

Abstract
Craniosynostoses are a heterogeneous group of disorders characterized by premature fusion of cranial sutures. Mutations in fibroblast growth factor receptors (FGFRs) have been associated with a number of such conditions. Nevertheless, the cellular mechanism(s) involved remain unknown. We analyzed cell proliferation and differentiation in osteoblasts obtained from patients with three genetically and clinically distinct craniosynostoses: Pfeiffer syndrome carrying the FGFR2 C342R substitution, Apert syndrome with FGFR2 P253R change, and a nonsyndromic craniosynostosis without FGFR canonic mutations, as compared with control osteoblasts. Osteoblasts from craniosynostotic patients exhibited a lower proliferation rate than control osteoblasts. P253R and nonsyndromic craniosynostosis osteoblasts showed a marked differentiated phenotype, characterized by high alkaline phosphatase activity, increased mineralization and expression of noncollagenous matrix proteins, associated with high expression and activation of protein kinase Cα and protein kinase Cε isoenzymes. By contrast, the low proliferation rate of C342R osteoblasts was not associated with a differentiated phenotype. Although they showed higher alkaline phosphatase activity than control, C342R osteoblasts failed to mineralize and expressed low levels of osteopontin and osteonectin and high protein kinase Cζ levels. Stimulation of proliferation and inhibition of differentiation were observed in all cultures on FGF2 treatment. Our results suggest that an anticipated proliferative/differentiative switch, associated with alterations of the FGFR transduction pathways, could be the causative common feature in craniosynostosis and that mutations in distinct FGFR2 domains are associated with an in vitro heterogeneous differentiative phenotype.
Craniosynostosis, the premature ossification of one or more sutures of the flat bones of the developing skull, is a relatively common defect of the cranial morphogenetic program, with a prevalence at birth of approximately 1:3000. It results in a wide spectrum of craniofacial anomalies, including abnormal head shape, protruding eyes, and midface underdevelopment. 1 Surgical treatment of craniosynostosis is frequently required to alleviate the skull deformity; however, in most cases reconstructive craniotomy is also directed to prevent its most severe consequences, ie, increased intracranial pressure, severe exorbitism, and obstructive apnea. 2 Craniosynostoses can occur as isolated cranial defect or as a feature of more than 100 syndromes, which are clinically distinguished on the basis of the suture(s) involved, the progression of their closure over time, the resulting craniofacial profile, and the pattern of cerebral, cardiac, genital, and limb involvement. 1,3 In about half of these conditions a genetic cause has been established or suggested; most of them are monogenic and are inherited in an autosomal dominant manner, with complete penetrance and variable expressivity. 3
Considerable advances have been made recently in the understanding of the molecular basis of craniosynostotic diseases. Mutations in three members of the fibroblast growth factor receptor (FGFR) family have been recently associated with a number of clinically distinct craniosynostotic conditions. 4,5 The FGFR family includes four cell surface tyrosine kinase receptors with a structure consisting of a glycosylated extracellular region characterized by three immunoglobulin-like (Ig-like) motifs, a single membrane-spanning segment, and an intracellular portion containing a split tyrosine kinase domain. 6 FGFRs bind to fibroblast growth factors (FGFs), which are known to regulate proliferation, survival, differentiation, and migration of a wide variety of cells. 7,8 Transduction of the FGF signals is mediated by receptor dimerization, followed by autophosphorylation in the dimer and phosphorylation of cellular substrates regulating the ras/mitogen-activated protein (MAP) kinases pathway, 9,10 the phospholipid turnover, and activation of protein kinase Cs (PKCs). 11 The great majority of craniosynostosis-associated FGFR mutations are spotted in two contiguous extracellular domains involved in FGF binding. 5 More precisely, two adjacent amino acidic changes (Ser252Trp and Pro253Arg) in the linker stretch between the second and third Ig-like domains account for the vast majority of cases of Apert syndrome. Homologous substitutions in FGFR1 (Pro252Arg) and FGFR3 (Pro250Arg) have been reported in the Pfeiffer syndrome and in a heterogeneous group of craniosynostotic conditions, respectively. Different mutations located in the third Ig-like domain of FGFR2 are associated with the Crouzon, Pfeiffer, and Jackson-Weiss syndromes. Among them, substitution of Cys-342 is the most recurrent mutation; the presence of an unpaired cysteine residue in this domain (by loss of Cys-278 or Cys-342 or by the introduction of an additional cysteine residue) has been observed in more than 50% of all cases. 5 All of the FGFR mutations are dominantly acting, and at least two distinct ways in which the FGF transduction pathway may be altered have been proposed. Functional studies focused on the role of mutations spotted in the third Ig-like domain in FGFR2 support that these mutations act by disrupting the intradomain disulfide bond formation, leading to constitutive receptor activation due to homodimerization between mutated receptors. 12-14 By contrast, FGFR mutations located in the linker seem to affect ligand binding stability, and they could affect either FGF binding affinity or specificity. 15
The FGF signaling plays a pivotal role in the control of intramembranous and endochondral ossification. 16 In particular, FGFs are involved in cranial bones’ normal growth as well as in their maintenance as discrete individual structures, separated from one another, during the intramembranous ossification of the skull. 16,17 Much evidence suggests that FGFs regulate both in vitro pre-osteoblast cell proliferation and osteoblast differentiation. 18-21 However, only a few data are available on the role of the FGFs/FGFRs network in the progression of undifferentiated cells toward differentiated osteoblasts, as well as in the control of suture growth and closure. 22-24 For this reason, the mechanisms leading to the premature closure of cranial sutures in craniosynostosis and underlying interference of FGFR mutations with cell proliferation or differentiation remain unclear.
The direct study of primary osteoblast cultures from patients affected by craniosynostosis is one approach to investigating the relationships between altered FGFR function and onset of the craniosynostotic condition. In vitro osteoblast cultures represent a useful tool to characterize possible alterations in cell function, differentiation, and metabolism. By such an approach, it has recently been shown that osteoblasts isolated from prematurely fused sutures are characterized by an increased maturation rate compared to osteoblasts from unaffected sutures of the same patient. 25 However, only a single study has focused on the proliferative/differentiative relationships in osteoblasts carrying a specific FGFR2 mutation 26 and no data are available on potential relationships between distinct mutational events in the FGFR or other genes and the cellular and molecular anomaly.
In this study, we analyzed possible heterogeneity in cell growth and differentiation among primary cultures of osteoblasts obtained from patients affected by three genetically and clinically distinct craniosynostotic disorders: the Pfeiffer type 2 syndrome associated with the Cys342Arg substitution in FGFR2, the Apert syndrome characterized by the Pro253Arg change in the same receptor, and a nonsyndromic craniosynostotic condition apparently not associated with mutations in the canonic FGFRs’ hot spots. Our results indicate that an alteration of the proliferative/differentiative pattern, induced by mutation of the FGFR transduction pathways, could be the causative common feature in craniosynostoses. Our data also suggest that mutations in distinct FGFR2 domains are associated with an in vitro heterogeneous osteoblastic differentiative phenotype.
Materials and Methods
Materials
Dulbecco’s modified Eagle’s medium (DMEM) nutrient MIX F-12, fetal bovine serum (FBS), penicillin, and streptomycin were purchased from Gibco BRL (Grand Island, NY). [Methyl-3H]-thymidine was purchased from DuPont-New England Nuclear (Boston, MA). Collagenase type IV, trypsin, ascorbic acid, β-glycerophosphate, dexamethasone, and heparin were obtained from Sigma Chemical Co. (St. Louis, MO). Micro BCA Protein Assay was obtained from Pierce (Rockford, IL). Goat or rabbit antibodies anti-β-actin and anti-protein kinase C isoforms were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Horseradish peroxidase-conjugated anti-rabbit and anti-goat IgG and ECL were obtained from Amersham (Arlington Heights, IL). Human recombinant FGF-2 (hr-FGF2) was obtained from Becton Dickinson Labware (Bedford, MA). AmpliTaq polymerase was purchased from Perkin-Elmer (Branchburg, NJ), T7 Sequenase 2.0 DNA sequencing kit from USB (Cleveland, OH), and BstUI and BsaAI endonucleases from New England Biolabs (Beverly, MA). Anti-osteopontin (LF-123), anti-bone sialoprotein (LF-83), and anti-osteonectin (Bon-I) antisera were generously provided by Dr. Larry Fisher of the Craniofacial and Skeletal Disease Branch, National Institute of Dental Research, National Institutes of Health (Bethesda, MD).
Clinical Evaluation
Three clinically distinct craniosynostotic conditions were considered in the study. The first patient (male, 3 months old) had severe craniosynostosis resulting in a “cloverleaf skull” malformation, midfacial hypoplasia, and severe ocular proptosis. Typical digit anomalies, ie, broad and medially deviated thumbs and great toes and partial bilateral cutaneous syndactily of the second and third toes, were also present. Such clinical features fit the Pfeiffer syndrome type 2 phenotype. 27 The patient affected by Apert syndrome (male, 2 months old) showed acrocephaly, proptosis, mid-facial hypoplasia, and severe bilateral bony and cutaneous syndactily of both hands and feet. The third patient (male, 9 months old) had a clinically unclassified nonsyndromic craniosynostosis, initially described as Crouzon syndrome, characterized by stenosis of the coronal suture with plagiocephaly, hypertelorism, and invaginated occipital bone. All cases were sporadic.
Bone Samples
Skull bone samples were obtained from the three infants during cranial surgery and consisted of bone fragments close to the sutures involved. Bone samples were also obtained from a control subject who underwent local bone surgery for an unrelated disease (male, 3 months old). Bone samples were obtained with parents’ signed consent. Samples were placed in cold Hanks’ solution supplemented with 100 μg/ml streptomycin and 100 units/ml penicillin and immediately processed.
Mutation Analysis
Genomic DNAs were extracted from peripheral blood leukocytes. Molecular screening was carried out by single strand conformational analysis on the FGFR2 third Ig-like domain (exons IIIa, IIIb, and IIIc) and transmembrane segment (TM) coding sequences and exons IIIa of both FGFR1 and FGFR3 genes. Polymerase chain reaction (PCR) conditions for amplifications of the FGFR2 exons coding the third Ig-like domain and exons IIIa of both FGFR1 and FGFR3 were performed as previously described. 28-30 PCR conditions for FGFR2 exons IIIb and TM were the same as those for FGFR2 exon IIIc amplification, by the oligonucleotide pairs previously reported. 31 The purified FGFR2 exon IIIa and exon IIIc PCR products were directly sequenced on both strands, by using the same oligonucleotides utilized during PCR. Mutations were confirmed by endonuclease digestion of the unpurified PCR products with BstUI (exon IIIa) and BsaAI (exon IIIc) according to the manufacturer’s specifications.
Osteoblast Cultures
Samples were processed by a modification of the sequential collagenase/trypsin digestion method. 32 Briefly, bone fragments were washed in sterile phosphate-buffered saline (PBS), minced, and treated with 1 mg/ml collagenase type IV and 0.25% trypsin for 30 minutes at 37°C with gentle agitation. The procedure was repeated three times; cells from the second and third digestions were collected by centrifugation and plated in 25-cm 2 flasks. At the end of digestions, fragments were also plated in 35-mm Petri dishes; cells proliferated from the bone fragments within 10 days of culture. At confluence, cells were trypsinized and amplified for characterization of the osteoblast phenotype and experiments. Osteoblasts were maintained in DMEM nutrient MIX F-12, supplemented with 100 μg/ml streptomycin and 100 units/ml penicillin and 10% heat-inactivated FBS at 37°C in a humidified incubator with 5% CO2. Medium was changed twice a week.
Cell Proliferation
Osteoblasts were plated at a density of 8 × 10 3 cells/well into 24-well plates and allowed to proliferate for 2 days. Cells were incubated with 1 μCi/ml 3H-thymidine for 12 hours. Osteoblasts were then washed twice in PBS, extracted with 1 ml of 1% sodium dodecyl sulfate (SDS) and precipitated with 10% thricloracetic acid. After centrifugation, pellets were dissolved in 1% SDS and aliquots transferred to vials containing 5 ml of scintillation fluid for counting the radioactivity in a β-counter Beckman LS 6500. To assay the effects of hr-FGF2 on cell growth, osteoblasts were seeded at 5 × 10 3 cells/well in 24-well plates, cultured in basal conditions until preconfluence, washed 3 times, and cultured for 24 hours in serum-free medium. Osteoblasts were then treated with hr-FGF2 (20 ng/ml) and heparin (50 μg/ml) for another 24 hours. Untreated cells were incubated in serum-free medium for 24 hours and processed under identical conditions.
Alkaline Phosphatase Detection
Alkaline phosphatase (ALP) activity was detected histochemically in monolayers cultured for 1 week in standard conditions. Osteoblasts were washed twice in 0.1 mol/L cacodylate buffer, pH 7.4, fixed for 10 minutes in 4% paraformaldehyde in 0.1 mol/L cacodylate buffer, and processed using the Fast Blue RR Salt and Mayer’s Hematoxylin (Sigma kit no. 85). Quantitative determinations of ALP were performed in cells lysated with 0.1% SDS in PBS, using a commercially available procedure and following the manufacturer’s instructions (Sigma kit no. 104). To study the effect of hr-FGF2 on ALP activity, osteoblasts after confluence were rinsed three times, cultured for 24 hours in serum-free medium, and treated for another 48 hours with or without hr-FGF2 (20 ng/ml) and heparin (50 μg/ml).
Analysis of in Vitro Mineralization
Cells were seeded in 24-well plates at a density of 10 4 cells/well and cultured in mineralization medium (DMEM-F12 with 10% FBS, supplemented with 100 μg/ml ascorbic acid, 10 mmol/L β-glycerophosphate) in presence or absence of 10−8 mol/L dexamethasone (DEX), for 6 weeks. Cells became confluent after 1 week of culture. Mineralization medium was changed twice a week. Cultures were fixed in 4% PFA in 0.1 mol/L cacodylate buffer, pH 7.4, for 10 minutes and washed twice in distilled water. For von Kossa’s staining, silver nitrate (5%) was added, 33 and plates were placed under an UV lamp (20 cm distance) for 60 minutes. Cultures were rinsed twice in distilled water, treated for 2 minutes with 5% sodium thiosulfate, rapidly rinsed twice in ethanol, and allowed to dry. When osteoblasts were treated for 2 weeks with hr-FGF2 (20 ng/ml) and heparin (50 μg/ml), after confluence cultures were washed twice and grown as described above except that FBS concentration was lowered to 0.1%.
Western Blot Analysis
Osteoblasts were grown in 90-mm culture Petri dishes in standard condition until confluence, then washed twice with cold PBS and scraped into 200 μl of ice-cold lysis buffer (20 mmol/L Tris, 1 mmol/L dithiothreitol, 1 mmol/L MgCl2, 0.1 mmol/L Na3VO4, 1 mmol/L NaF, 1% SDS, 0.3 mol/L Urea) containing 10 μg/ml aprotinin, 10 μg/ml leupeptin, 2 μg/ml benzamidin, 0.1 mmol/L phenylmethylsulfonyl fluoride. Samples were solubilized in 4× loading buffer (10% glycerol, 8% SDS, 0.05% bromophenol blue, 10% β-mercaptoethanol) and boiled at 90°C for 3 minutes. Fifty micrograms of proteins, determined using a Micro BCA Protein assay, were resolved by SDS polyacrylamide gel electrophoresis and transferred to nitrocellulose at 35 V overnight. Blots were saturated with 5% nonfat dry milk dissolved in Tris-buffered saline containing 0.1% Tween 20 (TTBS) for 2 hours at room temperature. Filters were probed with the appropriated antibody diluted in TTBS containing 1% milk overnight at 4°C. Rabbit polyclonal anti-osteopontin (LF-123), anti-osteonectin (Bon-1), and anti-bone sialoprotein (LF-83) and goat anti-β-actin antibodies were used at 1:500 dilution. 34 Blots were extensively washed in TTBS and incubated with horseradish peroxidase-conjugated anti-rabbit or anti-goat antibodies for 1 hour at room temperature and washed 3 times in TTBS. Protein bands were visualized by enhanced chemiluminescence (ECL) kit. Densitometric analysis was performed using Multianalyst software with a BioRad GS-700 Imaging Densitometer. To study the effects of FBS, osteoblasts after confluence were incubated for 72 hours with or without 10% FBS. To study the effect of hr-FGF2, osteoblasts were washed twice, cultured in serum-free medium for 24 hours, and treated with or without hr-FGF2 (20 ng/ml) and heparin (50 μg/ml) for an additional 48 hours.
PKC Isoenzymes Analysis
Preparation of cytosol or membrane fractions was carried out as previously described. 35 Briefly, cells were scraped in lysis buffer (20 mmol/L Tris-HCl, pH 7.5, 1 mmol/L EDTA, pH 8.0, 1 mmol/L EGTA, 2 mmol/L dithiothreitol, 2 mmol/L phenylmethylsulfonyl fluoride, 25 μg/ml leupeptin, 6 μg/ml aprotinin, 10 mmol/L benzamidin) on ice and transferred to Eppendorf tubes, then sonicated on ice for two bursts at setting 5 of a heat system sonicator. Cell lysates were centrifuged in a TL-100 Beckman ultracentrifuge at 200,000 × g for 50 minutes at 4°C and supernatants (cytosolic fraction) were recovered and precipitated in 10 volumes of cold acetone at −20°C overnight. Pellets (membrane fractions) were gently resuspended in 1 ml of lysis buffer containing 1% Triton-X 100, incubated on ice for 20 minutes, sonicated, and centrifuged for 50 minutes at 200,000 × g to eliminate nonprotein components. Supernatants were then precipitated overnight in cold acetone. The following day samples were centrifuged in a Sorvall centrifuge at 10,000 × g for 30 minutes at 4°C and pellets were resuspended in 50 μl of 10 mmol/L Tris-HCl, pH 7.5, and stored at −80°C until analysis. Twenty micrograms of cytosolic and membrane fractions were resolved by SDS polyacrylamide gel electrophoresis followed by immunoblotting according to the procedure described for the Western blot analysis of matrix proteins. Specific rabbit antibodies anti-protein kinase Cα, ε, and ζ isoenzymes were used.
Statistical Analysis
Data are expressed as means ± SE. Statistical significance between data points was determined using the two-tailed Student’s t-test. P <0.05 was considered statistically significant. Experiments were performed in triplicate at least 3 times.
Results
Parameters of osteoblast proliferation and differentiation were investigated in primary osteoblast cultures from skull bone fragments of three genetically and phenotypically distinct craniosynostotic disorders and from an unaffected sex- and age-matched control individual. More precisely, we considered two craniosynostotic conditions associated with mutations in the FGFR2, ie, the Apert and Pfeiffer type 2 syndromes, and a nonsyndromic craniosynostotic condition characterized by the absence of canonic FGFR mutations.
Mutational Analysis
Direct sequencing of the FGFR2 exon IIIa in the Apert patient showed the occurrence of a de novo C-to-G transversion at nucleotide 937. Such a missense mutation results in an arginine-for-proline substitution at codon 253 in the linker stretch between the second and third Ig-like domain of the receptor (hereinafter P253R) (data not shown). The whole third Ig-like domain and the transmembrane segment coding sequences of FGFR2, as well as exons IIIa of both FGFR1 and FGFR3 genes, were screened by single strand conformational analysis in the two patients affected by Pfeiffer type 2 syndrome and nonsyndromic craniosynostosis. An altered migration pattern was found for the FGFR2 exon IIIc PCR product in the Pfeiffer patient. Direct sequencing revealed a T-to-C transition at nucleotide 1203, resulting in a substitution of arginine-for-cysteine at codon 342 in the third Ig-like domain of the receptor (C342R, hereinafter) (data not shown). The presence of this mutation was confirmed by restriction analysis, because the T-to-A change in FGFR2 exon IIIc destroys a restriction site for BsaAI. By contrast, no mutation in the canonic regions indicated as hot spots for FGFR1, FGFR2, and FGFR3 genes was detected in the patient with nonsyndromic craniosynostosis (noncanonic mutation, NCM, hereinafter) (data not shown). Mutational and clinical data are summarized in Table 1 ▶ .
Table 1.
Mutational and Clinical Data of the Craniosynostotic Patients Included in the Study
| Craniosynostotic conditions | Clinical features | FGFR mutations and locations |
|---|---|---|
| Apert syndrome | Acrocephaly, proptosis, mid-facial hypoplasia, severe bilateral bony and cutaneous syndactily of both hands and feet | P253R |
| FGFR2, linker between the II and III Ig-like domains | ||
| Pfeiffer type II syndrome | Cloverleaf skull, mid-facial hypoplasia, severe ocular proptosis, digit anomalies | C342R |
| FGFR2, III Ig-like domain | ||
| Nonsyndromic craniosynostosis | Coronal stenosis, plagiocephaly, hypertelorism and invaginated occipital bone | No canonic FGFR mutations (NCM) |
Primary Osteoblast Cultures and Cell Morphology
Cell cultures consisted of homogeneous osteoblast populations as identified by ALP activity, which is an early marker of osteoblast phenotype 36 (see below). Osteo-blasts from the second to the fourth passage with no detectable changes in cell phenotype were used.
By phase contrast microscopy, control osteoblasts appeared with a typical fibroblast-like morphology (Figure 1A) ▶ , characteristic of the proliferating osteoblastic cell. 37,38 At confluence, a few cell aggregates appeared and became multilayered to form nodules. NCM osteoblasts showed a typical fusiform aspect but they reached only 70% confluence in cultures and formed a higher number of multilayered nodules than control cells (Figure 1B) ▶ . P253R osteoblasts showed flattened and rounded shape and formed several multilayered nodules even at partial confluence (about 60%) (Figure 1C) ▶ . C342R osteoblast shape appeared similar to control osteoblasts and formed just a few number of multilayered nodules; however, as observed in the NCM and P253R cell cultures, they reached only partial confluence (60%) (Figure 1D) ▶ .
Figure 1.
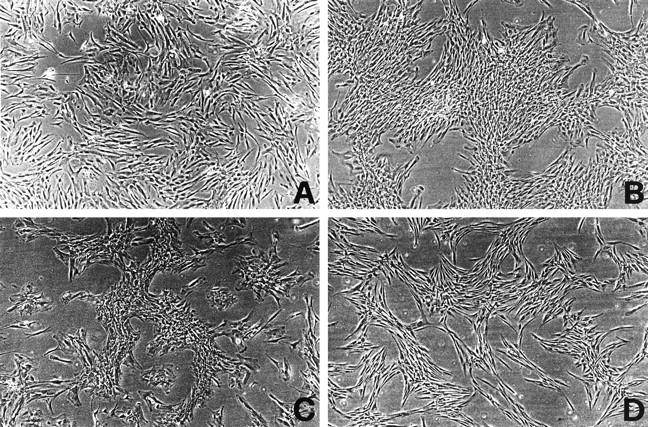
Phase contrast microphotographs representing comparative morphology of primary osteoblasts cultured for 2 weeks (second passage). A: Control osteoblasts; B: NCM osteoblasts; C: P253R osteoblasts; D: C342R osteoblasts. Original magnification, ×120.
Cell Proliferation
In vitro cell proliferation was measured by 3H-thymidine uptake in basal conditions. As shown in Figure 2 ▶ , proliferation rates of NMC, P253R, and C342R osteoblasts were significantly lower than those of control osteoblasts. More precisely, DNA synthesis was about 40% in NCM osteoblasts and 20% in P253R and C342R osteoblasts compared to control osteoblasts.
Figure 2.

In vitro cell proliferation comparison between control osteoblasts and osteoblasts isolated from distinct craniosynostotic patients. Data represent pool of three independent experiments performed in triplicate, and are expressed as means ± SE. *P <0.001 versus control cells.
ALP Activity
ALP activity represents an early marker of osteoblastic differentiation. This enzyme is believed to play a primary role in bone mineralization, and its expression during osteoblast differentiation has been inversely related with cell proliferation. 36,39,40 As shown in Figure 3A ▶ , control osteoblast cultures showed about 50% of cells stained for ALP; the remaining cell population appeared only weakly positive. NCM and C342R osteoblast cultures showed a heterogeneous ALP staining, with a high number of cells strongly positive (Figure 3, B and D) ▶ . The majority of the P253R osteoblasts exhibited an intense and uniform staining for ALP (Figure 3C) ▶ . Moreover, these cells were also characterized by a marked ALP staining in subconfluent conditions, whereas control cells showed an appreciable staining only in confluent cultures (data not shown). ALP activity was also measured using biochemical assay to quantify the above-observed differences (Figure 4) ▶ . As expected, ALP activity in osteoblasts isolated from craniosynostotic patients was significantly higher in respect to control osteoblasts.
Figure 3.
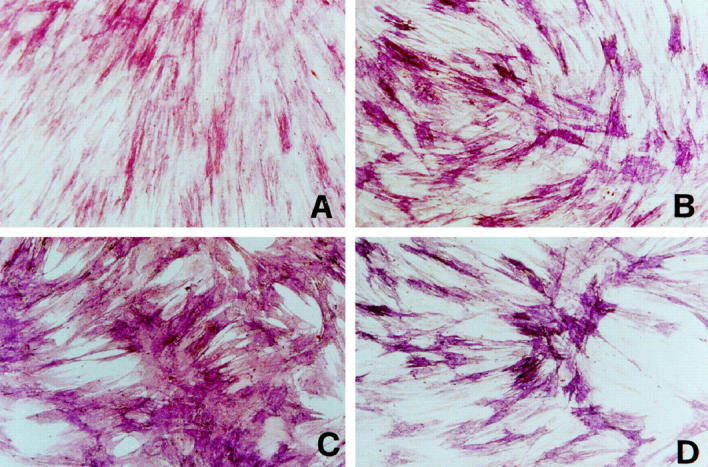
Comparative histochemical analysis of ALP activity in control osteoblasts and in osteoblasts isolated from craniosynostotic patients. Cells were cultured for one week in standard conditions before analysis. A: Control osteoblasts; B: NCM osteoblasts; C: P253R osteoblasts; D: C342R osteoblasts. Microphotographs are representative of at least four experiments performed between the second and third passages. Original magnification, ×200.
Figure 4.

Biochemical analysis of ALP activity expressed as mean of Sigma units/mg total proteins, assayed in control osteoblasts and in osteoblasts isolated from craniosynostotic patients. Data represent pool of three independent experiments performed in triplicate, and are expressed as means ± SE. *P <0.001 versus control cells.
Analysis of Mineralization Pattern by von Kossa’s Staining
Osteoblast cell cultures maintained in continuous presence of glucocorticoids and with appropriate substrates, produce abundant extracellular matrix that can mineralize in vitro. 41,42 To evaluate differences in matrix mineralization, osteoblasts were maintained in culture for 6 weeks in appropriate medium with or without DEX.
Figure 5 ▶ shows the mineralization profiles observed in 2-week (Figure 5, A, C, E, and G) ▶ and 6-week (Figure 5, B, D, F, and H) ▶ DEX-stimulated cultures. In control osteoblast cultures few mineralized nodules were apparent after 2 weeks (Figure 5A) ▶ , and appreciable matrix mineralization was detected only after 6 weeks (Figure 5B) ▶ . NCM and P253R osteoblasts showed a substantial increase in matrix mineralization relative to control cells; a high number of mineralized nodules were observed even after 2 weeks of culture (Figure 5, C and E) ▶ , and increased progressively strongly after 6 weeks (Figure 5, D and F) ▶ . Surprisingly, DEX-stimulated C342R osteoblast cultures failed to produce mineralized matrix throughout the 6 weeks of cultures (Figure 5, G and H) ▶ . All osteoblasts cultured without DEX failed to form mineralized nodules at any time (data not shown).
Figure 5.
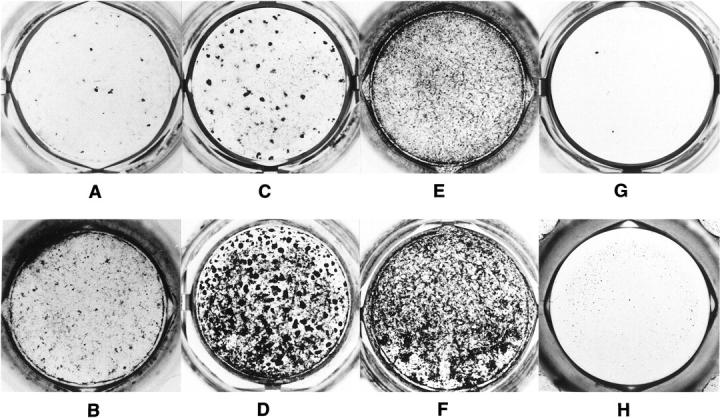
Comparison of in vitro mineral deposition profile in osteoblast cultures isolated from control individual and patients with different craniosynostotic disorders. Osteoblasts were cultured in 24 wells/plate for 6 weeks in presence of dexamethasone. After 2 (A, C, E, and G) and 6 (B, D, F, and H) weeks of culture, plates were fixed and mineralization was detected by von Kossa’s staining. Control osteoblasts produced appreciable mineralization only after 6 weeks (A and B); NCM (C and D) and P253R (E and F) osteoblasts showed positive mineralization profiles after 2 weeks that sensibly increased after 6 weeks of culture. In C342R osteoblast cultures no mineralized nodules were detected during 6 weeks of culture (G and H). Photographs are representative of three distinct experiments.
The increased mineralization in the NCM and P253R osteoblast cultures suggests that these cells be characterized by a more differentiated phenotype compared with control osteoblasts. By contrast, the absence of C342R osteoblast mineralization suggests that these cells do not reach the late differentiative stage of osteoblast maturation in vitro.
Expression of Noncollagenous Matrix Proteins
In vivo and in vitro expression of noncollagenous matrix proteins is correlated with the differentiative stage of osteoblasts. 40,43 We investigated by Western blot analysis, the synthesis levels of osteopontin (OPN), bone sialoprotein (BSP), and osteonectin (ONC); these matrix proteins are expressed in a specific temporal sequence during the switch toward the mature osteoblast phenotype.
As shown in Figure 6A ▶ , NCM and P253R osteoblasts maintained in 10% FBS showed enhanced expression of OPN and BSP and a weak increase of ONC compared to control osteoblasts. The increased expression of OPN in the NCM and P253R osteoblasts was also observed in serum-free conditions. By contrast, OPN and ONC expression in the C342R osteoblasts cultured in 10% FBS was found to be lower than the one observed in the NCM, P253R, and control osteoblasts. However, C342R cells exhibited a relatively higher BSP synthesis in comparison to control osteoblasts. Semiquantification by means of densitometric analysis showed that NCM and P253R osteoblasts were characterized by a significant increase in expression of all three matrix proteins, expressed as ratio of specific protein expression to β-actin (Figure 6B) ▶ . Moreover, these results suggest that NCM and P253R osteoblasts are characterized by a more differentiated phenotype than control osteoblasts, and that the reduced proliferation rate observed in the C342R osteoblasts does not seem to be coupled to an anticipated progression of osteoblast differentiation.
Figure 6.
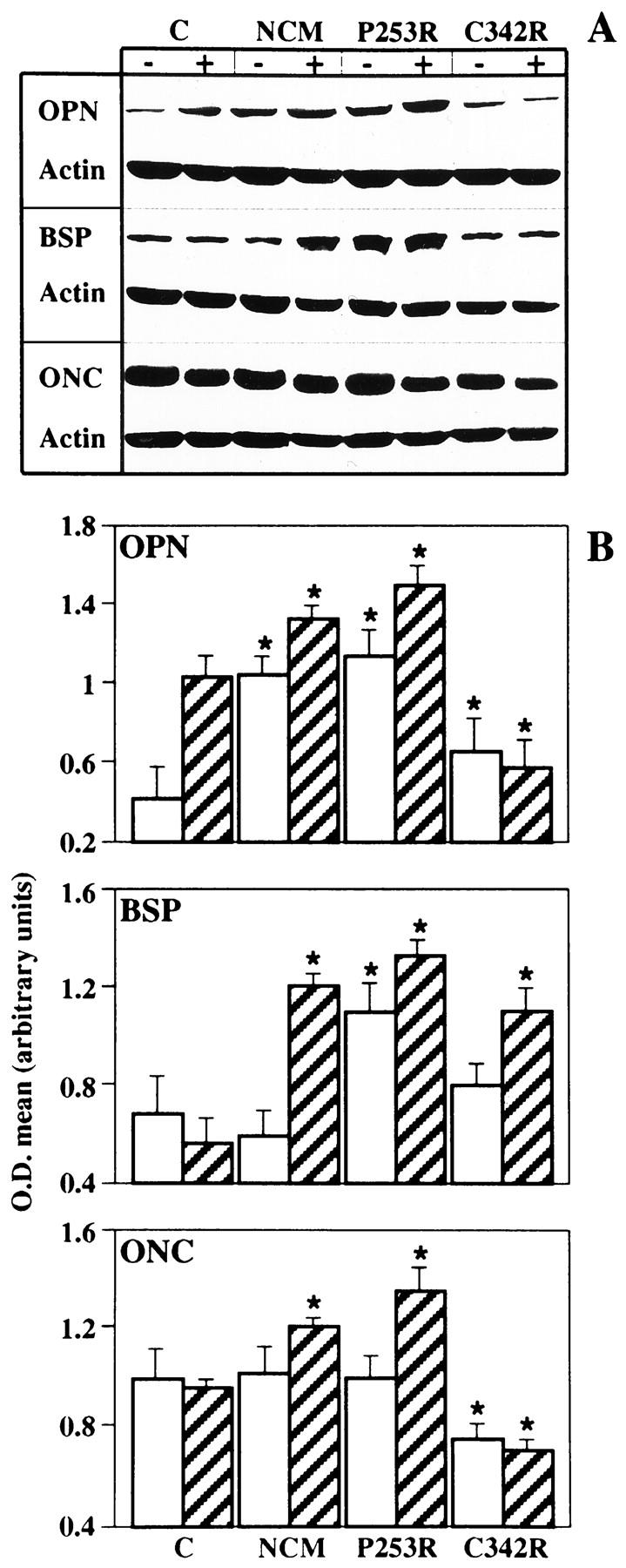
Changes in noncollagenous matrix protein expression in osteoblast cultures obtained from control individual and patients with different craniosynostotic disorders by Western blotting analysis. A: Composite autoradiograms from representative experiments. Western blotting analysis was performed using anti-OPN, anti-BSP, anti-ONC, and anti-β-actin (Actin) antibodies in osteoblasts cultured with (+) or without (−) 10% FBS for 72 hours. B: Bar graphs represent quantification of protein levels by densitometric scanning of autoradiograms obtained from multiple experiments. Striped bars and white bars refer to cultures with and without 10% FBS, respectively. Values are means ± SE and are represented as ratios of the specific protein to β-actin to compensate for any loading differences. *P <0.05 versus control cells.
PKC Isoenzyme Characterization
The FGFR transduction pathway involves the phospholipase C-γ (PLCγ)-mediated phosphoinositide hydrolysis and, consequently, the activation of protein kinase type C (PKC) isoenzymes, which essentially consists of their translocation from the cytosol compartment to the membrane. 44,45 Recently we observed that proliferating and differentiated osteoblasts exhibit differential PKC isoenzymes expression and activation profiles 38 (our unpublished data). To investigate the expression and activation patterns of PKC isoenzymes, osteoblasts were grown to subconfluence and cytosol and membrane fractions were immunodetected for expression of PKCα, PKCε, and PKCζ isoenzymes, which are known to be expressed in osteoblasts. 38,46,47
Control osteoblasts showed the majority of PKCα expression in the cytosol fraction, whereas only a small amount was observed in the membrane fraction (Figure 7) ▶ . Such a distribution profile has been found to be associated with a proliferating osteoblast phenotype. 38 Both NCM and P253R osteoblasts exhibited peculiar profiles, unambiguously distinct from those observed in control osteoblasts. NCM cells were characterized by a substantial increase of PKCα expression, particularly in the cytosolic fraction, whereas the P253R osteoblasts showed a higher level of activated membrane-bound PKCα. On the contrary, C342R osteoblasts had significantly lower PKCα expression levels than control, NCM, and P253R osteoblasts. High PKCε expression levels have been observed in nonproliferating differentiated osteoblasts (our unpublished results). Highest PKCε expression was found in the NCM and P253R osteoblast membrane fractions, whereas it was again strongly attenuated in C342R osteoblasts (Figure 7) ▶ . Finally, we observed that PKCζ isoenzyme was expressed in osteoblasts with undifferentiated phenotype (unpublished data); PKCζ was found to be weakly expressed in control osteoblasts and to be increased in the cytosolic fraction of the NCM and P253R osteoblasts, although no differences among these cells were observed in the membrane-bound isoenzyme levels. By contrast, PKCζ expression was greatly increased and membrane-translocated in the C342R osteoblasts (Figure 7) ▶ .
Figure 7.
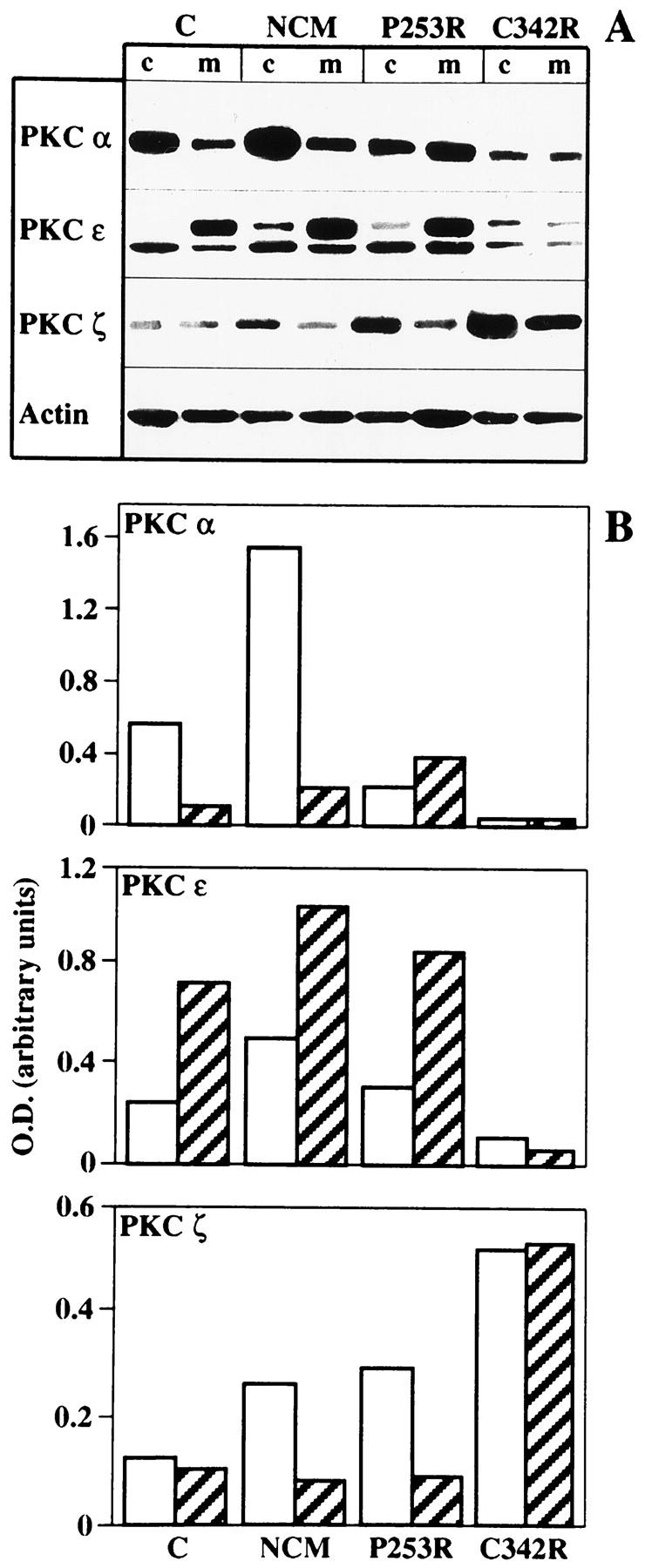
Differential expression and activation of PKCα, PKCε, and PKCζ isoenzymes in control osteoblasts and in osteoblasts isolated from patients with different craniosynostotic disorders by Western blotting analysis. Cytosol (c) and membrane (m) fractions were separated from subconfluent cultures. A: Composite autoradiograms from a single representative experiment. B: Bar graphs of PKCα, PKCε, and PKCζ isoenzymes represent quantification of protein levels by densitometric scanning of autoradiograms obtained from a single representative experiment of three distinct experiments performed. White bars and striped bars refer to the cytosol fraction and membrane fraction, respectively. Values are represented as ratios of PKC isoenzyme to β-actin to compensate for any loading differences.
On the whole, the differential PKC isoenzyme profiles found in osteoblasts from craniosynostotic patients suggest the presence of a disruption of the PKC intracellular pathway, which is downstream of the FGFR signaling.
FGF2 Effects on Osteoblast Proliferation and Differentiation
FGF2 exerts a mitogenic effect on osteoblasts and modulates osteoblast differentiation. 20,21 To study the effects of such growth factor on the proliferation/differentiation pattern of osteoblasts obtained from craniosynostotic patients, we evaluated, in serum-free cultures, hr-FGF2 modulation of DNA synthesis, ALP activity, BSP, OPN, and ONC expression, and extracellular matrix mineralization.
As shown in Figure 8 ▶ , FGF2 stimulated cell proliferation in control, NCM, P253R, and C342R osteoblasts, albeit to different extents. A twofold increase in the proliferation rate of the FGF2-treated control, NCM, and P253R osteoblasts versus the untreated counterparts was observed, whereas a fourfold increase was found in the FGF2-treated C342R osteoblasts.
Figure 8.

Effect of treatment with hr-FGF2 (20 ng/ml) and heparin (50 μg/ml) on cell proliferation of osteoblasts isolated from control individual and patients with different craniosynostotic conditions. Cells were serum-free cultured before treatment for 24 hours. FGF2 was added to the medium culture for 24 hours. Data are representative of three distinct experiments performed in quadruplicate and are expressed as cpm/well means ± SE. *P <0.001 versus untreated cells.
Because of such proliferative effect, we wondered whether FGF2 could regulate osteoblast differentiation. In all osteoblast cultures, 48 hours of FGF2 treatment apparently did not exert any significant effect on ALP activity as assayed histochemically and biochemically, nor on OPN, OCN, and BSP expression levels as assayed by Western blot analysis (data not shown). However, long-term FGF2 treatment totally inhibited DEX-stimulated matrix mineralization of control, NCM, and P253R osteoblasts cultured in 0.1% FBS (Figure 9, I, J and K) ▶ . It should be noted that matrix mineralization in control and NCM osteoblast cultures was observed in DEX-stimulated 0.1% FBS conditions (Figure 9, E and F) ▶ , although lower compared to 10% FBS cultures (Figure 9, A and B) ▶ . Interestingly, P253R osteoblasts showed a high FBS-independent degree of mineralization (Figure 9, C and G) ▶ . As expected, C342R osteoblasts failed to mineralize in all DEX-stimulated culture conditions (Figure 9, D, H, and L) ▶ .
Figure 9.
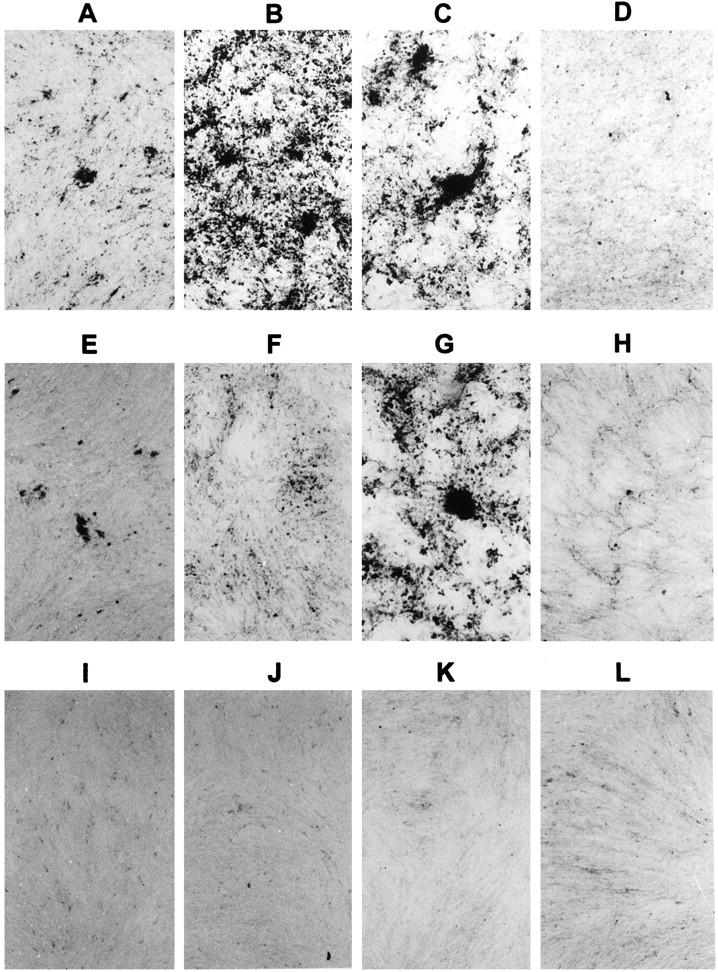
Effect of treatment with hr-FGF2 (20 ng/ml) and heparin (50 μg/ml) on in vitro mineralization of control (A, E, and I), NCM (B, F, and J), P253R (C, G, and K), and C342R (D, H, and L) osteoblast cultures. At the end of cultures, plates were fixed and mineralization was detected by von Kossa’s staining. Cultures were serum-deprived for 24 hours before treatment. During the 2 weeks of culture, FGF2 and heparin were continuously added in the mineralization medium supplemented with 0.1% FBS (I, J, K, and L). FGF2 treatment inhibited mineralization in all samples. Osteoblasts were also cultured in mineralization medium without growth factor as a control, supplemented with 0.1% FBS (E, F, G, and H) and 10% FBS (A, B, C, and D). Note the increased mineralization in P253R in 0.1% FBS cultures (G). Microphotographs were representative of three distinct experiments performed in triplicate. Original magnification, ×50.
These findings indicate that, regardless of the presence or absence of mutations in FGFR2, FGF2 stimulates osteoblast proliferation and that, similarly to what was observed in control osteoblasts, the FGF2-induced increase in cell proliferation rate of NCM and P253R osteoblasts is associated with a negative modulation of the late osteoblast differentiation in long-term cultures.
Discussion
In the present study, we reported a proliferative/differentiative profile characterization of primary osteoblast cultures isolated from patients affected by genetically and clinically distinct craniosynostotic disorders. Our aim was to evaluate the presence of possible heterogeneity in such proliferative and differentiative patterns to contribute to an elucidation of the effects of different FGFR mutations (or mutations involving other genes) on osteoblast function. Three different craniosynostoses were considered: the Apert syndrome associated with the Pro253Arg substitution in FGFR2 (P253R), one of the two common nucleotidic changes associated with this syndrome; 48 the Pfeiffer type 2 syndrome with the Cys342Arg change in the same receptor (C342R), the most common mutation associated with Crouzon, Pfeiffer, and Jackson-Weiss syndromes, accounting for about 40% of total diagnosed cases; 5 and a nonsyndromic craniosynostotic condition apparently not associated with mutations in the canonic FGFR hot spots (NCM). Interestingly, the P253R and C342R amino acidic substitutions are located in two distinct but contiguous functional FGFR2 domains involved in ligand binding, ie, the linker segment between the second and third Ig-like domains and the third Ig-like domain, respectively. This gave us the opportunity to evaluate the effect of different FGFR2 mutations on the osteoblast function. Parameters of osteoblastic proliferation and differentiation were investigated in these osteoblast cultures and compared with osteoblasts from an age-matched, unaffected individual.
Under our experimental conditions, osteoblasts from craniosynostotic patients showed a lower proliferation rate compared to control cells. Consistent with a nonproliferating phenotype, these osteoblast cultures failed to reach confluence and were organized to form cellular clusters. Furthermore, a high number of multilayered nodules were peculiar to NCM and P253R cultures. The low proliferation we observed in the P253R osteoblasts apparently is not in agreement with the absence of alteration in basal cell proliferation in osteoblasts isolated from infants and fetuses with Apert syndrome associated with S252W mutation recently reported. 26 It must be pointed out that the low proliferative rate in osteoblasts from craniosynostotic patients was not constitutively controlled. Potential proliferative capacity was verified by FGF2 treatment. FGF2 stimulated cell proliferation of all pathological osteoblast cultures in a similar manner to that observed in control osteoblasts, according to the previous data on the S252W osteoblasts. 26 Moreover, the FGF2-dependent increase in cell proliferation in NCM, P253R, and control osteoblasts was apparently coupled to an inhibition of the late osteoblast differentiative phenotype onset, as indicated by the failure of these cultures to mineralize in vitro in the presence of this growth factor.
We found substantial differences in the expression of parameters of cell differentiation among osteoblasts from the three craniosynostotic patients. In the NCM and P253R osteoblasts the decreased proliferation rate was associated with a marked differentiated osteoblastic phenotype, characterized by higher ALP activity and expression of noncollagenous matrix proteins relative to control cells; they also exhibited a striking increase in extracellular matrix mineralization rate. Such mature phenotype was also associated with high expression and activation of PKCα and PKCε isoenzymes. Accordingly, a distinct PKC expression pattern characterized by an increased expression and concomitant activation of PKCα and PKCε in differentiated osteoblasts was recently observed 38,47 (our unpublished data). The results presented here suggest that an alteration in this specific intracellular signaling exists in these distinct craniosynostotic conditions. We are investigating the biological meaning of these data because this altered pattern may represent either the pivotal alteration in these cells or the consequence of perturbed intracellular signaling linked to FGFR mutations.
Differences between NCM and P253R osteoblasts concerned ALP activity, considerably higher in the P253R osteoblasts, as well as expression of PKCα and cell proliferation rate, both increased in NCM osteoblasts. The differentiated phenotype observed in the NCM and P253R osteoblasts is in agreement with the increased differentiated phenotype described in osteoblasts isolated from patients with nonsyndromic craniosynostoses 25 and from patients affected by Apert syndrome carrying the S252W substitution. 26
Unlike the NCM and P253R osteoblasts, the low proliferation rate of the C342R osteoblasts was found to be associated with a less differentiated phenotype; these cells were totally unable to mineralize bone matrix and more responsive to FGF2 mitogenic stimulation. Furthermore, C342R osteoblasts showed relatively low OPN and ONC expression levels. According with such “defective” differentiative phenotype, C342R osteoblasts exhibit low levels of PKCα and PKCε isoenzymes but high levels of cytosolic and membrane-bound PKCζ. These findings suggest that the FGFR2 C342R amino acidic substitution might have a role in the modulation of a late step of the intracellular machinery involved in the in vitro osteoblast differentiation events.
The phenotypic differences we observed between P253R and C342R osteoblasts support the existence of at least two distinct mechanisms responsible for the alteration of the FGFs/FGFR2 transduction pathway and the premature ossification of the skull sutures. Our findings agree with evidence from a number of previous experiments based on transfection studies in a variety of cell lines, as well as on mRNA microinjections in Xenopus laevis embryos, which demonstrated that FGFR2 missense mutations located in the third Ig-like domain and in the linker segment between the second and third Ig-like domains have two different perturbing roles in receptor function. 12-15 The former, as the C342R substitution here considered, would result in constitutive FGFR2 homodimerization by intermolecular disulfide bonding and, consequently, in FGF-independent receptor activation. 12-14 Differently, P253R substitution and the adjacent S252W change are believed to affect FGF binding stability, affinity, or specificity. 15 Nevertheless, the causative cellular mechanisms responsible for this striking difference in the control and modulation of in vitro osteoblast differentiation remain unclear. Osteoblast differentiation is a series of events, modulated by sequential activation of genes that initially promote cell proliferation and then biosynthesis, organization, and mineralization of bone extracellular matrix. 43,49,50 In particular, two independent series of signaling mechanisms control the transition from a proliferative undifferentiated phenotype to a matrix secreting osteoblastic cell and the progression toward the late differentiative stage in which the expression of genes involved in matrix maturation and mineralization is observed. 51 It is possible to hypothesize that the perturbing action of the C342R (or other amino acidic substitutions in the third Ig-like domain) and P253R (or S253P) mutations in the FGFR2 might have a distinct effect on these two differentiative transition events. It must be pointed out that the “gain of function” mechanism proposed for mutated FGFR2 12-14 can be reverted in vitro, because the reduced proliferation rate associated with the perturbing action of a mutated FGFR2 is overcome by FGF2 stimulation.
Although no mutation was found in the canonic hot spots of the FGFRs in the NCM osteoblasts, these cells showed an osteoblastic mature phenotype similar to the one characterizing osteoblasts carrying the P253R change. These findings suggest that an alternative molecular alteration might be responsible for a premature proliferation/differentiation switch that drives the NCM osteoblasts toward a mature secreting phenotype. Differences observed in our in vitro cultures between NCM and P253R osteoblasts, ie, the higher ALP activity and increased mineralization profile of P253R osteoblasts, could be related to differences in pathology severity.
Taken together, our data demonstrate that these genetically and clinically distinct craniosynostotic conditions are associated with a decreased osteoblast proliferation and a differentiative phenotype peculiar to each genotype and that mutations in distinct FGFR2 domains are associated with an in vitro heterogeneous osteoblastic phenotype. Our results suggest that the unorthodox FGFR2 activation is directly involved in the negative modulation of the preosteoblast proliferation and in the stimulation of the progression of the osteoblast differentiation. In accordance with these observations, the activated FGFR2 could be involved in the differentiation progression of the proliferating osteogenic stem cells, and consequently an alteration of the FGFR2-coupled signal could induce an early proliferation/differentiation switch that could anticipate the suture closure. In vivo, during fetal mouse skull development, FGFR2 transcripts are most abundant at the level of the proliferation layer adjacent to the osteogenic front and are mutually exclusive with the osteopontin transcripts, indicating that FGFR2 operates in a cellular stage that precedes osteoblast differentiation. 22 Furthermore, it has recently been shown that beads soaked with FGF4 and implanted in the skull of neonate mice accelerate suture closure when placed at the osteogenic front during suture morphogenesis, with a mechanism involving increased osteoblast proliferation and differentiation. 23 The present findings highlight the complexity of the FGF/FGFR network and indicate that different FGFs and FGFRs might be involved in distinct cellular events in modulating osteogenic stem cell functions. A deeper understanding of the cellular mechanisms involved in craniosynostosis will require determining which FGF is locally secreted and which FGFR is expressed during the progression of cell differentiation toward mature osteoblast.
Acknowledgments
We cordially thank the families who took part in the study. We thank Dr. Larry Fisher for kind gift of anti-human osteopontin, bone sialoprotein, and osteonectin antibodies. We also wish to thank Dr. Paola Manduca for her kind help with Western blotting analysis of noncollagenous matrix proteins and Veronica Bordoni for her helpful assistance with experiments.
Footnotes
Address reprint requests to Dr. Marco Tartaglia, Laboratorio di Biologia Cellulare, Istituto Superiore di Sanità, Viale Regina Elena, 299–00161 Rome, Italy. E-mail: mtartaglia@iss.it.
Supported by the Istituto Superiore di Sanità research project “Risk factors for mother and child health” (to P. A. B.), the Consiglio Nazionale delle Ricerche research project “Gene expression and regulation of bone remodeling: osteoblast differentiation and function” (to A. T.), and a contribution of the Istituto Pasteur-Fondazione Cenci Bolognetti. S. M. was supported by an Eli Lilly grant.
References
- 1.Cohen MM, Jr: Craniosynostosis: diagnosis, evaluation and management. 1986. Raven Press, New York
- 2.Di Rocco C, Velardi F: Surgical management of craniosynostosis. Galli G eds. Craniosynostosis. 1984, :pp 181-248 CRC Press, Boca Raton, FL [Google Scholar]
- 3.McKusick VA: Mendelian inheritance in man. Catalogs of Human Genes and Genetic Disorders. 1994, Johns Hopkins University Press, Baltimore
- 4.Bonaventure J, Rousseau F, Legeai-Mallet L, Le Merrer M, Munnich A, Maroteaux P: Common mutations in the fibroblast growth factor receptor 3 (FGFR3) gene account for achondroplasia, hypochondroplasia, and thanatophoric dwarfism. Am J Med Genet 1996, 63:148-154 [DOI] [PubMed] [Google Scholar]
- 5.Wilkie AOM: Genes and mechanisms. Hum Mol Genet 1997, 6:1647-1656 [DOI] [PubMed] [Google Scholar]
- 6.Jaye M, Schlessinger J, Dionne CA: Fibroblast growth factor receptor tyrosine kinases: molecular analysis and signal transduction. Biochem Biophys Acta 1992, 1135:185-190 [DOI] [PubMed] [Google Scholar]
- 7.Basilico C, Moscatelli D: The FGF family of growth factors, and oncogenes. Adv Cancer Res 1992, 59:115-165 [DOI] [PubMed] [Google Scholar]
- 8.Fernig DJ, Gallager JT: Fibroblast growth factors and their receptor: an information network controlling tissue growth, morphogenesis and repair. Prog Growth Factor Res 1994, 5:353-377 [DOI] [PubMed] [Google Scholar]
- 9.Nakafuku M, Satoh T, Kaziro Y: Differentiation factors, including nerve growth factor, fibroblast growth factor and interleukin-6, induce an accumulation of an active Ras GTP complex in rat pheochromocytoma PC12 cells. J Biol Chem 1992, 267:19448-19454 [PubMed] [Google Scholar]
- 10.Wang JK, Xu H, Li H-C, Goldfarb M: Broadly expressed SNT-like proteins link FGF receptor stimulation to activators of RAS. Oncogene 1996, 12:721-729 [PubMed] [Google Scholar]
- 11.Burgess WH, Dionne CA, Kaplow J, Mudd R, Friesel R, Zilberstein A, Schlessinger J, Jaye M: Characterization and cDNA cloning of phospholipase C-γ, a major substrate for heparin growth factor 1 (acidic fibroblast growth factor) activated tyrosine kinase. Mol Cell Biol 1990, 10:4770-4777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Neilson KM, Friesel RE: Constitutive activation of fibroblast growth factor receptor-2 by a point mutation associated with Crouzon syndrome. J Biol Chem 1995, 270:26037-26040 [DOI] [PubMed] [Google Scholar]
- 13.Mangasarian K, Li Y, Mansukhani A, Basilico C: Mutation associated with Crouzon syndrome causes ligand-independent dimerization and activation of FGF receptor-2. J Cell Physiol 1997, 172:117-125 [DOI] [PubMed] [Google Scholar]
- 14.Robertson SC, Meyer AN, Hart KC, Galvin BD, Webster MK, Donoghue DJ: Activating mutations in the extracellular domain of the fibroblast growth factor receptor 2 function by disruption of the disulphide bond in the third immunoglobulin-like domain. Proc Natl Acad Sci USA 1998, 95:4567-4572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anderson J, Burns HD, Enriquez-Harris P, Wilkie AOM, Heath JK: Apert syndrome mutations in fibroblast growth factor receptor 2 exhibit increased affinity for FGF ligand. Hum Mol Genet 1998, 7:1475-1483 [DOI] [PubMed] [Google Scholar]
- 16.Mundolos S, Olsen BR: Heritable diseases of the skeleton. Part I: molecular insights into skeletal development-transcription factors and signalling pathways. FASEB J 1997, 11:125-132 [DOI] [PubMed] [Google Scholar]
- 17.Canalis E: The hormonal and local regulation of bone formation. Endocrin Rev 1983, 4:62-67 [DOI] [PubMed] [Google Scholar]
- 18.Hurley MM, Kessler M, Gronowicz G, Raisz LG: The interaction of heparin and basic fibroblast growth factor on collagen synthesis in 21-day fetal ray calvariae. Endocrinology 1992, 130:2675-2682 [DOI] [PubMed] [Google Scholar]
- 19.Tang K-S, Capparelli C, Stein JL, Stein GS, Lian JB, Huber AC, Braverman LE, DeVito WJ: Acidic fibroblast growth factor inhibits osteoblast differentiation in vitro: altered expression of collagenase, cell growth-related, and mineralization-associated genes. J Cell Biochem 1996, 61:152-166 [DOI] [PubMed] [Google Scholar]
- 20.Martin I, Muraglia A, Campanile G, Cancedda R, Quarto R: Fibroblast growth factor-2 supports ex vivo expansion and maintenance of osteogenic precursors from human bone marrow. Endocrinology 1997, 138:4456-4462 [DOI] [PubMed] [Google Scholar]
- 21.Debiais F, Hott M, Graulet AM, Marie PJ: The effects of fibroblast growth factor-2 on human neonatal calvaria osteoblastic cells are differentiation stage specific. J Bone Miner Res 1998, 13:645-654 [DOI] [PubMed] [Google Scholar]
- 22.Iseki S, Wilkie AOM, Heath JK, Ishimaru T, Eto K, Morris-Kay GM: Fgfr2, and osteopontin domains in the developing skull vault are mutually exclusive, and can be altered by locally applied FGF2. Development 1997, 124:3357-3384 [DOI] [PubMed] [Google Scholar]
- 23.Kim H-J, Rice DPC, Kettunen PJ, Thesleff I: FGF-, BMP-, and Shh-mediated signalling pathways in the regulation of cranial suture morphogenesis, and calvarial bone development. Development 1998, 125:1241-1251 [DOI] [PubMed] [Google Scholar]
- 24.Most D, Levine JP, Chang J, Sung J, McCarthy JG, Schendel SA, Longaker MT: Studies in cranial suture biology: up-regulation of transforming growth factor-β 1 and basic fibroblast growth factor mRNA correlates with posterior frontal cranial suture fusion in the rat. Plast Reconstr Surg 1998, 101:1431-1440 [DOI] [PubMed] [Google Scholar]
- 25.De Pollack C, Renier D, Hott M, Marie PJ: Increased bone formation and osteoblastic cell phenotype in premature cranial suture ossification (craniosynostosis). J Bone Min Res 1996, 11:401-407 [DOI] [PubMed] [Google Scholar]
- 26.Lomri A, Lemonnier J, Hott M, de Parseval N, Lajeunie E, Munnich A, Renier D, Marie PJ: Increased calvaria cell differentiation and bone matrix formation induced by fibroblast growth factor receptor 2 mutations in Apert syndrome. J Clin Invest 1998, 101:1310-1317 [PMC free article] [PubMed] [Google Scholar]
- 27.Cohen MM, Jr: Pfeiffer syndrome update, clinical subtypes and guideline for differential diagnosis. Am J Med Genet 1993, 45:300-307 [DOI] [PubMed] [Google Scholar]
- 28.Tartaglia M, Valeri S, Velardi F, Di Rocco C, Battaglia PA: Trp290Cys mutation in the exon IIIa of the fibroblast growth factor receptor 2 (FGFR2) gene is associated with Pfeiffer syndrome. Hum Genet 1997, 99:602-606 [DOI] [PubMed] [Google Scholar]
- 29.Tartaglia M, Di Rocco C, Lajeunie E, Valeri S, Velardi F, Battaglia PA: Jackson-Weiss syndrome: identification of two novel FGFR2 missense mutations shared with Crouzon and Pfeiffer craniosynostotic disorders. Hum Genet 1997, 101:47-50 [DOI] [PubMed] [Google Scholar]
- 30.Tartaglia M, Saulle E, Bordoni V, Battaglia PA: Polymorphism at position 882 of the fibroblast growth factor receptor 3 (FGFR3) gene detected by SSCP analysis. Mol Cell Probes 1998, 12:335-337 [DOI] [PubMed] [Google Scholar]
- 31.Przylepa KA, Paznekas W, Zhang M, Golabi M, Bias W, Bamshad MJ, Carey JC, Hall BD, Stevenson R, Orlow SJ, Cohen MM, Jr, Jabs EW: Fibroblast growth factor receptor 2 mutations in Beare-Stevenson cutis gyrata syndrome. Nat Genet 1996, 13:492-494 [DOI] [PubMed] [Google Scholar]
- 32.Robey Gehron P, Termine JD: Human bone cells in vitro. Calcif Tissue Int 1985, 37:453-460 [PubMed] [Google Scholar]
- 33.Bills CE, Eisenberg H, Pallante SL: Complexes of organic acids with calcium phosphate: the von Kossa stain as clue to the composition of bone mineral. Johns Hopkins Med J 1971, 128:194-207 [PubMed] [Google Scholar]
- 34.Fisher LW, Stubb JT, 3rd, Young MF: Antisera and cDNA probes to human and certain animal model bone matrix noncollagenous proteins. Acta Orthop Scand (suppl) 1995, 266:61-65 [PubMed] [Google Scholar]
- 35.Wodgett JR, Hunter T: Immunological evidence for two physiological forms of protein kinase C. Mol Cell Biol 1987, 7:85-96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Whyte MP: Hypophosphatasia and the role of alkaline phosphatase in skeletal mineralization. Endocr Rev 1994, 15:439-461 [DOI] [PubMed] [Google Scholar]
- 37.Wergedal JE, Baylink DJ: Characterization of cells isolated from human bone. Proc Soc Exp Biol Med 1984, 176:60-69 [DOI] [PubMed] [Google Scholar]
- 38.Migliaccio S, Wetsel WC, Fox WM, Washburn TF, Korach KS: Endogenous protein kinase-C activation in osteoblasts-like cells modulates responsiveness to estrogens and estrogen receptor levels. Mol Endocrinol 1993, 7:1133-1143 [DOI] [PubMed] [Google Scholar]
- 39.Fedarko NS, Bianco P, Vetter U, Gehron Robey P: Human bone cell enzyme expression and cellular heterogeneity: correlation of alkaline phosphatase enzyme activity with cell cycle. J Cell Physiol 1990, 144:115-121 [DOI] [PubMed] [Google Scholar]
- 40.Cowles EA, DeRome ME, Pastizzo G, Brailey LL, Gronowicz GA: Mineralization and expression of matrix proteins during in vivo bone development. Calcif Tissue Int 1998, 62:74-82 [DOI] [PubMed] [Google Scholar]
- 41.Bellows CG, Aubin JE, Heersche JNM: Physiological concentrations of glucocorticoids stimulate formation of bone nodules from isolated rat calvaria cells in vitro. Endocrinology 1987, 121:1985-1992 [DOI] [PubMed] [Google Scholar]
- 42.Gundle R, Bradley J, Joyner C, Francis M, Beresford J: Bone formation in vivo and in vitro by cultured adult human bone-derived cells. J Bone Min Res 1993, 8:S374 [Google Scholar]
- 43.Stein GS, Lian JB: Molecular mechanisms mediating proliferation/differentiation interrelationship during progressive development of the osteoblast phenotype. Endocr Rev 1993, 14:424-442 [DOI] [PubMed] [Google Scholar]
- 44.Hug H, Sarre TF: Protein kinase C isoenzymes: divergence in signal transduction? Biochem J 1993, 291:329-343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jaken S: Protein kinase C isoenzymes and substrates. Curr Biol 1996, 8:168-173 [DOI] [PubMed] [Google Scholar]
- 46.Migliaccio S, Bernardini S, Wetsel WC, Korach KS, Faraggiana T, Teti A: Protein kinase C modulates estrogen receptors in differentiated osteoblastic cells in vitro. Steroid 1998, 63:352-354 [DOI] [PubMed] [Google Scholar]
- 47.Sanders JL, Stern PH: Expression and phorbol ester-induced down-regulation of protein kinase C isozymes in osteoblasts. J Bone Min Res 1996, 11:1862-1872 [DOI] [PubMed] [Google Scholar]
- 48.Wilkie AOM, Slaney SF, Oldridge M, Poole MD, Ashworth GJ, Hockley AD, Hayward RD, David DJ, Pulleyn LJ, Rutland P, Malcom S, Winter RM, Reardon W: Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat Genet 1995, 9:165-172 [DOI] [PubMed] [Google Scholar]
- 49.Aubin JE, Turksen K, Heersche JNM: Osteoblastic cell lineage. Noda M eds. Cellular and Molecular Biology of the Bone. 1993, :pp 1-45 Academic Press, San Diego [Google Scholar]
- 50.Owen TA, Aronow M, Shalhoub V, Barone LM, Wilming L, Tassinari MS, Kennedy MB, Pockwinse S, Lian JB, Stein GS: Progressive development of rat osteoblast phenotype in vitro: reciprocal relationships in expression of genes associated with osteoblast proliferation and differentiation during formation of the bone extracellular matrix. J Cell Physiol 1990, 143:420-430 [DOI] [PubMed] [Google Scholar]
- 51.Stein GS, Lian JB, Van Wijnen AJ, Montecino M: Transcriptional control of osteoblast growth and differentiation. Physiol Rev 1996, 76:593-629 [DOI] [PubMed] [Google Scholar]