

Abstract
To define a possible role for changes in the regulation of antigen presentation in fulminant hepatic failure (FHF), we studied the expression of co-stimulatory molecules CD80 (B7-1), CD86 (B7-2), and CD40 along with their ligands CD28 and CD154. We analyzed the liver tissue from patients with FHF (n = 18), chronic liver disease (n = 30), and acute hepatitis (n = 3) and from normal controls (n = 9) by immunohistochemistry and examined the temporal relationship between CD80/CD86 and CD40 expression and disease in the mouse models of galactosamine-lipopolysaccharide and galactosamine-tumor-necrosis-factor-induced FHF. In human controls, faint CD80/CD86 immunoreactivity was restricted to Kupffer cells, and CD40 expression was expressed on bile ducts, macrophages, and sinusoidal endothelial cells (SECs). In FHF, immunoreactivity for CD80 and CD86 was observed on significantly higher numbers of cells, including SECs. Increased CD80/CD86 expression corresponded to increased numbers of CD28-positive lymphocytes. The expression of CD40 was also clearly elevated on virtually all cell types in FHF. In both murine models, CD40 and CD80/CD86 expression was up-regulated before tissue damage could be detected. Our data suggest that up-regulated expression of co-stimulatory molecules might lead to an excessive antigen presentation in FHF as an early step in the pathogenesis before the onset of tissue damage.
Hepatitis B virus infection or drug toxicity can initiate a sequence of severe inflammatory reactions that may lead to rapid organ failure and death. Although the precise pathogenesis is poorly understood, enhanced immune activation, including aberrant antigen presentation and T-cell activation, may essentially contribute to hepatocyte destruction in fulminant hepatic failure (FHF). A balance in the interaction between antigen-presenting cells (APCs) and lymphocytes is of special importance to the liver, as the hepatic immune system is confronted with a high number of antigens that reach the liver via the portal tract and that normally do not elicit immune responses.
Recently, it has become evident that a sufficient immune response depends on an efficient T cell activation via co-stimulatory molecules. 1-4 The interaction of CD80 (B7-1) and CD86 (B7-2) with their counter-receptors on T lymphocytes (CD28 and CD152) provides a particularly potent co-stimulatory signal, which amplifies the response of T cells. Although antigen presentation followed by co-stimulation induces full T-cell activation, such antigen presentation that lacks co-stimulatory signals results in tolerance or anergy for the specific antigen. 5-7 CD80 is a 44- to 60-kd member of the immunoglobulin superfamily with a limited expression on professional APCs such as macrophages, dendritic cells, and activated B cells. CD86 is a 75- to 115-kd cell surface glycoprotein with 25% amino acid homology to CD80. It also has a restricted expression on APCs.
Regulation of co-stimulation may also involve the CD40 molecule. CD40 is a 45- to 48-kd glycoprotein, which interacts with its natural ligand CD154 (gp39) on lymphocytes and which can be expressed on a great variety of different cells. 8-10 A favored hypothesis suggests that recognition of the MHC-antigen complex by T cells leads to the induction of CD154 in these T cells. Subsequently, the simultaneous engagement of the MHC-antigen complex by the T-cell receptor and CD40 by CD154 up-regulates the expression of the co-stimulatory molecules CD80 and CD86 in the APCs. 11-14
Following the hypothesis that the delicate balance between hepatic immune tolerance and immune stimulation could be deranged in FHF, we studied the expression of the co-stimulatory and immunoactivating molecules CD80, CD86, and CD40 as well as their ligands in patients with FHF. Furthermore, the time cascade of CD80/CD86 and CD40 expression was examined after inducing of experimental FHF in BALB/c mice by exposure to either galactosamine and lipopolysaccharide (GalN/LPS) or galactosamine and tumor necrosis factor (GalN/TNF).
Materials and Methods
Patients
Explant tissue from 18 patients with FHF was included in the study. All patients fulfilled the King’s College criteria 15 for urgent transplantation. The etiology of hepatic failure was hepatitis B (n = 10), cryptogenic (n = 5), 5-methoxy-3,4-methylene-dioxymethamphetamine (MDMA) toxicity (n = 1), and autoimmune hepatitis (n = 2). The liver tissue from 30 patients with chronic liver disease (CLD); —nine cases of chronic persistent hepatitis B, five of chronic active hepatitis B, six of chronic hepatitis C, five of primary biliary cirrhosis, three of primary sclerosing cholangitis, and two of autoimmune chronic active hepatitis) as well as from two patients with fulminant Wilson’s disease and from three patients with acute hepatitis (HAV, n = 2; HCV, n = 1)—were included as disease controls. Non-tumor-bearing liver tissue obtained during the resection of hepatic malignancies in four patients and wedge biopsies from five donor livers at the time of hepatectomy served as normal controls (NCs).
Immunostaining Procedures
Liver tissue was immediately embedded in Tissue Tek OCT compound (Miles Laboratories, Naperwille, IL) and snap-frozen in liquid nitrogen. Frozen tissue was kept at −80°C until examined. Sections of 5 to 7 μm were stained by an indirect immunoperoxidase technique. In brief, endogenous peroxidase activity was blocked by 0.03% H2O2/NaNO3 (peroxidase blocking reagent, Dako, Carpinteria, CA). The sections were incubated with mouse monoclonal primary antibodies in phosphate-buffered saline (PBS)/1% fetal calf serum (FCS) in a moist chamber at room temperature for 90 minutes. After washing in PBS, peroxidase-coupled goat anti-mouse antibody (Dianova, Hamburg, Germany) was applied for 30 minutes. Any bound antibody was detected with 3-amino-9-ethylcarbazole (Sigma Chemicals, Munich, Germany). All sections were then counterstained with Meyer’s hematoxylin.
The results of the indirect immunoperoxidase technique were confirmed by a double-step immunoenzymatic labeling using immune complexes of alkaline phosphatase and monoclonal anti-phosphatase (APAAP). After incubating sections with the primary antibody, rabbit anti-mouse antibodies were applied to the slides for 25 minutes (Dako). Thereafter, APAAP complexes were detected with naphthol-AS biphosphate/neufuchsin (Sigma Chemicals). 16
Monoclonal Antibodies
To ensure the specificity of monoclonal antibodies (MAbs) we stained two different antibodies for the detection of CD40, CD86, and CD28 expression. Both 5C3 (Dianova) and anti-CD40 (Genzyme, Cambridge, MA) react with the 45- to 48-kd type 1 integral membrane glycoprotein CD40 found on peripheral blood and tonsillar B cells. Both Fun-1 (Dianova) and IT2.2 (Dianova) recognize different epitopes on the 75-kd cell surface protein CD86 (B7-2) expressed primarily on monocytes and activated B cells. Both Leu 28 (Becton Dickinson, Heidelberg, Germany) and anti-CD28 (Pharmingen, San Diego, CA) detect the disulfide-linked 44-kd CD28 antigen. CD8+ lymphocytes were stained with Leu 2a (Becton Dickinson). MAb BB1 (CD80/B7-1) (Becton Dickinson) reacts with a 44- to 60-kd protein on B7-transfected mouse cells identical to CD80. MAb TRAP1 (Pharmingen) reacts with CD154 (CD40 ligand, gp39), a 39-kd type II membrane glycoprotein expressed on activated T cells.
We performed immunohistochemistry on mice with rat anti-mouse CD80 (1G10, Pharmingen, San Diego, CA), rat anti-mouse CD86 (GL1, Pharmingen), rat anti-mouse CD40 (3/23, Pharmingen), and rat anti-mouse CD8 (53-6.7, Pharmingen).
As isotype controls, irrelevant mouse monoclonal antibodies (IgG1 and IgG2; Becton Dickinson) were used in human and rat MAbs (Pharmingen) were used in the mouse models.
Serial Sections and Double Stainings
To identify different parenchymal and nonparenchymal cell types, we carried out serial liver slides and double stainings. We used anti-cytokeratin (KL-1; Immunotech, Hamburg, Germany; positive reaction to hepatocytes and bile ducts), anti-CD31 (JC/70A; Dako; positive reaction to endothelial cells) and anti-liver sinusoidal endothelium (HM15/3; Monosan, Uden, The Netherlands; positive reaction to SECs), CD68 (PGM1; Dako; positive reaction to macrophages), and anti-smooth muscle actin (1A4; Dako; positive reaction to Ito cells). We performed double stainings with FITC-conjugated anti-CD31 and anti-CD68 antibodies. After performing the immunoperoxidase reaction, we incubated the sections for 20 minutes with irrelevant IgG1 and IgG2 antibodies to block any possible free binding site of the secondary antibody before the FITC-conjugated antibody was applied for 30 minutes. Analysis of double-labeling experiments was done by bright-field and fluorescent photomicrographs on a Leica DMLB fluorescence microscope with a MPS60 photo camera.
d-Galactosamine-LPS and d-Galactosamine-TNF Mouse Model
Six- to eight-week-old pathogen-free male BALB/c mice were obtained from the Animal Research Institute of the Medizinische Hochschule Hannover. All of the experiments were started between 8 and 10 a.m. and were performed in agreement with the German Legal requirements. Either 10 μg/kg LPS or 10 μg/kg TNF-α were injected intravenously with 700 mg/kg d-galactosamine. The livers of the animals were removed 4, 8, and 12 hours after treatment and frozen in liquid nitrogen for further analysis. To confirm lethality of the chosen GalN/LPS and GalN/TNF doses, one mouse each was treated without further intervention and died between 12 and 24 hours.
Statistical Analysis
CD80-, CD86-, CD28-, CD40-, and CD8-positive cells were counted in at least six randomly selected fields at 400-fold magnification. CD154-positive cells were counted in 100 visual fields at 400-fold magnification. The results for each patient are given as the mean of all evaluated visual fields. The results of each patient group are given as mean ± SD. Using the SPSS PC+ software package, we analyzed the differences between the groups for statistical significance by the nonparametric Mann-Whitney U test. Pearson’s correlation coefficients were also calculated.
Results
Histology
In patients with FHF, several patterns of hepatocyte damage could be discerned. Six patients (1, 2, 3, 7, 9, and 18) had a complete loss of hepatocytes in their specimens, whereas CD31-positive, CD68-negative SECs were preserved. In 11 patients with FHF, 75% to 95% of hepatocytes were destroyed. Single surviving hepatocytes were found in 5 of these 11 patients (4, 8, 12, 14, and 16). In the six other patients (5, 6, 10, 11, 13, and 15), surviving hepatocytes formed islets of intact hepatic structure, which were sharply limited by dense infiltrates of macrophages (Figure 1C) ▶ . No inflammatory cells were seen within these islets. One patient (17) had an intact hepatic structure with dense mononuclear infiltrates. Two patients with fulminant hepatitis from Wilson’s disease showed complete cirrhosis with minimal necrosis and only very few inflammatory infiltrates. These two patients were studied as a separate group.
Figure 1.
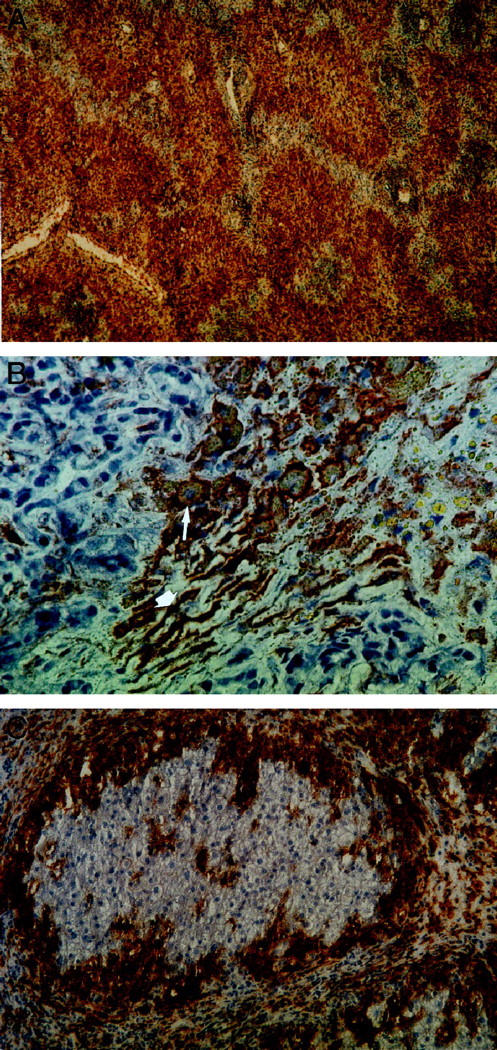
A and B: Hepatic CD86 expression in a patient with fulminant HBV (patient 2, MAb Fun-1). Cell types with typical morphological characteristics of macrophages (B, thin arrows) and of sinusoidal endothelial cells (thick arrows) are positive for CD86. Magnification, ×40 (A) and ×400 (B). C: Hepatic CD86 expression in a patient with toxic FHF (patient 9, MAb Fun-1). Islets of surviving hepatocytes showed absence of infiltrating cells and a normal expression of co-stimulatory molecules, exclusively on Kupffer cells, within the islets. The islets are surrounded by a dense wall of CD86-positive macrophages. In areas of liver necrosis outside the islets, macrophages and SECs, are CD86 positive. An identical pattern could be demonstrated for CD80 expression (not shown). Magnification, ×200.
Hepatic Expression of CD28, CD80, and CD86 in Human Specimens
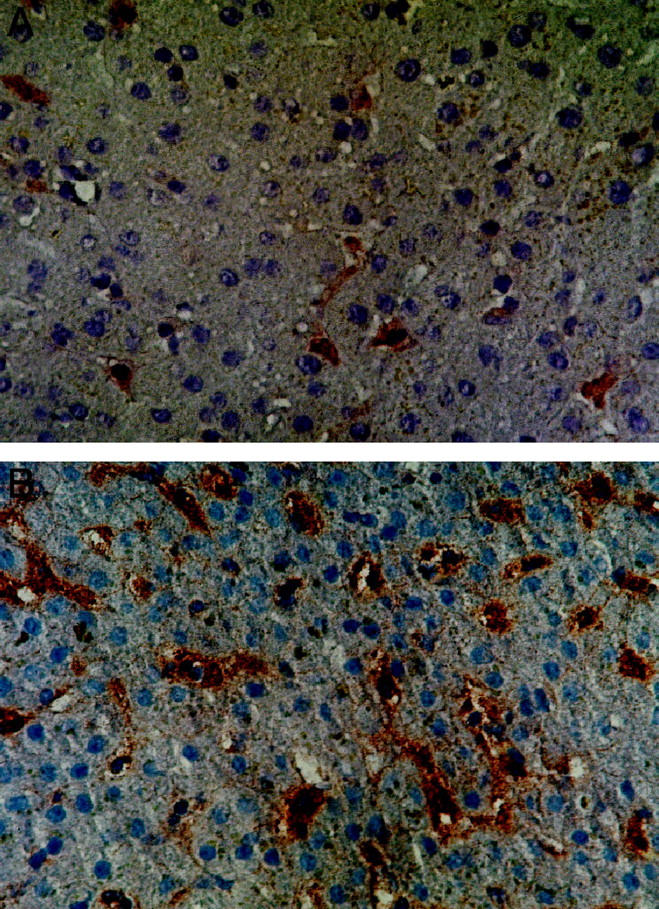
In all examined patients with FHF, a markedly increased number of both CD80- and CD86-positive cells was noted (data are given as number of cells per visual field (VF) at 400-fold magnification: CD80, 108 ± 37 cells/VF; CD86, 112 ± 36 cells/VF) as compared with patients with CLD (CD80, 23 ± 9 cells/VF, P < 0.0001; CD86, 22 ± 9 cells/VF, P < 0.0001) and NCs (CD80, 16 ± 7 cells/VF; CD86, 14 ± 6 cells/VF, P < 0.0001) (Figures 1, A–C and 3, A and B ▶ ▶ ; Table 1 ▶ ). CD80/CD86 expression of patients with fulminant Wilson’s disease, acute hepatitis, or chronic active hepatitis was not different from CLD. In FHF, most of these cells were CD68-positive macrophages, which constituted the predominant cell type in the liver infiltrates of FHF. In contrast, infiltrates in chronic liver disease were dominated by lymphocytes with only few interposed CD80- and CD86-positive macrophages. FHF was unique in that there was strong immunoreactivity of CD80/CD86 that was also found on a part of the SECs, as identified by morphological characteristics and positive reaction to the monoclonal antibody CD31 in serial sections (Figure 2, A–C) ▶ and double-staining experiments (Figure 2, D and E) ▶ . No CD80 or CD86 immunoreactivity could be observed on hepatocytes, bile ducts, or Ito cells (data now shown). In FHF with islets of surviving hepatocytes, SECs within the islets were consistently negative for CD80 and CD86, as were all the SECs in the specimens from CLD and NCs (Figures 1C and 3, A and B ▶ ▶ ).
Figure 3.
CD86 expression in a normal control (A, MAb Fun-1) and in a patient with chronic HBV infection (B). Only macrophages are CD86 positive in both normal controls and chronic liver disease. The number of positive Kupffer cells is higher and staining is stronger in chronic HBV compared with normal controls. Magnification, ×400.
Table 1.
CD80, CD86, CD28, CD40, CD154, and CD8 Staining
| Disease | n | Number of positive stained cells per high-power field | |||||
|---|---|---|---|---|---|---|---|
| CD80 macrophages and SECs | CD86 macrophages and SECs | CD28 lymphocytes | CD40 total number of cells | CD154 lymphocytes | CD8 lymphocytes | ||
| Fulminant hepatic failure | |||||||
| 1 HBV | 1 | 175 | 180 | 66 | 181 | 0.96 | 66 |
| 2 HBV | 1 | 129 | 139 | 46 | 196 | 0.71 | 46 |
| 3 HBV | 1 | 117 | 124 | 50 | 210 | –* | 50 |
| 4 HBV | 1 | 115 | 109 | 62 | 241 | 0.46 | 62 |
| 5 HBV | 1 | 173 | 166 | 64 | 233 | 0.09 | 64 |
| 6 HBV | 1 | 117 | 126 | 65 | 180 | 0.47 | 65 |
| 7 HBV | 1 | 109 | 122 | 55 | 159 | 0.56 | 55 |
| 8 HBV | 1 | 95 | 99 | 36 | 147 | 0.45 | 41 |
| 9 HBV | 1 | 146 | 153 | 26 | 77 | 0.6 | 43 |
| 10 HBV | 1 | 138 | 143 | 52 | 87 | 0.32 | 40 |
| 11 toxic (MDMA) | 1 | 132 | 125 | 38 | 211 | 0.51 | 36 |
| 12 autoimmune hepatitis | 1 | 75 | 77 | 64 | 204 | 0.28 | 77 |
| 13 autoimmune hepatitis | 1 | 52 | 73 | 28 | 100 | 0.05 | 57 |
| 14 cryptogenic | 1 | 103 | 101 | 44 | 125 | 0.15 | 53 |
| 15 cryptogenic | 1 | 85 | 91 | 67 | 278 | 0.21 | 67 |
| 16 cryptogenic | 1 | 85 | 81 | 25 | 68 | 0.46 | 32 |
| 17 cryptogenic | 1 | 29 | 31 | 16 | 20 | 0.06 | 8 |
| 18 cryptogenic | 1 | 82 | 80 | 45 | 93 | 0.2 | 31 |
| Total (mean± SD) | 18 | 108± 37† | 112± 36† | 48± 15† | 156± 68† | 0.39± 0.23† | 57± 12† |
| Fulminant Wilson’s disease | |||||||
| 19 | 1 | 26 | 28 | 13 | 55 | 0.08 | 24 |
| 20 | 1 | 37 | 37 | 26 | 45 | 0.1 | 52 |
| Total (mean± SD) | 2 | 32± 6 | 33± 5 | 20± 7 | 50± 5 | 0.09± 0.01 | 38± 14 |
| Acute hepatitis | |||||||
| 21 (HAV) | 1 | 34 | 39 | 16 | 116 | –* | 27 |
| 22 (HCV) | 1 | 39 | 46 | 15 | 38 | –* | 35 |
| 23 (HAV) | 1 | 15 | 24 | 19 | 95 | –* | 35 |
| Total (mean± SD) | 3 | 29± 10 | 36 ± 9 | 17± 2 | 83± 33 | –* | 32± 4 |
| Chronic liver disease | |||||||
| 24–37 HBV | 14 | 29± 9 | 28± 9 | 11± 8 | 66± 24 | 0.05± 0.05 | 18± 11 |
| 38–43 HCV | 6 | 18± 6 | 18 ± 6 | 12± 6 | 51± 11 | 0.05± 0.07 | 16± 7 |
| 44–53 autoimmune (5 PBC, 3 PSC, 2 AIH) | 10 | 18± 6 | 18± 6 | 10± 10 | 50± 19 | 0.05± 0.05 | 10± 7 |
| Total (mean± SD) | 30 | 23 ± 9 | 22± 9 | 12± 8 | 57± 22 | 0.05± 0.05 | 15± 8 |
| Normal controls | |||||||
| 54–62—total (mean± SD) | 9 | 16± 7 | 14± 6 | 1.4± 1 | 28 ± 17 | 0.003± 0.004 | 1.8± 1.4 |
Results are given as the mean of at least six high-power fields for CD80, CD86, CD28, CD40, and CD8. CD154-positive cells were counted in 100 high-power fields.
*As only needle biopsies were available, only an inadequate number of high-power fields could be evaluated.
†The number of CD80-, CD86-, CD28-, CD40-, CD154-, and CD8-positive cells is significantly enhanced in FHF compared with chronic liver disease and compared with normal controls (P < 0.001; CD154, P < 0.005).
Figure 2.
Assignment of CD86 expression to macrophages and SEC (patient 1). Serial sections were stained for CD86 (A, MAb Fun-1), CD68 (B, MAb JC/70A), and CD31 (C, MAb PGM1). As shown by the (thin arrows), expression of CD86 can be allocated to macrophages (CD86+, CD68+, CD31−) and as shown by the thick arrows also to SECs (CD86+, CD68−, CD31+). Double stainings with CD31-FITC (D) and CD86 (E) demonstrate CD86-positive (thick arrows) and CD86-negative (thin arrows) SECs. Magnification, ×400 (A to C) and ×1000 (D and E).
A close correspondence was found between the numbers of CD80/CD86-positive cells in the liver tissue and the number of infiltrating CD28-positive lymphocytes (r = 0.83, P < 0.001), which was higher in FHF (48 ± 15 cells/VF) than in CLD (12 ± 8 cells/VF, P < 0.0001) or NCs (1.4 ± 1 cells/VF, P < 0.001; see Figure 6 ▶ ). Interestingly, in the islets of surviving hepatocytes, no infiltration of CD28-positive lymphocytes was noted.
Figure 6.

Correlation between the number of CD40-positive cells to CD8- and CD28- as well as CD86- to CD28-positive cells.
Hepatic Expression of CD40, CD154, and CD8
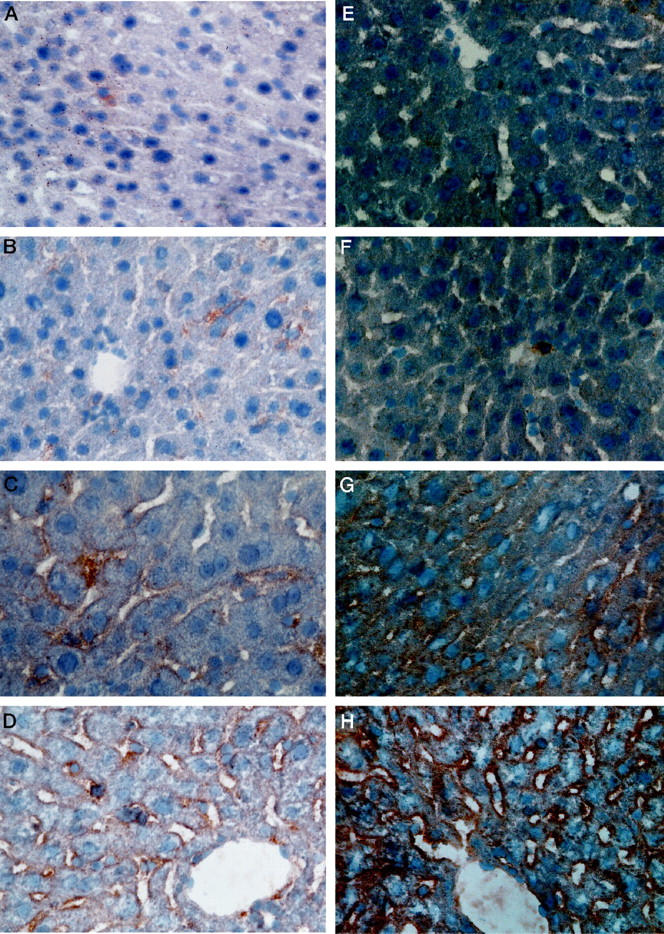
Significantly higher numbers of CD40-positive cells were found in FHF than in control groups (FHF, 156 ± 68 cells/VF; CLD, 50 ± 5 cells/VF, P < 0.001; NC, 28 ± 17 cells/VF, P < 0.001), comprising macrophages, lymphocytes, SECs, Kupffer cells, hepatocytes, and bile duct epithelia. CD40 immunoreactivity was not found on hepatocytes in NCs, whereas 32 ± 38% of hepatocytes were positive in CLD, and 63 ± 25% of hepatocytes were positive in FHF. Immunoreactivity for CD40 was detected on bile ducts and bile duct proliferates in FHF (100 ± 0%), in CLD (89 ± 23%), and in NCs (48 ± 18%). Sinusoidal CD40 immunoreactivity was observed in eight of nine NCs as well as in all specimens from patients with FHF and CLD. Infiltrating macrophages and Kupffer cells were consistently CD40 positive in all groups (Figure 4) ▶ . In contrast to abundant hepatic CD40 immunoreactivity, numbers of CD154-positive lymphocytes were comparatively low (Table 1) ▶ . However, still significantly more CD154-positive lymphocytes were detected in FHF (0.53 ± 0.32 cells/VF) than in CLD (0.05 ± 0.05 cells/VF, P < 0.005) and NCs (0.003 ± 0.004 cells/VF, P < 0.005).
Figure 4.

CD40 expression in a liver specimen of a patient with chronic HBV (MAb 5C3). Hepatocytes, Kupffer cells, SECs, and bile duct epithelia stain strongly positive for CD40 in the patient with chronic HBV. Magnification, ×400.
Overall, the number of lymphocytes with cytotoxic/suppressor (CD8+) phenotype were higher in FHF (57 ± 12 cells/VF) than in CLD (15 ± 8 cells/VF, P < 0.001) or NCs (1.8 ± 1.4 cells/VF, P < 0.001). We found a close correlation between the number of total CD40-positive cells in the liver specimens and the counts of CD8- as well as CD28-positive lymphocytes, respectively (r = 0.84, P < 0.001; r = 0.84, P < 0.001; see Figure 6 ▶ ).
Animal Models
In the animal models, mice were killed 4, 8, and 12 hours after injection of GalN/LPS or GalN/TNF, respectively. Microscopic examination showed preserved liver architecture throughout the whole study period. However, 12 hours after treatment, focal edema and hepatocyte swelling was observed. The numbers of infiltrating macrophages and lymphocytes did not reach the extent seen in human fulminant hepatic failure. With respect to CD80, CD86, and CD40 expression, untreated mice matched the physiological findings in healthy man; a faint CD80, CD86, and CD40 expression was exclusively restricted to some Kupffer cells. SECs, bile ducts, and hepatocytes were negative for CD80, CD86, and CD40. Only a low number of CD8-positive lymphocytes was observed in the control livers (0.4 per visual field).
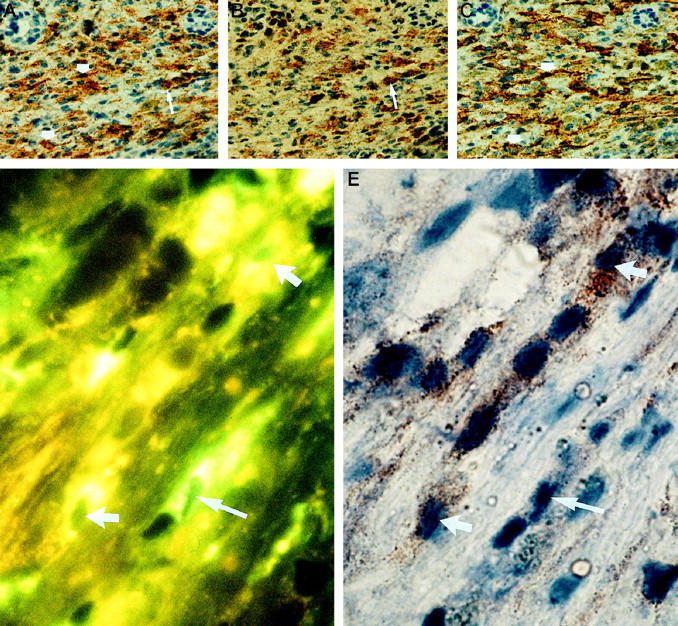
In both murine models, a time-dependent up-regulation of CD80 and CD86 expression was noted (Figure 5, A–D) ▶ . The numbers of CD80- and CD86-positive Kupffer cells increased 4 hours after induction of liver failure in each model. At 8 hours, additional expression of CD80 and CD86 was noted on SECs in focal areas. At 12 hours, the numbers of CD80- and CD86-positive cells had further increased, and focal edema and swelling of hepatocytes became apparent particularly in areas with CD80/86-positive SECs. However, CD86 staining proved stronger than CD80 staining in each cell type throughout the study. In parallel to CD80/CD86 expression, the CD40 immunoreactivity increased on Kupffer cells, and an increase in immunoreactivity also became detectable 8 hours after induction of liver failure (Figure 5, E–H) ▶ .
Figure 5.
CD86 (A to D, MAb GL1) and CD40 (E to H, MAb 3/23) expression in mice without (A and E) and 4 (B and F), 8 (C and G) and 12 (D and H) hours after treatment with GalN/TNF. Sinusoidal CD40 and CD86 expression is induced 8 hours after treatment with GalN/TNF. Similar expression could be observed in the GalN/LPS model (not shown).
The induction of CD80, CD86, and CD40 was accompanied by a rising number of CD8-positive lymphocytes (NCs, 0.4 cells/VF; GalN/LPS at 4 hours, 0.4 cells/VF; 8 hours, 0.7 cells/VF; 12 hours, 1 cell/VF; GalN/TNF at 4 hours, 0.5 cells/VF; 8 hours, 0.7 cells/VF; 12 hours, 1.1 cell/VF).
Discussion
Our data lend strength to the hypothesis that insufficient co-stimulation by liver-specific APCs may be the basis for the physiological down-regulation of the hepatic immune response. We have demonstrated that only few Kupffer cells and no SECs express the co-stimulatory molecules CD80 and CD86 in normal individuals and even in patients with acute or chronic active inflammatory liver disease. Antigens reaching the liver via the portal blood usually do not elicit an immune response. 17,18 This observation may be explained by the specific way antigen is presented in the liver by Kupffer cells and SECs, which can take up, process, and present antigens 19 but are only poor activators of T lymphocytes, 20 probably due to the lack of co-stimulation by SECs.
In FHF, this situation is changed dramatically. Our results demonstrate a marked increase in the numbers of CD80- and CD86-positive cells in FHF not only including macrophages but also SECs. Thus, antigens reaching the liver in FHF are presented not only by a higher number of macrophages but also by SECs. An expression of abundant co-stimulatory molecules may indicate that now both cell types have become competent in efficient co-stimulation. Thus, portal antigens may no longer be tolerated but instead trigger potent immunoactivation that contributes to further hepatocyte damage in FHF. In line with these observations, we found a significant increase in lymphocytes that express the CD80/CD86 counter-receptor CD28. Furthermore, there was a close correlation between the total number of CD80/CD86-positive cells and CD8-positive lymphocytes. Interestingly, within islets of surviving hepatocytes, a normal pattern of CD80 and CD86 expression exclusively on Kupffer cells was linked with a complete absence of CD28-positive and CD8-positive lymphocytes. It is tempting to speculate that these hepatocytes survived because control of immune activation had been maintained locally within the islets.
A collapse of liver tissue in FHF is unlikely to account for the higher numbers of CD80/CD86-positive cells, because their numbers were still increased when corrected for the number of portal tracts per visual field (data not shown).
Nevertheless, our data in human livers cannot exclude the possibility that expression of co-stimulatory molecules may follow extensive hepatocellular necrosis, as serial specimens are not available in FHF in man. Thus, we examined the temporal relationship of CD80 and CD86 expression in the GalN/LPS and in the GalN/TNF mouse models of FHF. In both of these well characterized mouse models we could demonstrate up-regulation of CD80 and CD86 expression on Kupffer cells and SECs. This up-regulation occurred before the onset of massive hepatocyte necrosis and hepatic infiltration by inflammatory cells. The results in the animal models therefore indicate that CD80 and CD86 up-regulation is involved as an early step in the pathogenesis of FHF.
The interaction of CD40 with its ligand CD154 is believed to be a key step in an up-regulation of CD80 and CD86 expression in APCs and has a strong impact on B-cell activation, the initiation of antigen-specific T-cell responses, and macrophage development. 8,21,22 In liver tissue from patients with FHF we found markedly enhanced expression of CD40 on macrophages, Kupffer cells, SECs, hepatocytes, and bile ducts as well as an increased frequency of CD154-positive lymphocytes. This observation of enhanced CD40 expression in humans is confirmed in the murine models, where we could demonstrate an early and strong expression of CD40 on macrophages and SECs that accompanies the induction of CD80 and CD86. Up-regulation of CD40 occurred before tissue damage but apparently did not precede CD80/CD86 induction. However, hepatocytes and bile ducts were CD40 negative in mice in contrast to humans where these also stained positive in cases of chronic active liver disease or FHF.
There is circumstantial evidence that expression of CD80/CD86 or CD40 conveys potent immunoactivating properties to the cells that express these molecules (overview in Ref. 7 ). Blocking these pathways with a CTLA-4Ig fusion protein or CD154 antibodies can prevent autoimmune disease, autoantibody production, antigen presentation, T cell activation, T cell-B cell interaction and clonal expansion of effector T-cell populations 19,23-29 as well as rejection in most transplant models. 30-34 Therefore, it is quite likely that also in human FHF, an up-regulation of CD80/CD86 and CD40 will result in a potent activation of immunocompetent cytotoxic effector cells. This concept is further supported by recent experiments by Adachi and co-workers. 35 Blocking the CD80/CD86-CD28 or the CD40-CD154 pathways with neutralizing CD80 and CD86 or CD154 antibodies prevented liver cell damage in their concanavalin A mouse model of fulminant hepatitis.
In summary, our observations provide evidence for abnormally up-regulated antigen presentation by nonparenchymal liver cells as an early step in the immune-mediated pathogenesis of FHF. In particular, expression of co-stimulatory molecules is markedly enhanced and may trigger and maintain the inflammatory processes leading to massive hepatocyte damage. Novel therapeutic strategies targeted at blocking these co-stimulatory molecules might therefore be a promising novel approach to the treatment and prevention of FHF in man.
Figure 7.

Number of CD86- and CD40-expressing cells after treatment with GalN/TNF or GalN/LPS.
Acknowledgments
We thank Prof. Fischer and Dr. Doubrowski, Department of Pathology, University of Bonn, for helpful discussion in analysis of our immunostainings and Dr. Müller and Dr. Wolff, Department of Surgery, University of Bonn, for providing material.
Footnotes
Address reprint requests to Dr. L. Leifeld, Department of Internal Medicine1, Sigmund Freud Strasse 25, D-53105 Bonn, Germany.
Supported in part by a grant from the BONFOR Forschungsförderung.
References
- 1.Linsley PA, Brady W, Grosmaire L, Aruffo A, Damle NK, Ledbetter A: Binding of the B cell activation antigen B7 to CD28 costimulates T cell proliferation and interleukin 2 mRNA accumulation. J Exp Med 1991, 173:721-730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grimmi CD, Freeman GJ, Gribben JG, Sugita K, Freedman AS, Morimoto C, Nadler LM: B-cell surface antigen B7 provides a costimulatory signal that induces T cells to proliferate and secrete interleukin 2. Proc Natl Acad Sci USA 1991, 68:6575-6579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen C, Gault A, Shen L, Nabavi N: Molecular cloning and expression of early T cell costimulatory molecule-1 and its characterization as CD86 molecule. J Immunol 1994, 152:4929-4936 [PubMed] [Google Scholar]
- 4.Lanier LL, O’Fallon S, Somoza C, Phillips JH, Linsley PS, Okumura K, Ito D, et al: CD80 (B7) and CD86 (B70) provide similar costimulatory signals for T cell proliferation, cytokine production, and generation of CTL. J Immunol 1995, 154:97–105 [PubMed]
- 5.Gaspari AA, Jenkins MK, Katz SI: Class II MHC-bearing keratocytes induce antigen-specific unresponsiveness in hapten-specific clones. J Immunol 1988, 141:2216-2220 [PubMed] [Google Scholar]
- 6.Schwartz RH: A cell culture model for T lymphocyte clonal anergy. Science 1990, 248:1349. [DOI] [PubMed] [Google Scholar]
- 7.Guinan EC, Gribben JG, Boussiotis VA, Freeman GJ, Nadler LM: Pivotal role of B7: CD28 pathway in transplantation tolerance and tumor immunity. Blood 1994, 84:3261-3282 [PubMed] [Google Scholar]
- 8.Hollenbaugh D, Mischel-Petty N, Edwards CP, Simon JC, Denfeld RW, Kiener PA, Aruffo A: Expression of functional CD40 by vascular endothelial cells. J Exp Med 1995, 182:33-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caux C, Massacrier C, Vanbervliet B, Dubois B, Kooten CV, Durand I, Banchereau J: Activation of human dendritic cells through CD40 cross-linking. J Exp Med 1994, 180:1263-1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fries KM, Sempowski GD, Gaspary AA, Blieden T, Looney RJ, Phipps RP: CD40 expression by human fibroblasts. Clin Immunol Immunopathol 1995, 77:42-51 [DOI] [PubMed] [Google Scholar]
- 11.Grewal IS, Flavell A: A central role of CD40 ligand in the regulation of CD4+ T-cell response. Immunol Today 1996, 17:410-414 [DOI] [PubMed] [Google Scholar]
- 12.Ranheim EA, Kipps TJ: Activated T cells induce expression of B7/BB1 on normal or leukemic B cells through a CD40-dependent signal. J Exp Med 1993, 177:925-935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kennedy MK, Mohler KM, Shanebeck KD, Baum PR, Picha KS, Otten-Evans CA, Janeway CA, et al: Induction of B cell costimulatory function by recombinant murine CD40 ligand. Eur J Immunol 1994, 24:116-123 [DOI] [PubMed] [Google Scholar]
- 14.Yellin MJ, Sinning J, Covey LR, Sherman W, Lee JJ, Glickman-Nir E, Sippel KC, et al: T lymphocyte T cell-B cell activating molecule/CD40-L molecules induce normal B cells or chronic lymphocytic leukemia B cells to express CD80 (B7/BB1) and enhance their costimulatory activity. J Immunol 1994, 153:666-674 [PubMed] [Google Scholar]
- 15.O’Grady JG, Alexander GJ, Hayllar KM, Williams R: Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989, 97:439-445 [DOI] [PubMed] [Google Scholar]
- 16.Cordell JL, Falini B, Erber WN, Ghosh AK, Abdulaziz Z, MacDonald S, Pulford KA, et al: Immunoenzymatic labeling of monoclonal antibodies using immune complexes of alkaline phosphatase and monoclonal anti-alkaline phosphatase (APAAP complexes). J Histochem Cytochem 1984, 32:219-229 [DOI] [PubMed] [Google Scholar]
- 17.Triger DR, Cynamon MH, Wright R: Studies on hepatic uptake of antigen. I. Comparison of inferior vena cava and portal vein routes of immunization. Immunology 1973, 25:941-950 [PMC free article] [PubMed] [Google Scholar]
- 18.Cantor HM, Dumont AE: Hepatic suppression of sensitization to antigen absorbed into the portal system. Nature 1967, 215:744. [DOI] [PubMed] [Google Scholar]
- 19.Lohse AW, Knolle PA, Bilo K, Uhrig A, Waldmann C, Meyer zum Büschenfelde KH: Antigen-presenting function and B7 expression of murine sinusoidal endothelial cells and Kupffer cells. Gastroenterology 1996, 110:1175-1181 [DOI] [PubMed] [Google Scholar]
- 20.Rubinstein D, Roska AK, Lipsky E: Liver sinusoidal lining cells express class II major histocompatibility antigens but are poor stimulators of fresh allogeneic T lymphocytes. J Immunol 1986, 137:1803-1810 [PubMed] [Google Scholar]
- 21.Yang Y, Wilson M: CD40 ligand dependent T cell activation: requirement of B7-CD28 signaling through CD40. Science 1996, 273:1862-1864 [DOI] [PubMed] [Google Scholar]
- 22.Armitage RJ, Tough TW, Macduff BM, Fanslow WC, Spriggs MK, Ramsdell F, Alderson MR: CD40 ligand is a T cell growth factor. Eur J Immunol 1993, 23:2326-2331 [DOI] [PubMed] [Google Scholar]
- 23.Griggs ND, Agersborg SS, Noelle RJ, Ledbetter JA, Linsley PS, Tung SK: The relative contribution of the CD28 and gp39 costimulatory pathways in the clonal expansion and pathogenic acquisition of self reactive T-cells. J Exp Med 1996, 183:801-810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Milich DR, Linsley PS, Hughes JL, Jones JE: Soluble CTLA-4 can suppress autoantibody production and elicit long term unresponsiveness in a novel transgenic model. J Immunol 1994, 153:429-435 [PubMed] [Google Scholar]
- 25.Nasir A, Ferbel B, Salminen W, Berth RK, Gaspari AA: Exaggerated and persistent cutaneous delayed-type hypersensitivity in transgenic mice whose epidermal keratinocytes constitutively express B7–1 antigen. J Clin Invest 1994, 84:892-898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Costimulator B7–1 confers antigen-presenting-cell function to parenchymal tissue and in conjunction with tumor necrosis factor α leads to autoimmunity in transgenic mice. Proc Natl Acad Sci USA 1994, 91:5138–5142 [DOI] [PMC free article] [PubMed]
- 27.Perrin PJ, Scott D, Quigley L, Albert PS, Feder O, Gray GS, Abe R, June CH, Racke MK: Role of B7: CD28/CTLA-4 in the induction of chronic relapsing experimental allergic encephalomyelitis. J Immunol 1995, 154:1481-1490 [PubMed] [Google Scholar]
- 28.Lenshow DJ, Ho SC, Sattar H, Rhee L, Gray G, Nabavi N, Herold KC, Bluestone JA: Differential effects of anti B7–1 and anti B7–2 monoclonal antibody treatment on the development of diabetes in the nonobese diabetic mouse. J Exp Med 1995, 181:1145-1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gimmi CD, Freeman GJ, Gribben JG, Gray G, Nadler LM: Human T-cell clonal anergy is induced by antigen presentation in the absence of B7 costimulation. Proc Natl Acad Sci USA 1994, 91:5138-5142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kirk AD, Harlan DM, Armstrong NN, Davis TA, Dong Y, Gray GS, Hong X, Thomas D, Fechner JH, Knechtle SJ: CTLA4-Ig and CD40 ligand prevent renal allograft rejection in primates. Proc Natl Acad Sci USA 1997, 94:8789-8794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Onodera K, Chandraker A, Schaub M, Stadlbauer TH, Korom S, Peach R, Linsley P, Sayegh MH, Kupiec-Weglinski JW: CD28–B7 T-cell costimulatory blockade by CTLA4Ig in sensitized rat recipients. J Immunol 1997, 159:1711-1717 [PubMed] [Google Scholar]
- 32.Levisetti MG, Padrid PA, Szot GL, Mittal N, Meehan SM, Wardrip CL, Gray GS, Bruce DS, Thistlewaite JR, Bluestone JA: Immunosuppressive effects of human CTLA4Ig in a non-human primate model of allogeneic pancreatic islet transplantation. J Immunol 1997, 159:5187-5191 [PubMed] [Google Scholar]
- 33.Sun H, Subbotin V, Chen C, Aitouche A, Valdivia LA, Sayegh MH, Linsley PS, Fung JJ, Starzl TE, Rao AS: Prevention of chronic rejection in mouse aortic allografts by combined treatment with CTLA-4Ig and anti-CD40 ligand monoclonal antibody. Transplantation 1997, 64:1838-1843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hale GA, Gottschalk R, Maki T, Monaco AP: Use of CTLA4-Ig in combination with conventional immunosuppressive agents to prolong allograft survival. Transplantation 1997, 64:897-900 [DOI] [PubMed] [Google Scholar]
- 35.Adachi M, Higushi H, Miura S, Ishi H: Blocking of CD40/CD40L and CD28/B7 interaction prevents the concanavalin A-induced liver injury in BALB/c mouse through the inhibition of interleukin 12 production. Hepatology 1998, 324A