

Abstract
Microglia are a key component of the inflammatory response in the brain and are associated with senile plaques in Alzheimer’s disease (AD). Although there is evidence that microglial activation is important for the pathogenesis of AD, the role of microglia in cerebral amyloidosis remains obscure. The present study was undertaken to investigate the relationship between β-amyloid deposition and microglia activation in APP23 transgenic mice which express human mutated amyloid-β precursor protein (βPP) under the control of a neuron-specific promoter element. Light microscopic analysis revealed that the majority of the amyloid plaques in neocortex and hippocampus of 14- to 18- month-old APP23 mice are congophilic and associated with clusters of hypertrophic microglia with intensely stained Mac-1- and phosphotyrosine-positive processes. No association of such activated microglia was observed with diffuse plaques. In young APP23 mice, early amyloid deposits were already of dense core nature and were associated with a strong microglial response. Ultrastructurally, bundles of amyloid fibrils, sometimes surrounded by an incomplete membrane, were observed within the microglial cytoplasm. However, microglia with the typical characteristics of phagocytosis were associated more frequently with dystrophic neurites than with amyloid fibrils. Although the present observations cannot unequivocally determine whether microglia are causal, contributory, or consequential to cerebral amyloidosis, our results suggest that microglia are involved in cerebral amyloidosis either by participating in the processing of neuron-derived βPP into amyloid fibrils and/or by ingesting amyloid fibrils via an uncommon phagocytotic mechanism. In any case, our observations demonstrate that neuron-derived βPP is sufficient to induce not only amyloid plaque formation but also amyloid-associated microglial activation similar to that reported in AD. Moreover, our results are consistent with the idea that microglia activation may be important for the amyloid-associated neuron loss previously reported in these mice.
Substantial evidence supports the view that processing of the amyloid-β precursor protein (βPP) and accumulation of the amyloid-β peptide (Aβ) in the brain of Alzheimer’s disease (AD) patients is crucial to the pathophysiology of the disease. 1,2 Senile amyloid plaques in AD brains are surrounded and infiltrated by activated microglia, which acquire an amoeboid morphology and express various proteins involved in the central nervous system inflammation. 3-5 The tight association of amyloid fibrils and microglia has suggested that microglia are somehow involved in either the formation or the phagocytosis of amyloid fibrils. 3,6-10 Activation of microglia is thought to induce an inflammatory response in the central nervous system and to be a mediator of the amyloid-associated neurodegeneration in AD brain. 11,12 The involvement of inflammation in the progress of AD is underlined by clinical studies showing an attenuation of AD symptoms by nonsteroidal anti-inflammatory drugs. 13,14
Recently, transgenic mice have been produced that overexpress mutant human βPP (APP23 line, Swedish double mutation) under the control of a neuron-specific Thy-1 promoter element. 15 These mice develop amyloid plaques, predominantly in neocortex and hippocampus, progressively with age. The plaques have most characteristics of human AD plaques, including fibrillar Aβ cores, and are surrounded by dystrophic neurites and activated glial cells. Region-specific amyloid-associated neurodegeneration including neuron loss, synapse deficits, and cholinergic alterations have been reported 16,45 in these mice. To study the involvement of microglia in amyloid plaque formation and neurodegeneration, we have analyzed the microglial response in both young and adult APP23 transgenic mice at light microscopic and ultrastructural levels.
Materials and Methods
Animals
The APP23 transgenic mice used in this study have been described previously. 15 The mice express mutated human βPP (Swedish double mutation) under a brain and neuron-specific murine Thy-1 promoter element. We used a total of 12 hemizygous and homozygous male APP23 mice between 14 and 18 months of age and 10 control mice from the F3-F5 generation (most of these animals have been used to assess neurodegeneration. 16,45 In addition, a group of young male 4- to 9-month-old hemizygous APP23 mice and littermate controls from the F6-F8 generation were used. APP23 mice were initially on a B6D2 background and subsequently have been backcrossed with B6 mice.
Tissue Preparation for Light Microscopy
Animals were injected with an overdose of pentobarbital and transcardially perfused with phosphate buffered saline (PBS) (pH 7.4) followed by 4% paraformaldehyde in PBS at room temperature. Brains were removed and postfixed in the same fixative overnight and placed in 30% sucrose in PBS for 2 days, all at 4°C. Brains were then frozen in 2-methylbutane at −25°C and serially sectioned on a freezing-sliding microtome at 10–40 μm.
Immunohistochemistry and Congo Red Staining
Immunohistochemical staining was performed according to a previously published protocol. 17 In brief, sections were preincubated for 30 minutes in 1% H2O2 followed by 0.3% Triton X-100 in Tris-buffered saline for 10 minutes and 5% blocking serum for 30 minutes. Sections were reacted overnight at 4°C with primary antibodies in 0.3% Triton and 2% serum. The avidin-biotin-peroxidase method (ABC Elite Kit, Vector Laboratories, Burlingame, CA) with diaminobenzidine (DAB) as the chromogen was used to visualize the antibodies (brown reaction product). In some experiments NiCl2 was added to the DAB leading to a black reaction product. Some sections were counterstained with cresyl-violet according to standard protocols.
The following antibodies were used: rat monoclonal anti-mouse CR3 (CD11b; Mac-1; diluted 1:2000; Serotec, Oxford, UK), mouse monoclonal antibody to phosphotyrosine (PT-66; diluted 1:500; Sigma, St. Louis, MO), and polyclonal antibody NT-11 to human Aβ (1:2000). 15
For double labeling, a sequential protocol was used. The first primary antibody (NT-11) was visualized with DAB alone and the second primary antibody (Mac-1) was visualized with Vector SP (Vector Laboratories). Congo Red staining was performed on Mac-1 immunostained sections according to standard protocols and viewed under cross-polarized light.
Semithin Sections and Ultrastructural Analysis
For electron microscopy, mice were transcardially perfused with PBS followed by 4% paraformaldehyde plus 1–2.5% glutaraldehyde in PBS. Mice used for immunoelectron microscopy were perfused with PBS followed by 4% paraformaldehyde containing 0.1% glutaraldehyde in PBS. Brains were removed and postfixed overnight in 4% paraformaldehyde. Sections were cut with a vibratome (60 μm). Sections used for immunoelectron microscopy were stained free-floating with the Mac-1 antibody under the same conditions as described above but without H2O2 and Triton X-100. Sections prepared for both electron and immunoelectron microscopy were postfixed with 0.5% OsO4 in PBS containing 6.86% sucrose for 30 minutes, dehydrated in ascending series of ethanol and acetone, and flat-embedded between glass slides and coverslips in EMbed-812 (Electron Microscopy Sciences, Fort Washington, PA). Using an ultramicrotome, semithin (2 μm) and ultrathin sections were cut from the tissue surface. Ultrathin sections were mounted on copper grids and some sections were contrasted with uranyl acetate and lead citrate and finally analyzed with a Zeiss EM 900 electron microscope.
Semiquantitative Analyses
To study early plaque formation serial 15-μm sections were cut from the brain of young 4- to 9-month-old APP23 mice. A sequence of sections was alternately immunostained for Mac-1 (microglia) and NT-11 (amyloid plaques). Early Aβ-immunoreactive deposits were identified and the corresponding microglia activation in adjacent sections was quantified. In reverse, activated microglia were identified and evidence for corresponding Aβ deposits was determined in adjacent sections. Microglia were considered activated when clustering of at least 2 microglia was observed and both cell body and processes were intensely immunoreactive and hypertrophic. The quantification (number of amyloid plaques with a corresponding microglial reaction, number of clusters of activated microglia with corresponding amyloid deposition) was carried out by two different raters and results were within 10% of each other.
Results
Activated Microglia Associated with Congophilic Plaques but not with Diffuse Amyloid Deposits
Consistent with previous reports in mice and humans, 18,19 immunostaining using an antibody to CD11b (Mac-1) revealed specific labeling of microglia in all white and gray matter regions of the APP23 mouse brain. In 14- to 18-month-old APP23 transgenic mice clusters of microglia intensely stained with Mac-1 were observed in all regions with plaque formation, primarily neocortex and hippocampus (Figure 1A) ▶ . The microglia clusters appeared as focal aggregations when the periphery of a plaque was sectioned and as clusters around an unstained center when the core of an amyloid plaque was sectioned. No such aggregations of Mac-1-positive microglia were observed in nontransgenic littermates (Figure 1B) ▶ . Similarly, using an antibody to phosphotyrosine, which is a specific marker for microglia in the central nervous system, 20,21 clusters of intensely stained microglia were observed in transgenic mice but not in littermate controls (Figure 1, C and D) ▶ . In contrast to anti-Mac-1 staining, antibodies to phosphotyrosine revealed only weak staining of microglia in nontransgenic control mice (Figure 1D) ▶ . Overall, the activation of microglia as characterized by swollen processes and cell bodies was restricted to the plaque areas. Based on morphological and immunohistochemical observations, no global activation of microglia in transgenic compared to nontransgenic brains was observed.
Figure 1.
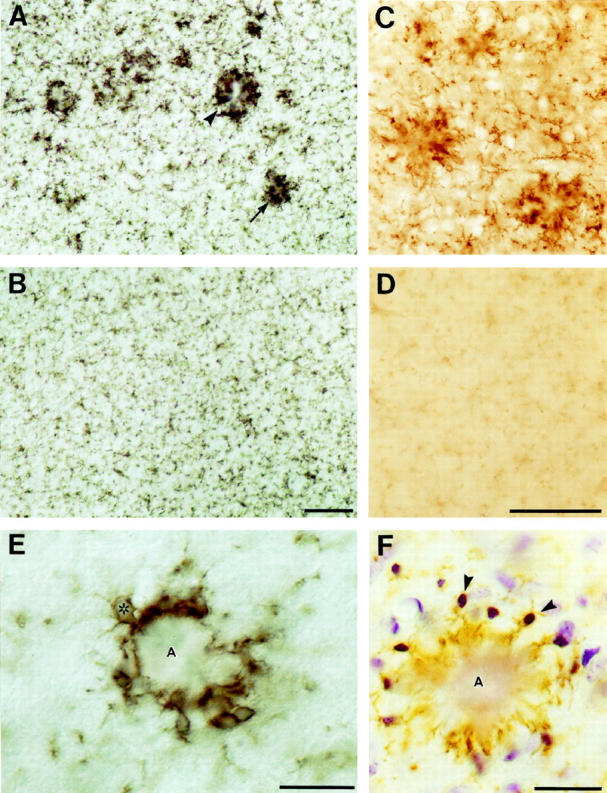
Activated microglia are associated with dense core amyloid plaques in neocortex of APP23 transgenic mice. A: Clusters of microglia immunostained with antibodies to Mac-1 in the frontal cortex (layer II/III) of a 16-month-old transgenic mouse. An unstained amyloid core surrounded by activated microglia (arrowhead) was detectable when the center of a plaque was sectioned while an aggregation of microglia (arrow) was observed when the periphery was sectioned. B: No such microglia aggregates were present in control mice. C and D : Similarly, antibodies to phosphotyrosine revealed labeling of clusters of microglia around amyloid plaques in APP transgenic (C) but not in control (D) mice. E: High-power light microscopy shows cell surface-staining of Mac-1-positive microglia cell bodies (one cell body is indicated by an asterisk) in close vicinity of the amyloid core (A). Microglia processes appear to cover nearly the entire surface of this dense core plaque. F: Mac-1-positive microglia (brown reaction product) counterstained with cresyl-violet reveals the numerous microglial nuclei at the periphery of an amyloid plaque (two are indicated by arrowheads). From the amyloid-facing pole of these microglia long and hypertrophic processes penetrate into the star-shaped amyloid core. Calibration bars are 100 μm (B, D) and 25 μm (E, F).
Microglial cell bodies were located in the close vicinity of the amyloid (Figure 1, E and F) ▶ . Depending on the size of the plaques, between 3 and ∼20 microglial cell bodies were observed at the periphery of amyloid plaques. Mac-1-positive microglial cells, with their processes penetrating deeply into the amyloid, appeared to cover nearly the entire surface of a plaque (Figure 1, E and F) ▶ . These amyloid-associated microglial cells revealed a polar morphology with thin processes away from the plaque and hypertrophic processes directed toward the plaque (Figure 1, E and F) ▶ . Mac-1-positive cell bodies were not observed within the center of amyloid plaques. Occasionally, a microglial cell body had the appearance of being trapped between a main plaque and satellite plaques, giving the impression that the microglia resides in the center of a large plaque.
Although the amyloid core in the center of such microglial clusters could easily be recognized in unstained sections using Nomarski optics, double labeling with Congo Red and Aβ antibodies was performed. Results revealed that virtually all congophilic amyloid plaques in 14- to 18-month-old APP23 mice were associated with clusters of activated microglia (Figure 2A) ▶ . When adjacent serial sections were immunolabeled alternately for Aβ and Mac-1 it was noted again that the vast majority (>90%) of compact amyloid deposits were surrounded by hypertrophic microglia (Figure 2, B and C) ▶ . Diffuse amyloid was observed in the striatum, the dentate gyrus molecular layer, and the cingulate and entorhinal cortices but overall accounted for less than 10% of the total amyloid burden in 14- to 18-month-old APP23 mice. 16 Comparison of adjacent sections that immunoreacted either with Aβ or Mac-1 revealed that diffuse amyloid was not associated with a detectable microglia activation (Figure 2, D–G) ▶ .
Figure 2.
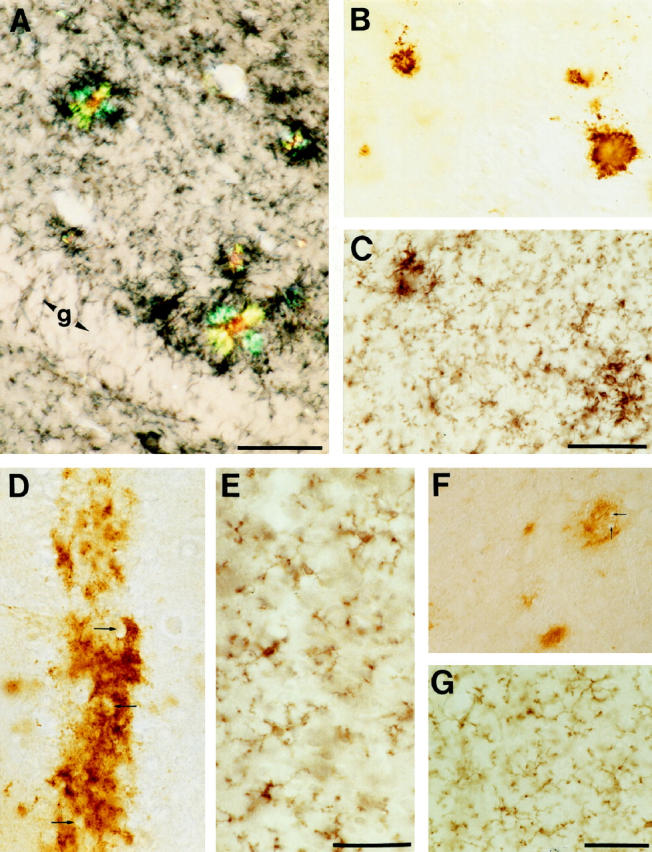
Activated microglia are associated with dense core and congophilic amyloid plaques but not with diffuse amyloid deposits. A: Double labeling of Mac-1 immunostaining (blue-black reaction product) and Congo red histochemistry revealed that virtually all birefringent plaques are surrounded by activated microglia. Shown is the molecular layer of the hippocampus of a 14 month-old APP23 transgenic mouse. g, granular cell layer. B and C: Similarly, serial sections alternately immunostained for Aβ and Mac-1 revealed that the vast majority of compact plaques were surrounded by intensely stained and hypertrophic Mac-1 positive microglia. D, E, F, and G: Occasionally, NT-11 positive diffuse amyloid was seen as a band in the neuron-rich layer II of the cingulate cortex (D) and in areas such as the striatum (F). Note that the diffuse amyloid spares cell bodies (arrows). No corresponding microglia reaction was observed in adjacent sections immunoreacted with Mac-1 (E and G). All calibration bars are 50 μm.
Microglia and Amyloid in Semithin and Ultrathin Sections
To study more closely the interaction between microglia and amyloid fibrils, semithin and ultrathin sections were cut from unstained and Mac-1-immunostained plastic embedded sections. In both semithin and ultrathin sections, specificity of Mac-1 staining to the surface of microglial cell bodies and processes was confirmed (Figures 3 and 4) ▶ ▶ .
Figure 3.
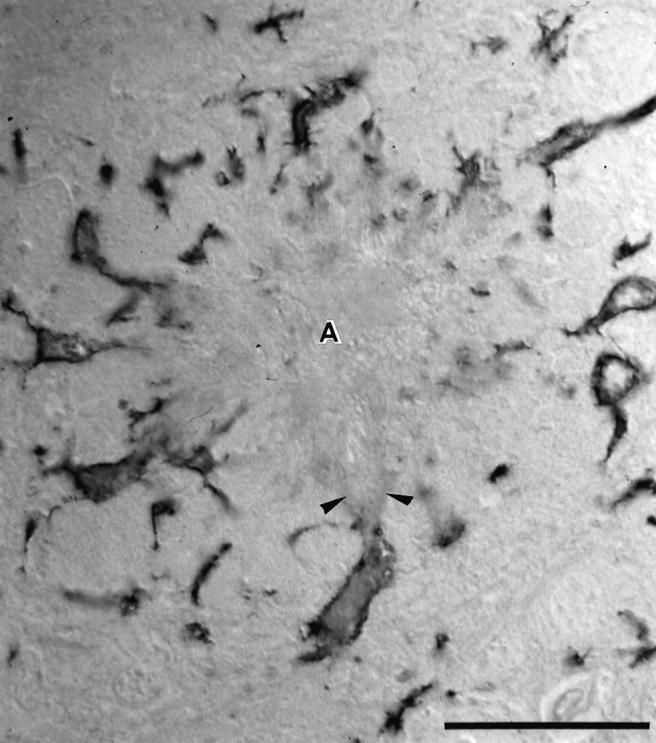
Morphology of an amyloid plaque surrounded by Mac-1-positive microglial cells in a semithin plastic section using Nomarski optics. The microglia cell bodies are located at the periphery of the amyloid plaque. At their amyloid-facing pole, Mac-1-positive membrane surfaces appear interrupted: here, amyloid has the appearance of streaming out (arrowheads) and forming the fingers of the amyloid star (A). Calibration bar is 50 μm.
Figure 4.
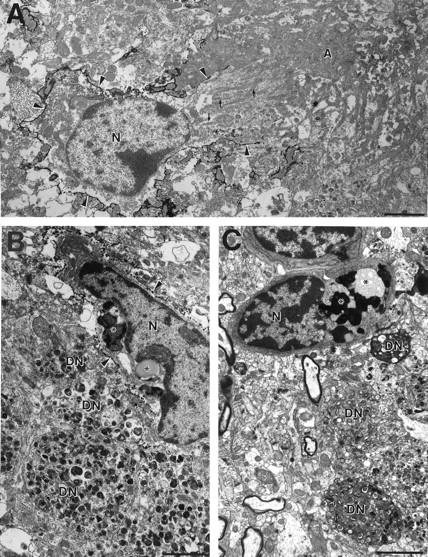
Electron micrograph of microglial cells in closest vicinity of amyloid fibrils and dystrophic neurites in 18-month-old APP23 transgenic mice. A: Mac-1 immunostaining revealed labeling of the microglial cell surface (arrowheads). At the amyloid-facing pole the cell membrane appears to be open and parallel running bundles of amyloid fibrils (small arrows) are observed in intimate relationship to the microglia nucleus (N). The nucleus itself is of regular shape. Toward the core of the amyloid plaque (A) amyloid bundles become more dense and less oriented. B and C: Microglia at the periphery of an amyloid plaque often show the typical characteristics of phagocytosis such as vacuoles, lipofuscin granules, and other inclusion bodies (asterisks) and are closely associated with dystrophic neurites (DN). Note the typical multilaminar and membranous electrodense bodies of the dystrophic neurites. B shows an immunoelectron micrograph using Mac-1 antibody, whereas C represents a normal (unstained) electron micrograph. Calibration bars, 2 μm.
In semithin sections, the star-shaped morphology of most amyloid plaques was clearly evident using Nomarski optics (Figure 3) ▶ . Microglia cell bodies were typically located at the periphery of the plaques, where the amyloid fibrils had the appearance of “streaming out” and forming amyloid stars. At least one Mac-1-positive hypertrophic process was always in direct contact with an amyloid finger. At the interfaces between amyloid and microglia, the Mac-1-positive microglial surface membrane appeared to be discontinuous (Figure 3) ▶ .
Electron and immunoelectron micrographs confirmed the observations obtained from semithin Mac-1-immunostained sections (Figures 4, 5, and 6) ▶ ▶ ▶ . Microglia were observed at the periphery of amyloid plaques, often among a group of dystrophic neurites and often in contact with amyloid fibrils. Similar to the observations at light microscopic levels, Mac-1-positive cell surface membranes appeared discontinuous in the region of communicating fibrils between microglial cells and amyloid deposits and parallel arrays of amyloid bundles were observed close to both microglial organelles and nucleus (Figure 4A) ▶ .
Figure 5.
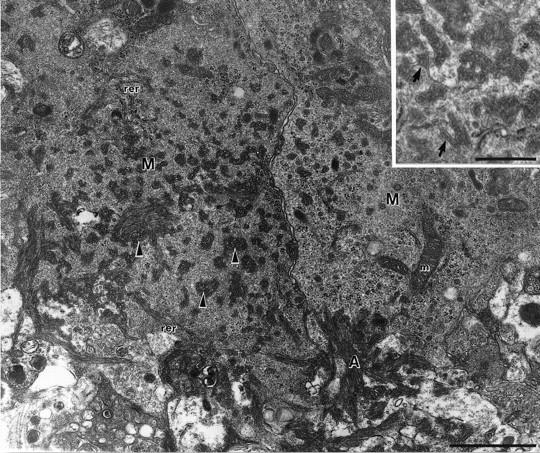
Electron micrograph of the interface between extracellular amyloid fibrils (A) and the cytoplasm of two microglial cells (M) in a 15-month-old APP23 transgenic mouse. Since a high concentration of glutaraldehyde was used in the fixative, the ultrastructure is better conserved compared to the immunoelectron micrographs shown in Figure 4, A and B ▶ . Within the microglia cytoplasm, pockets filled with parallel oriented amyloid fibrils (arrowheads) are observed. Cellular organelles such as rough endoplasmic reticulum (rer) and mitochondria (m) are in close vicinity of the amyloid fibrils. Insert: Amyloid bundles in the microglia cytoplasm surrounded by an incomplete membrane (arrows). Calibration bars, 1 μm and 0.5 μm (insert).
Figure 6.
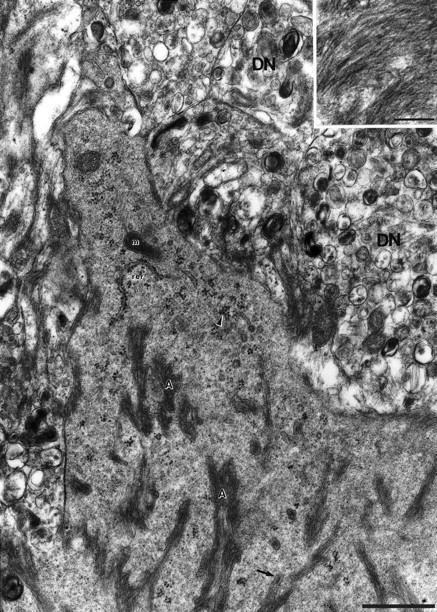
High-power electron micrograph of microglia cytoplasm with amyloid bundles (A) in a 15-month-old APP23 transgenic mouse. The cytoplasm reveals a dense-granular appearance which is rich in free polyribosomes (arrowhead) and rough endoplasmic reticulum (rer). Mitochondria (m) are also present. Occasionally a membrane surrounding the amyloid bundles is evident (arrow). Insert: Oriented amyloid fibrils in high magnification. DN, dystrophic neurite. Calibration bars, 1 μm and 0.2 μm (insert).
Only fixation employing 0.1% glutaraldehyde revealed reliable Mac-1 staining at the ultrastructural level. Tissue treated with higher concentrations (1–2.5%) revealed an improved ultrastructure, particularly of the microglia cytoplasm. In such tissue parallel bundles of amyloid fibrils were observed within the microglial cytoplasm (Figure 5) ▶ . Sometimes, a membrane surrounding these bundles was detectable. The microglia cytoplasm had a characteristic dense granular appearance with prominent endoplasmic reticulum and numerous free polyribosomes (Figures 5 and 6) ▶ ▶ .
At the amyloid-microglia interface lysosomes, vacuoles, lipofuscin, and other inclusion bodies indicative of microglial phagocytosis were absent 22 (Figures 5 and 6) ▶ ▶ . Microglia cells with the typical characteristics of phagocytosis were observed but these microglia were not usually the ones in contact with the amyloid fibrils (Figures 4A, 5, and 6) ▶ ▶ ▶ . Rather, microglia with the typical features of phagocytosis were the ones preferentially associated with dystrophic boutons at the plaque periphery (Figure 4, B and C) ▶ . Dystrophic boutons contained the typical membranous dense bodies but no paired helical filaments were observed.
Because tissue prepared for immunoelectron microscopy revealed a suboptimal preservation of the ultrastructure, it was difficult to determine unequivocally whether membranes surrounding amyloid fibrils in the microglia cytoplasm were part of the Mac-1-positive cell surface membrane or part of the subcellular membrane system. Therefore, it is possible that these membrane-bound bundles of amyloid within the microglia cytoplasm may be transversely sectioned deep infoldings of the microglia cell surface that are filled with amyloid (Figures 5 and 6) ▶ ▶ .
Early Amyloid Deposits in Young Mice Associated with Activated Microglia
To investigate the role of microglia in early amyloid deposition we studied the distribution and appearance of microglia in young hemizygous 4- to 9-month-old APP23 mice. The earliest plaques in hemizygous APP23 mice appear at about 6 months of age. 15 A series of serial 15-μm sagittal sections was cut through the brains and alternately immunostained for Mac-1 (microglia) and NT11 (amyloid) (Figure 7) ▶ . Additional sections were stained for Congo red. Results revealed that the early plaques were preferentially deposited in the frontal cortex and subiculum and ranged from tiny focal amyloid aggregates (<5 μm) to medium-sized plaques (15–30 μm). Interestingly, the tiniest and earliest plaques already appeared to have a dense core, were Congo red-positive, and were surrounded by hypertrophic microglial processes stained intensely with Mac-1 (Figure 7) ▶ . This microglial activation around small plaques was confined to only 2 or 3 microglial cells. Our semiquantitative results revealed that in 85% of the amyloid deposits, a corresponding microglial response was found in adjacent sections. In a reverse analysis, in 78% of the observed microglia aggregations an amyloid plaque was detected on adjacent sections. It should also be noted that microglia clusters consisting of 2 to 4 microglia (always without hypertrophic cell bodies and processes) have been observed in nontransgenic littermates.
Figure 7.

Early amyloid microdeposits in young APP23 transgenic mice are of dense core nature and are surrounded by intensely Mac-1-immunostained microglia. Serial sagittal sections in young (4- to 9-month-old) transgenic mice were immunostained alternately for Mac-1 and Aβ, respectively. Three series of 3 serial sections are shown: A-C, parietal cortex layer V/VI; D-F, frontal cortex, layer III; G-I, ventral orbital cortex immunostained for Mac-1 (A, C, D, F, G, I) and Aβ (B, E, H). The amyloid deposits in each series are associated with a notable clustering of intensely stained Mac-1 immunoreactive microglia in either the preceding or the following section of the series. Note that the smallest amyloid plaques with a diameter of <5 μm were already dense-core in nature (small arrowheads in H) and associated with a few activated microglia. Abbreviations: cc, corpus callosum; ac, anterior commissure. Calibration bar, 100 μm (I). All prints have the same magnification.
Because the smallest detectable amyloid deposits had a diameter of <5 μm and our mean postprocessing section thickness was 9.5 μm (as measured with a focus drive attached to the microscope), it is possible that the amyloid deposit and the corresponding microglia reaction were confined to the same section and evaded our quantification. Thus, it is likely that the percentage of plaques with a corresponding microglia reaction is in fact even higher. We also performed double labeling on the same sections but noted that the amyloid reaction product often masked identification of activated microglia processes closest to the amyloid.
Discussion
Our results in APP23 transgenic mice demonstrate a tight association of activated microglia with congophilic plaques but not with diffuse amyloid deposits. We have used Mac-1 and phosphotyrosine as microglia markers and have shown an up-regulation of these proteins on microglia near amyloid plaques. While microglial processes frequently penetrated into the amyloid plaques, microglial cell bodies were located at the periphery and preferentially at the points of the amyloid star. At the ultrastructural level, bundles of amyloid fibrils, sometimes surrounded by an incomplete membrane, were found within the microglial cytoplasm. Interestingly, these microglia did not show typical characteristics of phagocytosis such as vacuoles, lysosomes, and other inclusion bodies. Phagocytosing microglia, however, were often found in contact with dystrophic neurites at the plaque periphery.
These findings in APP23 mice are very similar to immunohistochemical and ultrastructural observations previously described in AD brain. 4,8,23,24 In AD, activation of microglia has been reported in association with neuritic plaques but not with diffuse amyloid. 5,25 In the present study, we have found diffuse amyloid deposits in noticeable amounts only in aged mice with a high overall plaque load. Diffuse amyloid was always accompanied by significant amounts of congophilic plaques. An exception was the striatum, where we found exclusively diffuse amyloid, paralleling observations in AD brain. 26 These results suggest that, at least in APP23 mice, diffuse amyloid deposits are not a necessary precursor of dense-core congophilic amyloid plaques. Moreover, even the earliest amyloid deposits in young APP23 mice were congophilic, compact, and surrounded by activated microglia.
The tight association of amyloid fibrils and microglia in AD brain has led to the suggestion that microglia are involved in either the phagocytosis or deposition of amyloid. 3-5,7 In culture, there is clear evidence that microglia are capable of taking up Aβ and amyloid fibrils via the receptor for advanced glycation end products (RAGE) and scavenger receptors. 27-29 However, our ultrastructural observations may be interpreted as having found microglia engaged in the formation of amyloid rather than in its phagocytosis. The absence of phagocytotic characteristics of the microglia at the amyloid-microglia interface is different from the subcellular alterations previously identified in amyloid phagocytosing microglia in culture and after stroke. 9,30 Moreover, in AD brain membrane-bound amyloid fibrils have been observed in direct contact with the endoplasmic reticulum of the microglia, thus implicating microglia as amyloid-producing cells. 4,7,8,24
In AD, microglia express βPP 31-33 and thus it is feasible that microglia in AD brain are both βPP-processing cells and amyloid-producing cells. 8 βPP or Aβ might also be taken up by microglia from the environment and subsequently processed into amyloid fibrils. 6,10 APP23 transgenic mice express βPP under the control of a brain- and neuron-specific Thy-1 promoter element and we have confirmed the exclusive neuronal expression of the human βPP protein 15 (Calhoun M, Wiederhold K, Staufenbiel M, Jucker M, unpublished manuscript). If microglia in APP23 mice are involved in amyloid deposition, our results then suggest that microglia take up neuron-derived human βPP or Aβ, which may then be processed into amyloid fibrils. Along this line, the possibility must be considered that in APP23 mice neuron-derived human βPP and/or Aβ activates microglia and may then stimulate the production of endogenous mouse Aβ. 33,34 Therefore, we cannot exclude that some of the amyloid in our transgenic mice is in fact mouse Aβ. However, our recent analysis of amyloid plaque formation in crosses of APP23 mice with APP-null mice (Sturchler-Pierrat C, Duke M, Wiederhold K, Sommer B, unpublished work) suggests that the vast majority of the deposited amyloid is human amyloid. Moreover, the ultrastructure of microglia in APP23 × APP-null mice is identical to the present observations (results not shown).
In APP23 mice, a discontinuous cell surface membrane was observed at the amyloid-facing pole of microglia and the same observation has been reported in AD brain. Such a consolidated microglia-amyloid space may give rise to leakage of cytotoxic and inflammatory microglial proteins into the surrounding neuropil. 10 However, an open microglia membrane clearly suggests amyloid-associated microglia cell death. In APP23 mice we have not observed convincing evidence for microglia cell death around amyloid plaques, although recent observations in AD brain suggest that amyloid-associated microglial cell death may indeed occur. 35
We have reported a significant correlation between neuron loss and amyloid plaque load in the CA1 region of hippocampus 16 in the exact same APP23 mice used in the present study. More than 90% of the amyloid deposits were congophilic in nature and here we report that these plaques are surrounded by activated microglia. Because plaque load, neuron loss, and activated microglia all seem to be related, these observations suggest that amyloid-associated microglia activation may play a role in CA1 neuron loss. Activated microglia release various cytokines and neurotoxins including reactive oxygen species 25,36-38 and persistent activation of microglia may give rise to chronic inflammation and neurodegeneration. 11,12
In PrP-APP transgenic mice, another mouse model with amyloid plaque formation, 39 a microglia activation very similar to the one observed in APP23 mice has been described at the light microscopic level. 40 With a plaque load significantly less than the one in APP23 mice, a nonsignificant neuron loss was reported in PrP-APP mice. 41 In contrast, in a third mouse model with amyloid plaque formation, PD-APP mice, 42 significant amounts of diffuse amyloid, no neuronal loss, and only occasional microglial clustering around amyloid plaques were reported. 43,44 The observations from the APP23, PrP-APP, and PD-APP transgenic mouse models suggest that amyloid-associated activated microglia may be important for the neurodegeneration in these mice and perhaps in AD.
In summary, the present results together with previous observations demonstrate that overexpression of human mutated βPP in neurons is sufficient to induce amyloid plaque formation, region-specific neurodegeneration, 15,16,45 and amyloid-associated microglia activation. Although the present observations cannot unequivocally determine whether microglia are causal, contributory, or consequential to cerebral amyloidosis, our results are most consistent with the idea that microglia are intimately involved in cerebral amyloidosis, by processing uptaken βPP or Aβ into amyloid fibrils and/or by ingesting neuron-derived βPP or amyloid fibrils by an uncommon phagocytotic mechanism. In any case, activation of microglia in APP23 mice seems to be important for the pathogenesis of AD-like pathology.
Acknowledgments
We thank M. Calhoun, M. Tolnay, D. Kurth, C. Mistl, C. Bauer, and H. Zysset (Institute for Pathology, University of Basel), D. Abramowski, C. Stürchler-Pierrat, and K. H. Wiederhold (Novartis Pharma), M. Graeber (Max Planck Institute for Neurobiology, Martinsried, Germany), and L. Walker (Parke-Davis, Ann Arbor, MI) for advice, assistance, and helpful comments on this manuscript.
Footnotes
Address reprint requests to Mathias Jucker, Ph.D., Institute of Pathology, University of Basel, Schönbeinstrasse 40, CH-4003 Basel, Switzerland. E-mail: mjucker@uhbs.ch.
Supported by grants 3130–44526.95 and 3100–46612.96 from the Swiss National Foundation.
References
- 1.Hardy J: Amyloid, the presenilins, and Alzheimer’s disease. Trends Neurosci 1997, 20:154-159 [DOI] [PubMed] [Google Scholar]
- 2.Selkoe DJ: Alzheimer’s disease: genotypes, phenotypes, and treatments. Science 1997, 275:630-631 [DOI] [PubMed] [Google Scholar]
- 3.Itagaki S, McGeer PL, Akiyama H, Zhu S, Selkoe D: Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J Neuroimmunol 1989, 24:173-182 [DOI] [PubMed] [Google Scholar]
- 4.Wegiel J, Wisniewski HM: The complex of microglial cells and amyloid star in three-dimensional reconstruction. Acta Neuropathol (Berl) 1990, 81:116-124 [DOI] [PubMed] [Google Scholar]
- 5.Mackenzie IR, Hao C, Munoz DG: Role of microglia in senile plaque formation. Neurobiol Aging 1995, 16:797-804 [DOI] [PubMed] [Google Scholar]
- 6.Cras P, Kawai M, Siedlak S, Mulvihill P, Gambetti P, Lowery D, Gonzalez-DeWhitt P, Greenberg B, Perry G: Neuronal and microglial involvement in β-amyloid protein deposition in Alzheimer’s disease. Am J Pathol 1990, 137:241-246 [PMC free article] [PubMed] [Google Scholar]
- 7.Perlmutter LS, Barron E, Chui HC: Morphologic association between microglia and senile plaque amyloid in Alzheimer’s disease. Neurosci Lett 1990, 119:32-36 [DOI] [PubMed] [Google Scholar]
- 8.Wisniewski HM, Vorbrodt AW, Wegiel J, Morys J, Lossinsky AS: Ultrastructure of the cells forming amyloid fibers in Alzheimer disease and scrapie. Am J Med Genet Suppl 1990, 7:287-297 [DOI] [PubMed] [Google Scholar]
- 9.Frackowiak J, Wisniewski HM, Wegiel J, Merz GS, Iqbal K, Wang KC: Ultrastructure of the microglia that phagocytose amyloid and the microglia that produce β-amyloid fibrils. Acta Neuropathol (Berl) 1992, 84:225-233 [DOI] [PubMed] [Google Scholar]
- 10.McGeer PL, Akiyama H, Kawamata T, Yamada T, Walker DG, Ishii T: Immunohistochemical localization of β-amyloid precursor protein sequences in Alzheimer and normal brain tissue by light and electron microscopy. J Neurosci Res 1992, 31:428-442 [DOI] [PubMed] [Google Scholar]
- 11.McGeer PL, McGeer EG: The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res Brain Res Rev 1995, 21:195-218 [DOI] [PubMed] [Google Scholar]
- 12.Rogers J, Webster S, Lue LF, Brachova L, Civin WH, Emmerling M, Shivers B, Walker D, McGeer P: Inflammation and Alzheimer’s disease pathogenesis. Neurobiol Aging 1996, 17:681-686 [DOI] [PubMed] [Google Scholar]
- 13.McGeer PL, Schulzer M, McGeer EG: Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer’s disease: a review of 17 epidemiologic studies. Neurology 1996, 47:425-432 [DOI] [PubMed] [Google Scholar]
- 14.Stewart WF, Kawas C, Corrada M, Metter EJ: Risk of Alzheimer’s disease and duration of NSAID use. Neurology 1997, 48:626-632 [DOI] [PubMed] [Google Scholar]
- 15.Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Burki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B: Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci USA 1997, 94:13287-13292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Calhoun M, Wiederhold K, Abramowski D, Phinney A, Probst A, Sturchler-Pierrat C, Staufenbiel M, Sommer B, Jucker M: Neuron loss in APP transgenic mice. Nature 1998, 395:755-756 [DOI] [PubMed] [Google Scholar]
- 17.Jucker M, Walker LC, Schwarb P, Hengemihle J, Kuo H, Snow AD, Bamert F, Ingram DK: Age-related deposition of glia-associated fibrillar material in brains of C57BL/6 mice. Neuroscience 1994, 60:875-889 [DOI] [PubMed] [Google Scholar]
- 18.Akiyama H, McGeer PL: Brain microglia constitutively express β-2 integrins. J Neuroimmunol 1990, 30:81-93 [DOI] [PubMed] [Google Scholar]
- 19.Long J, Kalehua A, Muth N, Hengemihle J, Calhoun M, Jucker M, Ingram D, Mouton P: Total number of astrocytes and microglia in hippocampus of C57BL/6 mice at different ages. Neurobiol Aging 1998, 19:497-503 [DOI] [PubMed] [Google Scholar]
- 20.Tillotson ML, Wood JG: Phosphotyrosine antibodies specifically label ameboid microglia in vitro and ramified microglia in vivo. Glia 1989, 2:412-419 [DOI] [PubMed] [Google Scholar]
- 21.Korematsu K, Goto S, Nagahiro S, Ushio Y: Microglial response to transient focal cerebral ischemia: an immunocytochemical study on the rat cerebral cortex using anti- phosphotyrosine antibody. J Cereb Blood Flow Metab 1994, 14:825-830 [DOI] [PubMed] [Google Scholar]
- 22.Peters A, Palay S, Webster H: The Fine Structure of the Nervous System. Third edition. 1991, :p 306 Oxford University Press, New York [Google Scholar]
- 23.Terry R, Gonatas N, Weiss M: Ultrastructural studies in Alzheimer’s preseniline dementia. Am J Pathol 1964, 44:269-297 [PMC free article] [PubMed] [Google Scholar]
- 24.Roher A, Gray EG, Paula-Barbosa M: Alzheimer’s disease: coated vesicles, coated pits and the amyloid- related cell. Proc R Soc Lond B Biol Sci 1988, 232:367-373 [DOI] [PubMed] [Google Scholar]
- 25.Giulian D, Haverkamp LJ, Yu JH, Karshin W, Tom D, Li J, Kirkpatrick J, Kuo LM, Roher AE: Specific domains of β-amyloid from Alzheimer plaque elicit neuron killing in human microglia. J Neurosci 1996, 16:6021-6037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gearing M, Wilson RW, Unger ER, Shelton ER, Chan HW, Masters CL, Beyreuther K, Mirra SS: Amyloid precursor protein (APP) in the striatum in Alzheimer’s disease: an immunohistochemical study. J Neuropathol Exp Neurol 1993, 52:22-30 [DOI] [PubMed] [Google Scholar]
- 27.DeWitt DA, Perry G, Cohen M, Doller C, Silver J: Astrocytes regulate microglial phagocytosis of senile plaque cores of Alzheimer’s disease. Exp Neurol 1998, 149:329-340 [DOI] [PubMed] [Google Scholar]
- 28.Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM: RAGE and amyloid-β peptide neurotoxicity in Alzheimer’s disease. Nature 1996, 382:685-691 [DOI] [PubMed] [Google Scholar]
- 29.Paresce DM, Ghosh RN, Maxfield FR: Microglial cells internalize aggregates of the Alzheimer’s disease amyloid β-protein via a scavenger receptor. Neuron 1996, 17:553-565 [DOI] [PubMed] [Google Scholar]
- 30.Wisniewski HM, Barcikowska M, Kida E: Phagocytosis of β/A4 amyloid fibrils of the neuritic neocortical plaques. Acta Neuropathol (Berl) 1991, 81:588-590 [DOI] [PubMed] [Google Scholar]
- 31.Banati RB, Gehrmann J, Czech C, Monning U, Jones LL, Konig G, Beyreuther K, Kreutzberg GW: Early and rapid de novo synthesis of Alzheimer β A4-amyloid precursor protein (APP) in activated microglia. Glia 1993, 9:199-210 [DOI] [PubMed] [Google Scholar]
- 32.Haass C, Hung AY, Selkoe DJ: Processing of β-amyloid precursor protein in microglia and astrocytes favors an internal localization over constitutive secretion. J Neurosci 1991, 11:3783-3793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bitting L, Naidu A, Cordell B, Murphy GM, Jr.: Beta-amyloid peptide secretion by a microglial cell line is induced by β-amyloid-(25–35) and lipopolysaccharide. J Biol Chem 1996, 271:16084–16089 [DOI] [PubMed]
- 34.Barger SW, Harmon AD: Microglial activation by Alzheimer amyloid precursor protein and modulation by apolipoprotein E. Nature 1997, 388:878-881 [DOI] [PubMed] [Google Scholar]
- 35.Wisniewski H, Wegiel J: Beta-protein fibrillogenesis and neuritic plaques. Calne D eds. Neurodegenerative Diseases. 1994, :pp 82-95 W.B. Saunders Company, Philadelphia [Google Scholar]
- 36.Meda L, Cassatella MA, Szendrei GI, Otvos L, Jr., Baron P, Villalba M, Ferrari D, Rossi F: Activation of microglial cells by β-amyloid protein and interferon- γ. Nature 1995, 374:647–650 [DOI] [PubMed]
- 37.McDonald DR, Brunden KR, Landreth GE: Amyloid fibrils activate tyrosine kinase-dependent signaling and superoxide production in microglia. J Neurosci 1997, 17:2284-2294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.El Khoury J, Hickman SE, Thomas CA, Loike JD, Silverstein SC: Microglia, scavenger receptors, and the pathogenesis of Alzheimer’s disease Neurobiol Aging 1998, 19:S81-84 [DOI] [PubMed] [Google Scholar]
- 39.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G: Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 1996, 274:99-102 [DOI] [PubMed] [Google Scholar]
- 40.Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, Cole GM: Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol 1998, 152:307-317 [PMC free article] [PubMed] [Google Scholar]
- 41.Irizarry MC, McNamara M, Fedorchak K, Hsiao K, Hyman BT: APPSw transgenic mice develop age-related Aβ deposits, and neuropil abnormalities, but no neuronal loss in CA1 J Neuropathol Exp Neurol 1997, 56:965-973 [DOI] [PubMed] [Google Scholar]
- 42.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagopian S, Johnson-Wood K, Khan K, Lee M, Leibowitz P, Lieberburg I, Little S, Masliah E, McConlogue L, Montoya-Zavala M, Mucke L, Paganini L, Penniman E, Power M, Schenk D, Seubert P, Snyder B, Soriano F, Tan H, Vitale J, Wadsworth S, Wolozin B, Zhao J: Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature 1995, 373:523-527 [DOI] [PubMed] [Google Scholar]
- 43.Masliah E, Sisk A, Mallory M, Mucke L, Schenk D, Games D: Comparison of neurodegenerative pathology in transgenic mice overexpressing V717F β-amyloid precursor protein and Alzheimer’s disease. J Neurosci 1996, 16:5795-5811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Irizarry MC, Soriano F, McNamara M, Page KJ, Schenk D, Games D, Hyman BT: Abeta deposition is associated with neuropil changes, but not with overt neuronal loss in the human amyloid precursor protein V717F (PDAPP) transgenic mouse. J Neurosci 1997, 17:7053-7059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Calhoun ME, Phinney AL, Abramowski D, Wiederhold KH, Probst A, Staufenbiel M, Sommer B, Jucker M: Neuron, synaptic bouton, and cholinergic fiber loss is associated with amyloid plaque formation in APP transgenic mice. Neurobiol Aging 1998, 19:S143 (abstr.)