavirus infection in rats. Adv Exp Med Biol 1987, 218:203-217 [DOI] [PubMed] [Google Scholar]
- 12.Lewandowski G, Hobbs MV: Evidence for deficiencies in intracerebral cytokine production, adhesion molecule induction, and T cell recruitment in herpes simplex virus type-2 infected mice. J Immunol 1998, 81:58-65 [DOI] [PubMed] [Google Scholar]
- 13.Monso-Hinard C, Lou JN, Behr C, Juillard P, Grau GE: Expression of major histocompatibility complex antigens on mouse brain microvascular endothelial cells in relation of susceptibility to cerebral malaria. Immunolgy 1997, 92:53-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grau GE, Tacchini-Cottier F, Vesin C, Milon G, Lou JN, Piguet PF, Juillard P: TNF-induced microvascular pathology: active role for platelets and importance of the LFA- 1/ICAM-1 interaction. Eur Cytokine Network 1993, 4:415-419 [PubMed] [Google Scholar]
- 15.Smith ME, McFarlin DE, Dhib-Jalbut S: Differential effect of interleukin-1β on Ia expression in astrocytes and microglia. J Neuroimmunol 1993, 46:97-104 [DOI] [PubMed] [Google Scholar]
- 16.Wong D, Dorovini-Zis K: Upregulation of intercellular adhesion molecule-1 (ICAM-1) expression in primary cultures of human brain microvessel endothelial cells by cytokines and lipopolysaccharide. J Neuroimmunol 1992, 39:11-21 [DOI] [PubMed] [Google Scholar]
- 17.McCarron RM, Wang L, Cowan EP, Spatz M: Class II MHC antigen expression by cultured human cerebral vascular endothelial cells. Brain Res 1991, 566:325-328 [DOI] [PubMed] [Google Scholar]
- 18.McCarron RM, Wang L, Racke MK, McFarlin DE, Spatz M: Cytokine-regulated adhesion between encephalitogenic T lymphocytes and cerebrovascular endothelial cells. J Neuroimmunol 1993, 43:23-30 [DOI] [PubMed] [Google Scholar]
- 19.Stins MF, Gilles F, Kim KS: Selective expression of adhesion molecules on human brain microvascular endothelial cells. J Neuroimmunol 1997, 76:81-90 [DOI] [PubMed] [Google Scholar]
- 20.Tanaka M, McCarron RM: The inhibitory effect of tumor necrosis factor and interleukin-1 on Ia induction by interferon-γ on endothelial cells from murine central nervous system microvessels. J Neuroimmunol 1990, 27:209-215 [DOI] [PubMed] [Google Scholar]
- 21.Male D, Pryce G: Synergy between interferons and monokines in MHC induction on brain endothelium. Immunol Lett 1988, 17:267-271 [DOI] [PubMed] [Google Scholar]
- 22.Loughlin AJ, Woodroofe MN, Cuzner ML: Regulation of Fc receptor and major histocompatibility complex antigen expression on isolated rat microglia by tumour necrosis factor, interleukin-1 and lipopolysaccharide: effects on interferon-γ induced activation. Immunology 1992, 75:170-175 [PMC free article] [PubMed] [Google Scholar]
- 23.Panek RB, Benveniste EN: Class II MHC gene expression in microglia: regulation by the cytokines IFN-γ, TNF-α, and TGF-β. J Immunol 1995, 154:2846-2854 [PubMed] [Google Scholar]
- 24.Andersson PB, Perry VH, Gordon S: Intracerebral injection of proinflammatory cytokines or leukocyte chemotaxins induces minimal myelomonocytic cell recruitment to the parenchyma of the central nervous system. J Exp Med 1992, 176:255-259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ballestas ME, Benveniste EN: Interleukin-1β- and tumor necrosis factor-α-mediated regulation of ICAM-1 gene expression in astrocytes requires protein kinase C activity. Glia 1995, 14:267-278 [DOI] [PubMed] [Google Scholar]
- 26.Rosenman SJ, Shrikant P, Dubb L, Benveniste EN, Ransohoff RM: Cytokine-induced expression of vascular cell adhesion molecule-1 (VCAM-1) by astrocytes and astrocytoma cell lines. J Immunol 1995, 154:1888-1899 [PubMed] [Google Scholar]
- 27.Satoh J, Kastrukoff LF, Kim SU: Cytokine-induced expression of intercellular adhesion molecule-1 (ICAM-1) in cultured human oligodendrocytes and astrocytes. J Neuropathol Exp Neurol 1991, 50:215-226 [DOI] [PubMed] [Google Scholar]
- 28.Grell M, Wajant H, Zimmermann G, Scheurich P: The type 1 receptor (CD120a) is the high-affinity receptor for soluble tumor necrosis factor. Proc Natl Acad Sci USA 1998, 95:570-575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grell M, Douni E, Wajant H, Löhden M, Clauss M, Maxeiner B, Georgopoulos S, Lesslauer W, Kollias G, Pfizenmaier K, Scheurich P: The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kd tumor necrosis factor receptor. Cell 1995, 83:793-802 [DOI] [PubMed] [Google Scholar]
- 30.Hof H, Emmerling P, Höhne K, Seeliger HRP: Infection of congenitally athymic (nude) mice with Toxoplasma gondii. Ann Microbiol (Inst Pasteur) 1976, 127b:503-507 [PubMed] [Google Scholar]
- 31.Suzuki Y, Remington JS: Regulation of resistance against Toxoplasma gondii infection by Lyt-2+ and Lyt-1+, L3T4+ T cells in mice. J Immunol 1988, 140:3943-3946 [PubMed] [Google Scholar]
- 32.Schlüter D, Kaefer N, Hof H, Wiestler OD, Deckert-Schlüter M: Expression pattern and cellular origin of cytokines in the normal and Toxoplasma gondii-infected murine brain. Am J Pathol 1997, 150:1021-1035 [PMC free article] [PubMed] [Google Scholar]
- 33.Suzuki Y, Orellana MA, Schreiber RD, Remington JS: Interferon-γ: the major mediator of resistance against Toxoplasma gondii. Science 1988, 240:516-518 [DOI] [PubMed] [Google Scholar]
- 34.Gazzinelli RT, Eltoum I, Wynn TA, Sher A: Acute cerebral toxoplasmosis is induced by in vivo neutralization of tumor necrosis factor-α and correlates with the down-regulated expression of inducible nitric oxide synthase and other markers of macrophage activation. J Immunol 1993, 151:3672-3681 [PubMed] [Google Scholar]
- 35.Haung S, Hendriks W, Althage A, Hemmi S, Bluethmann H, Kamijo R, Vilcek J, Zinkernagel RM, Aguet M: Immune responses in mice that lack the interferon-γ receptor. Science 1993, 259:1742-1745 [DOI] [PubMed] [Google Scholar]
- 36.Rothe J, Lesslauer W, Lötscher H, Lang Y, Koebel P, Köntgen F, Althage A, Zinkernagel R, Steinmetz M, Bluethmann H: Mice lacking the tumor necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature 1993, 364:798-802 [DOI] [PubMed] [Google Scholar]
- 37.Erickson SL, de Sauvage FJ, Kikly K, Carver Moore K, Pitts Meek S, Gillett N, Sheehan KC, Schreiber RD, Goeddel DV, Moore MW: Decreased sensitivity to tumour necrosis factor but normal T-cell development in TNF receptor-2-deficient mice. Nature 1994, 372:560-563 [DOI] [PubMed] [Google Scholar]
- 38.Deckert-Schlüter M, Albrecht S, Hof H, Wiestler OD, Schlüter D: Dynamics of the intracerebral and splenic cytokine mRNA production in Toxoplasma gondii-resistant and -susceptible congenic strains of mice. Immunology 1995, 85:408-418 [PMC free article] [PubMed] [Google Scholar]
- 39.Deckert-Schlüter M, Rang A, Weiner D, Huang S, Wiestler OD, Hof H, Schlüter D: Interferon-γ receptor deficiency renders mice highly susceptible to toxoplasmosis by decreased macrophage activation. Lab Invest 1996, 75:827-841 [PubMed] [Google Scholar]
- 40.Deckert-Schlüter M, Bluethmann H, Rang A, Hof H, Schlüter D: Critical role of TNF receptor type 1 (p55), but not of TNF receptor type 2 (p75), in murine toxoplasmosis. J Immunol 1998, 160:3427-3436 [PubMed] [Google Scholar]
- 41.Schlüter D, Bertsch D, Frei K, Hübers SB, Wiestler OD, Hof H, Deckert-Schlüter M: Interferon-γ antagonizes transforming growth factor-β2-mediated immunosuppression in murine toxoplasmosis. J Neuroimmunol 1998, 81:38-48 [DOI] [PubMed] [Google Scholar]
- 42.Hughes CC, Male DK, Lantos PL: Adhesion of lymphocytes to cerebral microvascular cells: effects on interferon-γ, tumour necrosis factor and interleukin-1. Immunology 1988, 64:677-681 [PMC free article] [PubMed] [Google Scholar]
- 43.Huynh HJ, Dorovini-Zis K: Effects of interferon-γ on primary cultures of human brain microvessel endothelial cells. Am J Pathol 1993, 142:1265-1278 [PMC free article] [PubMed] [Google Scholar]
- 44.Wong D, Dorovini-Zis K: Expression of vascular cell adhesion molecule-1 (VCAM-1) by human brain microvessel endothelial cells in primary culture. Microvasc Res 1995, 49:325-339 [DOI] [PubMed] [Google Scholar]
- 45.Rudin W, Eugster HP, Bordmann G, Bonato J, Müller M, Yamage M, Ryffel B: Resistance to cerebral malaria in tumor necrosis factor-α/β-deficient mice is associated with a reduction of intercellular adhesion molecule-1 upregulation and T helper type 1 response. Am J Pathol 1997, 150:257-266 [PMC free article] [PubMed] [Google Scholar]
- 46.Hess DC, Bhutwala T, Sheppard JC, Zhao W, Smith J: ICAM-1 expression on human brain microvascular endothelial cells. Neurosci Lett 1994, 168:201-204 [DOI] [PubMed] [Google Scholar]
- 47.Nansen A, Christensen JP, Ropke C, Markder O, Scheynius A, Thomsen AR: Role of interferon-γ in the pathogenesis of LCMV-induced meningitis: unimpaired leucocyte recruitment, but deficient macrophage activation in interferon-γ knock-out mice. J Neuroimmunol 1998, 86:202-212 [DOI] [PubMed] [Google Scholar]
- 48.Sean Riminton D, Körner H, Strickland DH, Lemckert FA, Pollard JD, Sedgwick JD: Challenging cytokine redundancy. Inflammatory cell movement and clinical course of experimental autoimmune encephalomyelitis are normal in lymphotoxin-deficient, but not tumor necrosis factor-deficient, mice. J Exp Med 1998, 187:1517-1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vass K, Lassmann H: Intrathecal application of interferon-γ: progressive appearance of MHC antigens within the rat nervous system. Am J Pathol 1990, 137:789-800 [PMC free article] [PubMed] [Google Scholar]
- 50.Xu J, Ling EA: Upregulation and induction of major histocompatibility complex I and II antigens on microglial cells in early postnatal rat brain following intraperitoneal injections of recombinant interferon-γ. Neuroscience 1994, 60:959-967 [DOI] [PubMed] [Google Scholar]
- 51.Grau V, Herbst B, van der Meide PH, Steiniger B: Activation of microglial and endothelial cells in the rat brain after treatment with interferon-γ in vivo. Glia 1997, 19:181-189 [DOI] [PubMed] [Google Scholar]
- 52.Woodroofe MN, Hayes GM, Cuzner ML: Fc receptor density, MHC antigen expression and superoxide production are increased in interferon-γ-treated microglia isolated from adult rat brain. Immunology 1989, 68:421-426 [PMC free article] [PubMed] [Google Scholar]
- 53.Williams K, Bar-Or A, Ulvestad E, Olivier A, Antel JP, Yong VW: Biology of adult human microglia in culture: comparisons with peripheral blood monocytes and astrocytes. J Neuropathol Exp Neurol 1992, 51:538-549 [DOI] [PubMed] [Google Scholar]
- 54.Shrikant P, Weber E, Jilling T, Benveniste EN: Intercellular adhesion molecule-1 gene expression by glial cells: differential mechanisms of inhibition by IL-10 and IL-6. J Immunol 1995, 155:1489-1501 [PubMed] [Google Scholar]
- 55.Hellendall RP, Ting JP: Differential regulation of cytokine-induced major histocompatibility complex class II expression and nitric oxide release in rat microglia and astrocytes by effectors of tyrosine kinase, protein kinase C, and cAMP. J Neuroimmunol 1997, 74:19-29 [DOI] [PubMed] [Google Scholar]
- 56.Körner H, Lemckert FA, Chaudhri G, Etteldorf S, Sedgwick JD: Tumor necrosis factor blockade in actively induced experimental autoimmune encephalomyelitis prevents clinical disease despite activated T cell infiltration to the central nervous system. Eur J Immunol 1997, 27:1973-1981 [DOI] [PubMed] [Google Scholar]
- 57.Frei K, Siepl C, Groscurth P, Bodmer S, Schwerdel C, Fontana A: Antigen presentation and tumor cytotoxicity by interferon-γ-treated microglial cells. Eur J Immunol 1987, 17:1271-1278 [DOI] [PubMed] [Google Scholar]
- 58.Renno T, Krakowski M, Piccirillo C, Lin JY, Owens T: TNF-α expression by resident microglia and infiltrating leukocytes in the central nervous system of mice with experimental allergic encephalomyelitis: regulation by Th1 cytokines. J Immunol 1995, 154:944-953 [PubMed] [Google Scholar]
- 59.Chabot S, Williams G, Yong VW: Microglial production of TNF-α is induced by activated T lymphocytes: Involvement of VLA-4 and inhibition by interferon-β-1b. J Clin Invest 1997, 100:604-612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Frei K, Fontana A: Immune regulatory functions of astrocytes and microglial cells within the central nervous system. Neuroimmune Networks: Physiology and Diseases. 1989, pp 127–136