

Abstract
To evaluate the roles of nitric oxide (NO) on human immunodeficiency virus (HIV) Tat-induced transactivation of HIV long terminal repeat (HIV-LTR), we examined the effect of NO in the regulation of nuclear factor (NF)-κB, a key transcription factor involved in HIV gene expression and viral replication. In the present study, we demonstrate that HIV Tat activates NF-κB and that this activation can be attenuated by endogenous or exogenous NO. Inhibition of endogenous NO production with the NO synthase (NOS) inhibitor l-NMMA causes a significant increase in Tat-induced NF-κB activity. In addition, NO attenuates signal-initiated degradation of IκBα, an intracellular inhibitor of NF-κB, and blocks the DNA binding activity of the NF-κB p50/p50 homodimer and p50/p65 heterodimer. To determine how NO is induced by HIV Tat, reverse transcription polymerase chain reaction was used to demonstrate the induction of NOS-2 and NOS-3 mRNA by Tat. Although a putative NF-κB binding site was identified in the −74 GGAGAGCCCCC −64 region of the NOS-3 gene promoter, gel mobility shift assays and site-directed mutation analyses suggest that the putative NF-κB site is not of primary importance. Rather, several Sp-1 sites adjoining the putative NF-κB binding site in the promoter region of NOS-3 gene are required for the induction of NOS-3 gene expression by Tat.
Nitric oxide (NO) is a free radical generated from l-arginine by nitric oxide synthases (NOS). In the mammalian species, three NOS enzyme forms exist, including inducible NOS (iNOS, or NOS-2), endothelial NOS (eNOS, or NOS-3), and neuronal NOS (nNOS, or NOS-1). 1,2 Whereas both NOS-1 and NOS-3 are considered to be constitutively expressed and result in physiological low output of NO, NOS-2 induced by environmental stimuli contributes to the pathological high output of NO. The diffusion of NO through lipid membranes without requirements for a special transporter or second messengers enables this free radical to play a versatile role in the regulation of intercellular and intracellular biochemical events.
As a nonspecific defense weapon, NO is considered a major ally of specific immune response against the invasion of microorganisms. Although antigen-specific T-cell-mediated immune response is essential for recovery from most primary viral infections, this response alone is insufficient to combat infection in the absence of early, nonspecific defense mechanisms. 3 It has been observed that inhibition of NO production worsens the course of viral or bacterial infection. 4 Treatment of mouse macrophages with interferon-γ has been shown to increase NO production concomitant with the inhibition of certain viruses, including ectromelia, vaccinia, herpes simplex virus, and vesicular stomatitis virus. 5-7 In addition, the NO-generating compound S-nitroso-N-acetylpenicillamine (SNAP) has been shown to exert a dose-dependent inhibition of encephalomyocarditis virus growth in L-929 cells 8 and Japanese encephalitis virus replication in a neuronal precursor cell line. 9 It has been reported that NO can inhibit late protein synthesis of Epstein-Barr virus and vaccinia virus without affecting early protein expression required for progeny viral DNA replication. 10,11 Therefore, the induction of NOS-2/NOS-3 and the resultant production of NO may be an important antiviral strategy to restrict viral dissemination early in the course of infection before specific T-cell-mediated immune responses are established.
With human immunodeficiency virus (HIV) infection, increased levels of NO are observed in the sera of HIV-infected individuals. 12 In vitro infection of human monocytes and brain astroglia cells with HIV results in a modest but significant increase in NO release. 13,14 However, the role of NO in HIV infection is still not fully understood. In our previous studies, we reported that NO is a potent inhibitor of signal-induced nuclear factor (NF)-κB activation. 15,16 This observation has now been confirmed by a number of studies showing that NO inhibits NF-κB by attenuating the DNA binding activity of NF-κB or by stabilizing IκBα, which blocks the activation of NF-κB. 17-22
NF-κB is a ubiquitous transcription factor that is responsible for the expression of a number of genes that are involved in inflammation, carcinogenesis, and tissue regeneration. 23,24 NF-κB is also involved in gene expression of viruses such as HIV and most members of SIV family. Activation of NF-κB by a regulating protein of HIV, Tat, has been well documented and is considered a pivotal step in HIV gene expression and viral replication. 25-29 In this report, we provide evidence indicating that NO may act as a negative regulator for HIV viral gene expression and replication through attenuation of HIV Tat protein-induced NF-κB activation.
Materials and Methods
Reagents
NO-generating compounds SNAP and sodium nitroprusside (SNP), the NOS inhibitor NG-monomethyl-l-arginine (l-NMMA), and recombinant bacterial expressed 20s proteasome were purchased from Calbiochem (San Diego, CA). All cell culture reagents were from Mediatech (Herndon, VA). Lipopolysaccharide, chloroquine, and N-acetyl-dl-penicillamine (NAP), an inactive analog of SNAP, were obtained from Sigma Chemical Co. (St. Louis, MO). Diethylaminoethylether-dextran was purchased from Pharmacia (Piscataway, NJ). Luciferase assay kit was from Promega (Madison, WI). Protein assay reagents were from Bio-Rad Laboratories (Hercules, CA).
Transient Transfection Assay
Mouse macrophage RAW264.7 cells were grown in Dulbecco’s modified Eagle’s Medium (DMEM) containing 10% fetal bovine serum in six-well tissue culture plates at a cell density of 5 × 10 6 cells/well for 2 days. The cells were transfected with 0 to 5.4 μg/ml pSV vector or pSV-Tat (amino acids 1 to 72) and HIV long terminal repeat (HIV-LTR)-luciferase reporter plasmid using the diethylaminoethylether-dextran method in the presence of 50 μg/ml chloroquine for 2 hours. The cells were then treated with 10% dimethylsulfoxide for 2 minutes and washed three times. The transfected cells were cultured in the complete medium for an additional 12 to 48 hours in the presence or absence of a NOS inhibitor, l-NMMA or NO-generating compounds SNP or SNAP or its inactive analog NAP. Cells were harvested at the end of incubation, washed twice with PBS (pH 7.6), and resuspended in lysis buffer (25 mmol/L Tris/HCl, 2 mmol/L EDTA, 2 mmol/L dithiothreitol (DTT), 10% glycerol, and 1% Triton X-100). Total protein concentration of each extract was quantitated using a Bio-Rad protein assay reagent. Luciferase activity was determined with a liquid scintillation counter (Beckman LS9000) using the luciferase assay kit as suggested by the manufacturer. Luciferase activity was expressed as relative luciferase activity normalized for transfection efficiency on the basis of β-galactosidase expression.
Electrophoretic Mobility Shift Assay (EMSA)
For nuclear protein extraction, cells were harvested and resuspended in hypotonic buffer A (10 mmol/L HEPES, pH 7.6, 10 mmol/L KCl, 0.1 mmol/L EDTA, 1 mmol/L DTT, 0.5 mmol/L phenylmethylsulfonyl fluoride) as previously described. 15,16 Briefly, cells were incubated in buffer A for 10 minutes on ice and then vortexed for 10 seconds. Nuclei were pelleted by centrifugation at 12,000 × g for 20 seconds and were resuspended in buffer C (20 mmol/L HEPES, pH 7.6, 25% glycerol, 0.4 mol/L NaCl, 1 mmol/L EDTA, 1 mmol/L DTT, 0.5 mmol/L phenylmethylsulfonyl fluoride) for 30 minutes on ice. The supernatants containing nuclear proteins were collected after centrifugation at 12,000 × g for 2 minutes and stored at −70°C. A 32P-labeled double-stranded oligonucleotide containing κB or a κB-like sequence was prepared for EMSA as described previously. 15,16 Briefly, single-stranded DNA was synthesized using a Millipore Cyclone Plus automated synthesizer. To prepare double-stranded DNA, the first-strand DNA was annealed with a complementary decameric primer to its 3′ tail in annealing buffer (100 mmol/L NaCl, 20 mmol/L Tris, pH 7.5, 0.1 mmol/L EDTA). The second strand was extended with DNA polymerase Klenow fragment in a reaction mixture containing 100 μCi of [32P]dCTP plus 5 mmol/L dATP, dGTP, and dTTP. For EMSA, 4 μg of nuclear protein was mixed with the labeled double-stranded probe and incubated at room temperature for 30 minutes. The reaction solution was subjected to electrophoresis on a native 5% polyacrylamide gel in 0.25X Tris-boric acid-EDTA buffer for 2 to 3 hours. The DNA binding proteins were visualized by autoradiography. The sequences of the oligonucleotides used for EMSA are as follows: consensus κB, 5′-CAACGGCAGGGGAATTCCCCTCTCCTT-3′; NOS-3 κB, 5′-CCAGCACTGGAGAGCCCCCTCCCATG-3′; consensus Sp-1, 5′-CAACGGCAGGGGGCGGGGCCTCCTCCTT-3′; mutated NOS-3 Sp-1, 5′-CCAGCACTGGAGAGCCCCCTttt ATG.
Griess Reaction
NO produced in cell culture medium was quantified spectrophotometrically as total nitrate/nitrite by Griess reaction as described previously. 16
In Vitro Proteolysis Reaction and Western Blot Assay
For the proteolysis assay, a GST tag in the GST-IκBα fusion protein (Santa Cruz Biotechnology, Santa Cruz, CA) was enzymatically removed by thrombin digestion as described previously. 30 The resulting recombinant IκBα was incubated with various concentrations of recombinant proteasome in 20 μl of reaction buffer (30 mmol/L Tris/HCl, pH 7.4, 30 mmol/L NaCl, 10 mmol/L CaCl2) at 68°C for 30 minutes. The reaction was terminated by adding 8 μl of SDS reducing buffer (3X, 350 mmol/L Tris/HCl, pH 6.8, 15% SDS, 10% glycerol, 3.6 mol/L β-mercaptoethanol, 0.01% bromophenol blue) and boiled for 5 minutes. For intracellular IκBα degradation study, whole-cell proteins were extracted. Both in vitro proteolysis reaction products and whole-cell extracts were subjected to 12% SDS-polyacrylamide gel electrophoresis (SDS-PAGE). Resolved proteins were transferred to a nitrocellulose membrane and incubated with affinity-purified rabbit polyclonal anti-IκBα serum raised against a peptide corresponding to amino acids 297 to 317 (mapping within the carboxyl-terminal domain of human IκBα molecule). After three 10-minute washes with PBS/Tween-20, the membranes were incubated with peroxidase-conjugated anti-rabbit immunoglobulin, and the antigen-antibody complexes were detected using ECL Western blotting detection reagents according to the manufacturer’s instructions.
Reverse Transcriptase Polymerase Chain Reaction (RT-PCR) Analysis
Total RNA was extracted from vector or Tat-transfected RAW264.7 cells using a QIAGEN RNeasy Mini Kit (Chatsworth, CA). One microgram of RNA was used for RT-PCR in the Promega Access RT-PCR system according to the manufacturer’s instructions. The resultant RT-PCR products were electrophoresed through 2% agarose gels and photographed using Polaroid film to measure the mRNAs for NOS-2, NOS-3, and GAPDH. The primers used for RT-PCR had the following sequences (5′ and 3′ primers, respectively): NOS-3, 5′-CTGTGTCCAACATGCTGCTGGAGATTG-3′ (corresponding to the encoding region 1008 to 1034 of the mouse NOS-3 gene) and 5′-TAAAGGTCTTCTTCCTGGTGATGCC-3′ (corresponding to the encoding region 1469 to 1493 of the mouse NOS-3 gene); NOS-2, 5′-GCCTCCCTCTGGAAAGA-3′ (corresponding to the region 1385 to 1401 of the mouse NOS-2 gene) and 5′-TCCATGCAGACAACCTT-3′ (corresponding to the region 1868 to 1884 of the mouse NOS-2 gene); GAPDH, 5′-CTGAACGGGAAGCTCACTGGCATGGCCTTC-3′ (corresponding to the region 710 to 739 of the mouse GAPDH gene) and 5′-CATGAGGTCCACCACCCTGTTGCTGTAGCC-3′ (corresponding to the region 995 to 1024 of the mouse GAPDH gene).
Plasmid Construction
Subcloning of the 116-bp 5′-flanking region of the human NOS-3 gene promoter into the upstream of chloramphenicol acetyl transferase (CAT) reporter gene was accomplished as previously reported. 31 Site-directed mutation of potential transcription factor binding sites was performed using a QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) and confirmed by DNA sequencing. CAT activity from the cells transfected with reporter plasmid was determined by the use of a CAT ELISA kit (Boehringer Mannheim, Indianapolis, IN).
Statistics
Data are expressed as means ± SEM. Statistical significance was determined by the two-tailed Student’s t-test for paired data and considered significant if P values were <0.05.
Results
Effect of Tat on NF-κB Activation and HIV-LTR Transactivation
The mouse macrophage cell line RAW264.7 is a useful model to examine the role of Tat in the transcriptional regulation of HIV-LTR. 32,33 Compared with human T cells or the Jurkat T cell line, RAW264.7 cells were much more effectively and easily transfected with Tat and HIV-LTR luciferase reporter plasmids. Whereas only 10% to 15% transfection efficiency was achieved for Tat in human blood T cells or Jurkat cells, the transfection efficiency was usually more than 35% in RAW264.7 cells (data not shown). Previous reports have suggested that the two randomly arranged κB elements of HIV-LTR are crucial for Tat-mediated transactivation of LTR in human T cells. 34 To determine whether similar results could be obtained with macrophages, we transfected RAW264.7 cells with Tat along with several luciferase reporter constructs. Figure 1 ▶ shows that both the NF-κB reporter construct containing two κB binding sites on the upstream of the luciferase gene and the wild-type HIV-LTR reporter construct produced a modest amount of luciferase activity after transient transfection into macrophages. In the presence of the Tat-expressing plasmid, this activity was increased to 10- and 18-fold, respectively. In contrast, the empty expression vector pSV showed no induction on this reporter gene. A κB-deletion construct of HIV-LTR (LTR-ΔκB) was threefold less active than wild-type HIV-LTR in the response to Tat induction. Figure 1B ▶ shows a dose-response curve demonstrating increasing luciferase activity in response to increasing amounts of the Tat expression plasmid.
Figure 1.
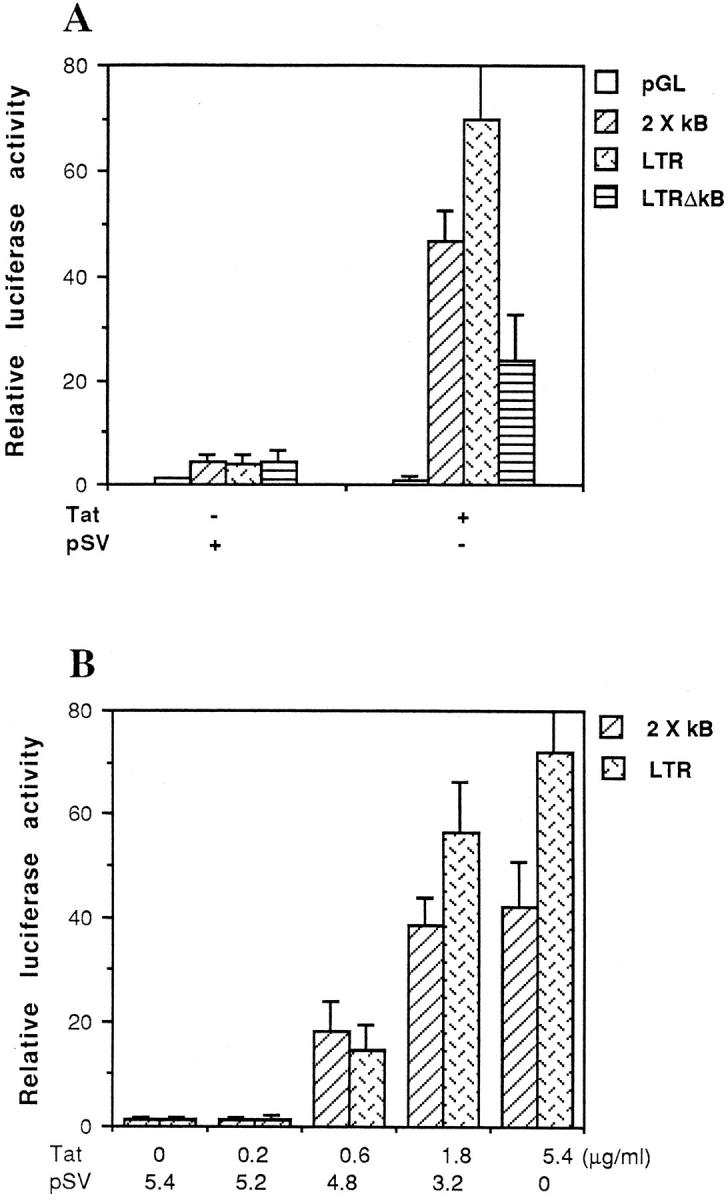
HIV Tat activates NF-κB-dependent gene expression. A: Luciferase reporter assay of RAW264.7 cells co-transfected with reporter constructs and empty expression vector (pSV) or expression vector for Tat as indicated. B: Dose-dependent transactivation of the 2 × κB and wild-type HIV-LTR reporter constructs by Tat. RAW264.7 cells were co-transfected with 2 μg of 2 × κB or wild-type HIV-LTR reporter construct, and 0, 0.2, 0.6, 1.8, or 5.4 μg of expression vector for Tat. Total DNA transfected was made up to 5.4 μg with empty expression vector (pSV). The results of luciferase were normalized to β-galactosidase expression. Data shown are from representative experiments where transfection was performed in triplicate.
Tat Induces Macrophage NO Production
Our preliminary studies have shown that NO can be induced from both mouse and human macrophage/monocytes by the bacterial endotoxin component lipopolysaccharide (LPS). 15,35 To investigate the capacity of HIV Tat to induce NO production in macrophages, we transiently transfected RAW264.7 cells with Tat-expressing plasmid or the empty transfection vector (pSV). Figure 2 ▶ shows that transient expression of HIV Tat induced a dose-dependent increase in NO production. However, no detectable NO induction was observed in vector transfected cells. The induction of NO by Tat was reduced by l-NMMA, an inhibitor of NOS. To rule out the possibility that a low transfection efficiency of vector was responsible for the low production of NO, β-galactosidase staining was performed. Results from this experiment showed a similar transfection efficiency (32% to 35%) for vector and Tat (data not shown).
Figure 2.

Tat induces NO production that can be blocked by the NOS inhibitor l-NMMA. RAW264.7 cells were transfected with increasing dose of expression vector for Tat as indicated in Figure 1B ▶ in the absence or presence of 200 μmol/L l-NMMA. At 48 h, cell culture supernatant was harvested and analyzed for NO production by the Griess reaction. Total DNA transfected was made up to 5.4 μg with empty expression vector (pSV). Data shown are from representative experiments where transfection was performed in triplicate.
NO Reduces Tat-Induced NF-κB Activation
As reported previously, NF-κB activity can be attenuated by endogenous or exogenous NO. 15-23 In the present study, we observed that l-NMMA, a competitive inhibitor of NOS, enhanced Tat-induced NF-κB activity at each time point selected in both the luciferase reporter gene activity assay and the gel shift assay (Figure 3, A and B) ▶ . This enhancement of NF-κB activity by l-NMMA was completely reversed by 5 mmol/L l-arginine, a substrate of NOS (data not shown). In a parallel set of experiments, exogenous NO generated from SNAP or SNP suppressed Tat-induced NF-κB activity in a dose-dependent manner (Figure 3C) ▶ . SNAP was a more potent inhibitor of NF-κB than SNP. The 50% inhibition values (ID50) for SNAP and SNP on NF-κB are 100 μmol/L and 250 μmol/L, respectively. No appreciable effect of NAP, the inactive analog of SNAP, on Tat-induced NF-κB activity was observed. To further define the role of NO on the inhibition of Tat-induced NF-κB activation, we determined the level of NF-κB induction by Tat in macrophages from both wild-type and NOS-2 gene knockout mice. In NOS-2 gene knockout macrophages, Tat induced a sustained activation of NF-κB, whereas the NO induction by Tat was marginal. In contrast, Tat induced a transient NF-κB activation in wild-type macrophages, and the induction of NO by Tat was quite pronounced (Figure 4) ▶ .
Figure 3.
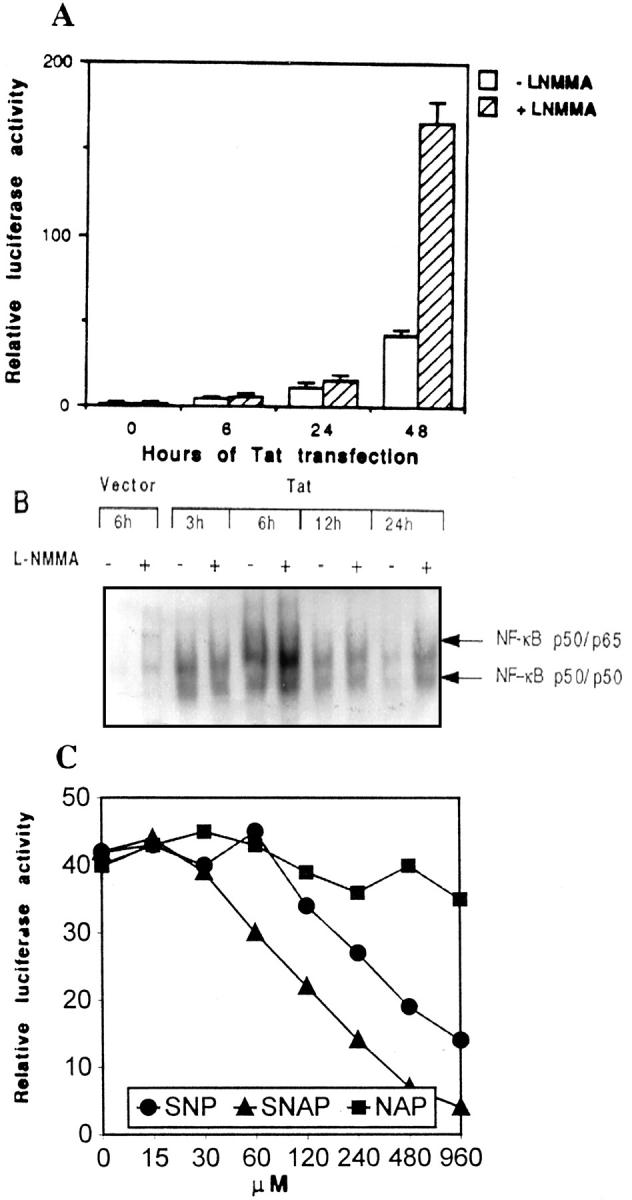
Inhibition of endogenous NO production by l-NMMA amplifies Tat-induced NF-κB activity. A: Luciferase reporter assay of RAW264.7 cells co-transfected with a 2 × κB luciferase reporter and expression vector for Tat in the absence or presence of 200 μmol/L NOS inhibitor l-NMMA for 6, 24, or 48 hours. B: EMSA with nuclear extracts from RAW264.7 cells and an oligonucleotide probe containing a NF-κB binding site. RAW264.7 cells were transfected with expression vector for Tat in the absence (−) or presence (+) of 200 μmol/L l-NMMA for 3, 6, 12, and 24 hours. C: Luciferase reporter assay of RAW264.7 cells co-transfected with a 2 × κB luciferase reporter and expression vector for Tat in the presence of various concentrations of SNAP, SNP, or NAP for 48 hours.
Figure 4.

Tat induces sustained NF-κB activation in NOS-2 gene knockout macrophages. EMSA was performed with nuclear extracts from mouse peritoneal macrophages and an oligonucleotide containing a NF-κB binding site. Peritoneal macrophages were harvested form wild-type or NOS-2 gene knockout B6 mice (iNOS-KO). Macrophages were immortalized by repeating passage and transfected with expression vector for Tat for different time periods as indicated. The lowest bands in the gel shift panel are nonspecific bands. Bottom numbers indicate relative NO production (μmol/L) from the cells transfected with Tat.
NO Inhibition of Tat-Induced HIV-LTR Promoter Is Mediated through κB Elements
To characterize the κB region of the LTR involved in NO inhibition of Tat-induced LTR promoter transactivation, the luciferase reporter plasmid containing either wild-type or κB deletion mutant HIV-LTR was co-transfected into macrophages with Tat expression plasmid in the absence or presence of exogenous NO. As expected, the wild-type LTR-luciferase construct was markedly induced by Tat, and this induction was strongly inhibited by SNAP. At 200 μmol/L, SNAP resulted in 70% inhibition of Tat-induced wild-type LTR-luciferase reporter gene activity (Figure 5) ▶ . In contrast, SNAP produced only 20% to 30% inhibition in cells transfected with the κB deleted LTR-luciferase reporter gene. Although prolonged culture (>72 hours) of cells in the presence of a high concentration of SNAP (500 μmol/L) resulted in cell death (data not shown), cell viability was not affected in the presence of 200 μmol/L SNAP at 48 hours of culture. This suggests that the inhibition of SNAP on Tat-induced LTR activity is not the result of a cytostatic effect.
Figure 5.

κB element-dependent inhibition of Tat-induced HIV-LTR transactivation by NO, as shown by luciferase reporter assay of RAW264.7 cells co-transfected with wild-type HIV-LTR or κB-mutated HIV-LTR (ΔκB) and an empty vector (pSV) or expression vector for Tat in the absence or presence of 200 μmol/L SNAP. Data shown are from representative experiments where transfections were performed in triplicate.
NO Inhibits Activation and DNA Binding of NF-κB
Previous observations from our laboratory using an electrophoresis mobility shift assay (EMSA) and the results from the experiments described above clearly show that suppression of endogenous NO production in macrophages by l-NMMA enhances NF-κB activity induced by Tat, LPS, or silica. 15,16 However, it is not clear from these results whether the inhibition of NF-κB by NO is through interference in NF-κB activation or by the inhibition of NF-κB binding to DNA, or both. The degradation of IκBα by proteasome is considered an obligatory step in the activation of NF-κB. 23 To determine whether NO is capable of impairing the proteolytic function of proteasomes, we evaluated the digestive ability of purified proteasome on recombinant IκBα. As shown in Figure 6, A and B ▶ , both NO-generating compounds, SNAP and SNP, reduced the degradative activity of proteasome. The turnover of intracellular IκBα was also examined after extracellular stimulation in the presence of exogenous NO. Figure 6C ▶ shows that LPS-induced IκBα degradation is retarded in the presence of NO (Figure 6C) ▶ . Although we observed a potent inhibition of NO on IκBα degradation by purified proteasome (Figure 6B) ▶ , only partial inhibition of cellular IκBα degradation by NO was measured. As shown in lanes 6 and 12 of Figure 6C ▶ , the resynthesis of IκBα was also inhibited by NO. This result differs from that for endothelial cells where NO stimulates overexpression of IκBα. 17 To investigate the effects of NO on NF-κB DNA binding activity, nuclear extracts from LPS-stimulated macrophages were incubated with SNAP. The DNA binding activity was determined with EMSA using 32P-labeled DNA fragments containing either the NF-κB binding site or the Sp-1 binding site. Although mild inhibition of Sp-1 binding was detected, a significant inhibition of NF-κB p50/p50 homodimer and p50/p65 heterodimer binding was observed. These observations collectively provide strong evidence indicating that the inhibition of NF-κB by NO occurs at multiple levels, including the activation level as well as the DNA binding level.
Figure 6.

Exogenous NO inhibits IκBα degradation and DNA binding of NF-κB. A: Recombinant IκBα (0.25 μg) was incubated with reaction buffer alone (None) or in recombinant 20s proteasome (0.0625, 0.125, 0.25, 0.5, and 1 μg) in 20 μl of reaction buffer at 68°C for 30 minutes. The reactions were terminated by adding 8 μl of SDS reducing buffer and boiling for 5 minutes. The protein bands were visualized by SDS-PAGE in 12% gel and Western blot analysis using the ECL system. B: Recombinant IκBα (0.25 μg) was incubated with reaction buffer alone (None) or 0.5 μg of recombinant 20s proteasome in the presence of various concentrations of SNAP (25, 50, 100, and 200 μmol/L) or SNP (100, 200, 400, and 800 μmol/L). The reaction condition was as described in A. C: RAW264.7 cells were treated with LPS (5 μg/ml) in the absence (lanes 1 to 6) or presence (lanes 7 to 12) of SNAP (200 μmol/L). Total cellular proteins were extracted at various time points as indicated and subjected to Western blot for the detection of IκBα protein. D and E: Nuclear proteins extracted from LPS-stimulated RAW264.7 cells were incubated with SNAP (0, 25, 50, 100, and 200 μmol/L) for 20 minutes followed by addition of 32P-labeled Sp-1 binding DNA probe (D) or NF-κB binding probe (E) and incubated for another 15 minutes and subjected to EMSA.
Tat Up-Regulated Transcription of NOS-2 and NOS-3 Genes
Increased levels of NO in the serum of HIV-infected individuals and excessive NO production with this disease has been suggested as responsible for the dementia seen in AIDS-afflicted patients, 12,13 yet little is known about the mechanisms responsible for increased NO in AIDS patients. To confirm our observation described above that Tat induced NO production by macrophages, RT-PCR assays were performed to evaluate the induction of NOS-2 and NOS-3 mRNA by Tat. As indicated in Figure 7 ▶ , both NOS-2 and NOS-3 are induced in the mouse macrophage cell line RAW264.7 cells by Tat transfection. We have also observed the induction of NOS-2 and NOS-3 mRNA by Tat in phorbol myristate acetate (PMA)-primed Jurkat T cells and in the THP-1 human monocyte cell line as well (data not shown). It has been suggested that Tat induced NOS-2 mRNA expression through the activation of NF-κB, as this nuclear transcription factor plays a crucial role in the transcription of NOS-2. 36,37 There is little evidence, however, to suggest that NF-κB is involved in the expression of NOS-3 gene.
Figure 7.

Tat induces NOS-3 (eNOS) and NOS-2 (iNOS) mRNA expression. Total RNA (1 μg) isolated from vector (V) or Tat (T) transfected RAW264.7 cells was used for RT-PCR analysis using specific primers for NOS-3 or NOS-2 (upper panel) and GAPDH (lower panel) as indicated in Materials and Methods.
The nucleotide sequence shown in Figure 8A ▶ is the 200-bp promoter region of the human NOS-3 gene. During the time we performed the experiments described in this report, the promoter region of murine NOS-3 had not been cloned and characterized. The sequence data of murine NOS-3 promoter were not available from GenBank until May 1998. As shown in Figure 8B ▶ , the 200-bp proximal promoter regions of human and murine are highly homologous. Both contain three AP-2-like elements, two Sp-1 sites that overlap each other, and a putative NF-κB binding site. Therefore, we believe that the 200-bp promoter region of human NOS-3 gene will be functionally equivalent with its murine counterpart in murine macrophages. In this 200-bp region, we were interested in the proximal 100-bp region for the regulation of NOS-3 transcription as suggested by the deletion mutant assays of Shizuta and co-workers. 31 To determine whether the putative κB site located in −74 to −64 (GGAGAGCCCC) is functional for binding by NF-κB, DNA fragments encompassing this region from −82 to −56 were synthesized and used as probe in the EMSA. Nuclear proteins used in the EMSA were extracted from Tat-transfected cells in the absence or presence of NOS inhibitor l-NMMA for 6 or 18 hours. We noted that the gel shift pattern was completely different from the probe encompassing a consensus NF-κB binding site (GGGGAATTCC; compare Figure 9, A and B ▶ ). The pattern was nearly identical to the probe containing a consensus Sp-1 site (GGGGCGGG; compare Figure 9, B and C ▶ ), except that the complex II is a singlet as opposed to the doublet seen in Figure 9C ▶ . An anti-Sp-1 antibody supershift assay confirmed that this band includes Sp-1 proteins (Figure 9B ▶ , lane 8). With respect to the Sp-1 binding in this DNA region, we searched the DNA sequence again and found an unidentified Sp-1 binding element located in the −68 CCCCCTCCC −60 region, which overlaps with the putative NF-κB binding site in the −74 to −64 region. When the 3′ CCC of this potential Sp-1 site was mutated into TTT, the Sp-1 binding activity was abolished and a single binding band was observed and identified as the NF-κB p50/p50 homodimer using an anti-p50 antibody supershift assay (Figure 9D) ▶ . It is tempting to speculate that the overlapping NF-κB and Sp-1 binding sites will be predominantly occupied by Sp-1 by virtue of its high abundance in nuclei or its high affinity for the cognitive DNA element.
Figure 8.

Proximal promoter region of NOS-3 gene. A: The 200-bp promoter region of human NOS-3 gene. The transcription factor binding sites for Sp-1, NF-κB, and AP-2 are underlined. B: Alignment of the 200-bp promoter sequence of human NOS-3 (upper line, GenBank ID AF032908) with the same region of murine NOS-3 (lower line, GenBank ID AF045940)
Figure 9.

Characterization of functional activity of putative κB motif in NOS-3 gene promoter regions. A: EMSA using the probe containing a consensus NF-κB binding site (underlined). B: EMSA using the probe from the −82 to −56 region of NOS-3 (eNOS) gene promoter, which contained a putative κB motif (underlined). C: EMSA using a Sp-1 probe that encompasses a consensus Sp-1 binding site (underlined). D: 3′ CCC of an unidentified Sp-1 binding site (−68 to −60) that overlapped with the putative κB motif in the −74 to −64 region of NOS-3 promoter was mutated to TTT and used as probe for EMSA. In all panels, the nuclear proteins were extracted from RAW264.7 cells transfected with vector or Tat in the absence or presence of 50 μmol/L l-NMMA for 6 or 18 hours. Lanes 6 to 8, nuclear protein extracted from Tat transfected cells was incubated with antibodies against NF-κB p50 (lane 6.), NF-κB p65 (lane 7), and Sp-1 (lane 8), respectively, for 30 minutes followed by EMSA. The bottom bands in each panel are nonspecific bands.
The results in Figure 10 ▶ provide further insight into which of the candidate response elements in the proximal 100-bp promoter region are functional or nonfunctional in regulating expression of NOS-3 gene by Tat. Mutation of the two overlapping Sp-1 sites located in the region of −97 to −83 (GGGGCGGGCGAGGG) by substituting −93 CGG −91 with TTT in the overlapping region significantly reduced Tat-induced CAT activity (by 40% to 60%). Mutation of the putative NF-κB site by changing −74 GG −73 to AA at the 5′ of this site, however, resulted in modest increase in Tat-induced CAT activity. The Tat-induced CAT activity was completely abolished when all of the Sp-1 sites were mutated. These results demonstrate that the Sp-1 sites in this proximal region are necessary for the induction of NOS-3 gene expression by Tat. The role of the putative NF-κB site in the induction of NOS-3 by Tat, however, appears less important due to the binding of p50/p50 homodimer, but not p50/p65 heterodimer, at this site. The role of p50/p50 homodimer on gene transcription is cell type dependent. In macrophages, NF-κB p50/p50 homodimer functions as a transcriptional repressor for most of the early response genes. In contrast, this homodimer acts as an activator in thymocytes for the expression of interleukin-2, tumor necrosis factor-β, and colony stimulating factor family genes. 38
Figure 10.

CAT activity assay of RAW264.7 cells transfected with a plasmid containing wild-type or mutated NOS-3 proximal promoter region (116 bp) in the presence of an empty vector (basal) or expression vector for Tat. Sp-1 sites or the NF-κB site are mutated using a QuikChange site-directed mutagenesis kit (Stratagene) and confirmed by DNA sequencing. CAT activity was determined 48 hours after transfection using 50 μg of whole cellular extract and a CAT ELISA kit. The values of basal and Tat induction were represented by the absorbance at 405 nm measured with a microplate reader and calibrated by transfection efficiency with β-galactosidase staining.
Discussion
In this report, we examined the effects of both endogenous and exogenous NO on HIV Tat protein-induced NF-κB activation. Our results suggest that two mechanisms are involved in the inhibition of Tat-induced NF-κB activation by NO. First, NO can inhibit NF-κB DNA binding activity. Second, NO can prevent the degradation of IκBα, the intracellular inhibitor of NF-κB, by impairing its digestion by proteasome. Our results also show that Tat can induce both NOS-2 and NOS-3 mRNA synthesis and hence increase NO production. The accumulation of NO may contribute to the inhibition of Tat-induced NF-κB activation in vivo.
The concept that HIV Tat induces NO production is not well documented. To that end, our results provide suggestive evidence that Tat can indeed induce NOS-2 and NOS-3 mRNA expression. These findings are supported by those of Hayman et al who demonstrated the neutralizing effect of a NOS inhibitor on the neurotoxicity of synthetic Tat protein. 39 Tat is a multifunctional protein affecting a number of kinases resulting in the activation of transcription factors, initiation of transcription, and elongation of RNA transcripts. It is not surprising therefore that Tat can also promotes NOS-2 and NOS-3 mRNA synthesis. The finding that accumulation of NO in Tat-transfected cells blocks Tat-induced NF-κB activity is consistent with reports suggesting that NO inhibits NF-κB both by influencing the binding of NF-κB to DNA and by preventing the degradation of IκBα. 15-23
Increased levels of NO have been noted in the sera of HIV-infected individuals. 12 In addition, the in vitro infection of human monocytes and brain astroglia cells with HIV results in a modest production of NO. 13,14 Both the HIV envelope glycoprotein gp120 and the regulatory protein Tat are considered responsible for the induction of NO through the activation of NOS-2 or NOS-3 genes. Rice et al noted previously that NO-related compounds could directly inhibit HIV infectivity by impairing a zinc finger transcription factor. 40,41 In addition, NO and its related reactive nitrogen species have been shown to suppress tumor necrosis factor-α induced HIV-LTR activity by inhibiting NF-κB, 42 the key transcription factor involved in HIV gene expression and viral replication. Deletion of the two κB sites in the HIV-LTR enhancer region (−109 to −79) or inhibition of NF-κB activation in HIV-infected cells results in impairment of HIV gene expression and viral replication. 43 Thus, it is important to determine whether increased levels of NO with HIV infection do compromise the replication of HIV and the development of AIDS.
The antiviral properties of NO have been evaluated in a number of viral infection studies. 5,6 Induction of NO has been implicated in resistance to several viruses, including ectromelia virus, vaccinia virus, herpes simplex virus type A, and hepatitis C virus. 5-7 Reducing endogenous NO availability by targeted disruption of the NOS-2 gene in mice is associated with enhanced susceptibility to influenza A virus, coxsackievirus, herpes simplex virus, and ectromelia virus. 44-47 In addition, treatment of animals with NOS inhibitors has been shown to increase titers of coxsackie B3 virus and produce high mortality rates. 48 The antiviral effects of NO have also been demonstrated with the use of NO-generating agents such as SNAP and SNP. In vitro studies have shown that NO-generating agents markedly inhibit viral replication, viral DNA or RNA synthesis, viral protein accumulation, and release of viral particles from cells infected with Japanese encephalitis virus, vesicular stomatitis virus, murine Friend leukemia virus, and Epstein-Barr virus. 9,49,50
NO exerts its antiviral effects through both direct and indirect pathways. The known ability of NO to inhibit ribonucleotide reductase may directly influence the replication of DNA viruses. 51 For RNA viruses that do not require ribonucleotide reductase for their RNA genome replication, NO may affect the viral life cycle by nitrosylating the cysteine residue in the active site of a viral protease required for viral assembly or replication. 52 The effects of NO on host cells provide an indirect antiviral strategy. NO and its related reactive nitrogen species can interact with either thiol-containing proteins or redox metal-containing proteins that control cellular functions. Cellular proteins that interact with NO include glyceraldehyde-3-phosphate dehydrogenase, cis-aconitase, mitochondrial respiratory enzymes, and several transcription factors, such as NF-κB. 53 The transmissibility of NO to bystander cells could further restrict the possible viral dissemination in adjacent cells. The net effect of NO on the pathogenesis of viral infection, however, may be either beneficial due to the direct antiviral role of NO or harmful due to the damaging effect of NO on host cells. 52
Macrophages are one of the first cells that come in contact with HIV and considered a major reservoir of HIV during subclinical infection. 54-56 Although T cells are rapidly killed after infection with HIV, macrophages infected with HIV can survive for weeks. Although we cannot conclude that high output of NO by macrophages is responsible for the prolonged survival of HIV-infected macrophages, it is tempting to speculate that the inhibition of NF-κB by NO may contribute to the maintenance of an asymptomatic status early in the infection with HIV. More extensive studies are in progress to understand the potential protective effect of NO on the early stage of HIV infection.
Acknowledgments
We thank Dr. Shao-Cong Sun for providing us with expression vectors for HIV Tat and HIV-LTR luciferase reporter gene. We also thank the members in Health Effects Laboratory Division of NIOSH for their careful reading and critique of the manuscript.
Footnotes
Address reprint requests to Dr. Fei Chen, Pathology and Physiology Research Branch, NIOSH, 1095 Willowdale Road, Morgantown, WV 26505. E-mail: lfd3@cdc.gov.
Supported by a Career Development Award to F. Chen under a cooperative agreement from the Centers for Disease Control and Prevention through the Association of Teachers of Preventive Medicine.
References
- 1.Nathan C: Inducible nitric oxide synthase: what difference does it make? J Clin Invest 1997, 100:2417-2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.MacMicking J, Xie QW, Nathan C: Nitric oxide and macrophage function. Annu Rev Immunol 1997, 15:323-350 [DOI] [PubMed] [Google Scholar]
- 3.Guidotti LG, Chisari FV: To kill or to cure: options in host defense against viral infection. Curr Opin Immunol 1996, 8:478-483 [DOI] [PubMed] [Google Scholar]
- 4.Tucker PC, Griffin DE, Choi S, Bui N, Wesselingh S: Inhibition of nitric oxide synthesis increases mortality in Sindbis virus encephalitis. J Virol 1996, 70:3972-3977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peterhans E: Reactive oxygen species and nitric oxide in viral diseases. Biol Trace Element Res 1997, 56:107-116 [DOI] [PubMed] [Google Scholar]
- 6.Karupiah G, Harris N: Inhibition of viral replication by nitric oxide and its reversal by ferrous sulfate and tricarboxylic acid cycle metabolites. J Exp Med 1995, 181:2171-2179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bi Z, Reiss CS: Inhibition of vesicular stomatitis virus infection by nitric oxide. J Virol 1995, 69:2208-2213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guillemard E, Geniteau-Legendre M, Kergot R, Lemaire G, Petit JF, Labarre C, Quero AM: Activity of nitric oxide-generating compounds against encephalomyocarditis virus. Antimicrobial Agents Chemother 1996, 40:1057-1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin YL, Huang YL, Ma SH, Yeh CT, Chiou SY, Chen LK, Liao CL: Inhibition of Japanese encephalitis virus infection by nitric oxide: antiviral effect of nitric oxide on RNA virus replication. J Virol 1997, 71:5227-5235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Melkova Z, Esteban M: Inhibition of vaccinia virus DNA replication by inducible expression of nitric oxide synthase. J Immunol 1995, 155:5711-5718 [PubMed] [Google Scholar]
- 11.Harris N, Buller RM, Karupiah G: Gamma interferon-induced, nitric oxide-mediated inhibition of vaccinia virus replication. J Virol 1995, 69:910-915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Torre D, Ferrario G: Immunological aspects of nitric oxide in HIV-1 infection. Med Hypotheses 1996, 47:405-407 [DOI] [PubMed] [Google Scholar]
- 13.Adamson DC, Wildemann B, Sasaki M, Glass JD, McArthur JC, Christov VI, Dawson TM, Dawson VL: Immunologic NO synthase: elevation in severe AIDS dementia and induction by HIV-1 gp41. Science 1996, 274:1917-1921 [DOI] [PubMed] [Google Scholar]
- 14.Bukrinsky M, Schmidtmayerova H, Zybarth G, Dubrovsky L, Sherry B, Enikolopov G: A critical role of nitric oxide in human immunodeficiency virus type 1-induced hyperresponsiveness of cultured monocytes. Mol Med 1996, 2:460-468 [PMC free article] [PubMed] [Google Scholar]
- 15.Chen F, Sun SC, Kuhn DC, Gaydos LJ, Demers LM: Essential role of NF-kappa B activation in silica-induced inflammatory mediator production in macrophages. Biochem Biophys Res Commun 1995, 214:985-992 [DOI] [PubMed] [Google Scholar]
- 16.Chen F, Kuhn DC, Sun SC, Gaydos LJ, Demers LM: Dependence and reversal of nitric oxide production on NF-kappa B in silica and lipopolysaccharide-induced macrophages. Biochem Biophys Res Commun 1995, 214:839-846 [DOI] [PubMed] [Google Scholar]
- 17.Spiecker M, Peng HB, Liao JK: Inhibition of endothelial vascular cell adhesion molecule-1 expression by nitric oxide involves the induction and nuclear translocation of IκBα. J Biol Chem 1997, 272:30969-30974 [DOI] [PubMed] [Google Scholar]
- 18.Taylor BS, Kim YM, Wang Q, Shapiro RA, Billiar TR, Geller DA: Nitric oxide down-regulates hepatocyte-inducible nitric oxide synthase gene expression. Arch Surg 1997, 132:1177-1183 [DOI] [PubMed] [Google Scholar]
- 19.Togashi H, Sasaki M, Frohman E, Taira E, Ratan RR, Dawson TM, Dawson VL: Neuronal (type I) nitric oxide synthase regulates nuclear factor kappa B activity and immunologic (type II) nitric oxide synthase expression. Proc Natl Acad Sci USA 1997, 94:2676-2680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park SK, Lin HL, Murphy S: Nitric oxide regulates nitric oxide synthase-2 gene expression by inhibiting NF-κB binding to DNA. Biochem J 1997, 322:609-613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khan BV, Harrison DG, Olbrych MT, Alexander RW, Medford RM: Nitric oxide regulates vascular cell adhesion molecule 1 gene expression and redox-sensitive transcriptional events in human vascular endothelial cells. Proc Natl Acad Sci USA 1996, 93:9114-9119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matthews JR, Botting CH, Panico M, Morris HR, Hay RT: Inhibition of NF-κB DNA binding by nitric oxide. Nucleic Acids Res 1996, 24:2236-2242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen F, Castranova V, Shi X, Demers LM: New insights into the role of NF-κB, a ubiquitous transcription factor in the initiation of diseases. Clin Chem 1999, 45:7-17 [PubMed] [Google Scholar]
- 24.Webber EM, Bruix J, Pierce RH, Fausto N: Tumor necrosis factor primes hepatocytes for DNA replication in the rat. Hepatology 1998, 28:1226-1234 [DOI] [PubMed] [Google Scholar]
- 25.Baldwin AS, Jr: The NF-κB and IκB proteins: new discoveries and insights. Annu Rev Immunol 1996, 14:649-683 [DOI] [PubMed] [Google Scholar]
- 26.Mhashilkar AM, Biswas DK, LaVecchio J, Pardee AB, Marasco WA: Inhibition of human immunodeficiency virus type 1 replication in vitro by a novel combination of anti-Tat single-chain antibodies and NF-κB antagonists. J Virol 1997, 71:6486-6494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beauparlant P, Kwon H, Clarke M, Lin R, Sonenberg N, Wainberg M, Hiscott J: Transdominant mutants of IκBα block Tat-tumor necrosis factor synergistic activation of human immunodeficiency virus type 1 gene expression and virus multiplication. J Virol 1996, 70:5777-5785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harhaj E, Blaney J, Millhouse S, Sun SC: Differential effects of IκB molecules on Tat-mediated transactivation of HIV-1 LTR. Virology 1996, 216:284-287 [DOI] [PubMed] [Google Scholar]
- 29.Alcami J, Lain de Lera T, Folgueira L, Pedraza MA, Jacque JM, Bachelerie F, Noriega AR, Hay RT, Harrich D, Gaynor RB: Absolute dependence on κB responsive elements for initiation and Tat-mediated amplification of HIV transcription in blood CD4 T lymphocytes. EMBO J 1995, 14:1552-1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen F, Lu Y, Kuhn DC, Maki M, Shi X, Sun SC, Demers LM: Calpain contributes to silica-induced IκBα degradation and nuclear factor-κB activation. Arch Biochem Biophys 1997, 342:383-388 [DOI] [PubMed] [Google Scholar]
- 31.Wariishi S, Miyahara K, Toda K, Ogoshi S, Doi Y, Ohnishi S, Mitsui Y, Yui Y, Kawai C, Shizuta Y: A SP1 binding site in the GC-rich region is essential for a core promoter activity of the human endothelial nitric oxide synthase gene. Biochem Biophys Res Commun 1995, 216:729-735 [DOI] [PubMed] [Google Scholar]
- 32.Murphy KM, Sweet MJ, Ross IL, Hume DA: Effects of the tat and nef gene products of human immunodeficiency virus type 1 (HIV-1) on transcription controlled by the HIV-1 long terminal repeat and on cell growth in macrophages. J Virol 1993, 67:6956-6964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sweet MJ, Hume DA: RAW264 macrophages stably transfected with an HIV-1 LTR reporter gene provide a sensitive bioassay for analysis of signalling pathways in macrophages stimulated with lipopolysaccharide, TNF-α or taxol. J Inflamm 1995, 45(2):126-135 [PubMed] [Google Scholar]
- 34.Munoz-Fernandez MA, Navarro J, Garcia A, Punzon C, Fernandez-Cruz E, Fresno M: Replication of human immunodeficiency virus-1 in primary human T cells is dependent on the autocrine secretion of tumor necrosis factor through the control of nuclear factor-κB activation. J Allergy Clin Immunol 1997, 100:838-845 [DOI] [PubMed] [Google Scholar]
- 35.Chen F, Kuhn DC, Gaydos LJ, Demers LM: Induction of nitric oxide and nitric oxide synthase mRNA by silica and lipopolysaccharide in PMA-primed THP-1 cells. APMIS 1996, 104:176-182 [DOI] [PubMed] [Google Scholar]
- 36.Kim IY, Stadtman TC: Inhibition of NF-κB DNA binding and nitric oxide induction in human T cells and lung adenocarcinoma cells by selenite treatment. Proc Natl Acad Sci USA 1997, 94:12904-12907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chao CC, Lokensgard JR, Sheng WS, Hu S, Peterson PK: IL-1-induced iNOS expression in human astrocytes via NF-κB. Neuroreport 1997, 8:3163-3166 [DOI] [PubMed] [Google Scholar]
- 38.Ishikawa H, Claudio E, Dambach D, Raventos-Suarez C, Ryan C, Bravo R: Chronic inflammation and susceptibility to bacterial infections in mice lacking the polypeptide (p)105 precursor (NF-κB1) but expressing p50. J Exp Med 1998, 187:985-996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hayman M, Arbuthnott G, Harkiss G, Brace H, Filippi P, Philippon V, Thomson D, Vigne R, Wright A: Neurotoxicity of peptide analogues of the transactivating protein tat from Maedi-Visna virus and human immunodeficiency virus. Neuroscience 1993, 53:1-6 [DOI] [PubMed] [Google Scholar]
- 40.Rice WG, Schaeffer CA, Graham L, Bu M, McDougal JS, Orloff SL, Villinger F, Young M, Oroszlan S, Fesen MR: The site of antiviral action of 3-nitrosobenzamide on the infectivity process of human immunodeficiency virus in human lymphocytes. Proc Natl Acad Sci USA 1993, 90:9721-9724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rice WG, Schaeffer CA, Harten B, Villinger F, South TL, Summers MF, Henderson LE, Bess JW, Jr, Arthur LO, McDougal JS: Inhibition of HIV-1 infectivity by zinc-ejecting aromatic C-nitroso compounds. Nature 1993, 361:473-475 [DOI] [PubMed] [Google Scholar]
- 42.Sekkai D, Aillet F, Israel N, Lepoivre M: Inhibition of NF-κB and HIV-1 long terminal repeat transcriptional activation by inducible nitric oxide synthase 2 activity. J Biol Chem 1998, 273:3895-3900 [DOI] [PubMed] [Google Scholar]
- 43.Kwon H, Pelletier N, DeLuca C, Genin P, Cisternas S, Lin R, Wainberg MA, Hiscott J: Inducible expression of IκBα repressor mutants interferes with NF-κB activity and HIV-1 replication in jurkat T cells. J Biol Chem 1998, 273:7431-7440 [DOI] [PubMed] [Google Scholar]
- 44.Karupiah G, Chen JH, Mahalingam S, Nathan CF, MacMicking JD: Rapid interferon γ-dependent clearance of influenza A virus and protection from consolidating pneumonitis in nitric oxide synthase-2-deficient mice. J Exp Med 1998, 188:1541-1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zaragoza C, Ocampo C, Saura M, Leppo M, Wei XQ, Quick R, Moncada S, Liew FY, Lowenstein CJ: The role of inducible nitric oxide synthase in the host response to Coxsackievirus myocarditis. Proc Natl Acad Sci USA 1998, 95:2469-2474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karupiah G, Chen JH, Nathan CF, Mahalingam S, MacMicking JD: Identification of nitric oxide synthase-2 as an innate resistance locus against ectromelia virus infection. J Virol 1998, 72:7703-7706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.MacLean A, Wei XQ, Huang FP, Al-Alem UA, Chan WL, Liew FY: Mice lacking inducible nitric oxide synthase are more susceptible to herpes simplex virus infection despite enhanced Th1 cell responses. J Gen Virol 1998, 79:825-830 [DOI] [PubMed] [Google Scholar]
- 48.Lowenstein CJ, Hill SL, Lafond-Walker A, Wu J, Allen G, Landavere M, Rose NR, Herskowitz A: Nitric oxide inhibits viral replication in murine myocarditis. J Clin Invest 1996, 97:1837-1843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Akarid K, Sinet M, Desforges B, Gougerot-Pocidalo MA: Inhibitory effect of nitric oxide on the replication of a murine retrovirus in vitro and in vivo. J Virol 1995, 69:7001-7005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kawanishi M: Nitric oxide inhibits Epstein-Barr virus DNA replication and activation of latent EBV. Intervirology 1995, 38:206-213 [DOI] [PubMed] [Google Scholar]
- 51.Kwon NS, Stuehr DJ, Nathan CF: Inhibition of tumor cell ribonucleotide reductase by macrophage derived nitric oxide. J Exp Med 1991, 174:761-768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saura M, Zaragoza C, McMillan A, Quick RA, Hohenadl C, Lowenstein JM, Lowenstein CJ: An antiviral mechanism of nitric oxide: inhibition of a viral protease. Immunity 1999, 10:21-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bolanos JP, Peuchen S, Heales SJ, Land JM, Clark JB: Nitric oxide-mediated inhibition of the mitochondrial respiratory chain in cultured astrocytes. J Neurochem 1994, 63:910-916 [DOI] [PubMed] [Google Scholar]
- 54.Perno CF, Crowe SM, Kornbluth RS: A continuing enigma: the role of cells of macrophage lineage in the development of HIV disease. J Leukocyte Biol 1997, 62:1-3 [DOI] [PubMed] [Google Scholar]
- 55.Magnani M, Rossi L, Fraternale A, Casabianca A, Brandi G, Benatti U, De Flora A: Targeting antiviral nucleotide analogues to macrophages. J Leukocyte Biol 1997, 62:133-137 [DOI] [PubMed] [Google Scholar]
- 56.Montaner LJ, Herbein G, Gordon S: Regulation of macrophage activation and HIV replication. Adv Exp Med Biol 1995, 374:47-56 [DOI] [PubMed] [Google Scholar]