

Abstract
Novel plaque-like “AMY” lesions were recently described in the brains of patients with Alzheimer’s disease (AD). Using three Aβ antibodies, we now document the co-occurrence of AMY immunoreactivity (IR) with amyloid β-peptide (Aβ) in the large majority of plaques in AD brain. AMY IR was detected in many compacted plaques, whereas its co-localization with early, diffuse Aβ deposits was rare. AMY IR overlapped considerably or fully with Aβ and, in more severely affected AD brains, decorated the periphery of some plaques. In a temporal series of 29 Down syndrome (DS) brains from patients aged 12 to 73 years, the earliest AMY IR was detected in some plaques at age 15, following the earliest appearance of Aβ plaques (age 12 years), and then accrued within a subset of Aβ deposits, namely, the more spherical, compacted plaques. Brains from DS patients 29 years and older showed AMY staining in many Aβ plaques, as seen in AD. Brains from eight monkeys aged 17 to 34 years and thirty APP transgenic mice aged 8 to 20 months showed Aβ IR but no AMY IR. We conclude that AMY IR represents an amyloid-associated antigen that co-deposits in most but not all Aβ plaques in AD and DS and that accumulation of the AMY antigen follows Aβ deposition in plaques.
Alzheimer’s disease (AD) is characterized neuropathologically by the presence of two principal brain lesions, amyloid plaques and neurofibrillary tangles. The earlier of the two lesions, the amyloid plaque, is formed by the progressive extracellular deposition in brain parenchyma of heterogeneous amyloid β-peptides (Aβ) proteolytically derived from the β-amyloid precursor protein (βAPP). 1 Because the βAPP gene is encoded on chromosome 21 and is overexpressed in trisomy 21 (Down syndrome (DS)), DS provides a temporal model for studying AD pathogenesis. 2-5 Aβ peptides ending at residue 42 (Aβ42), as opposed to the more abundantly produced Aβ peptides ending at residue 40 (Aβ40), have been shown to be the initially deposited species in AD and DS brain, 6,7 with the earliest deposits detected immunohistochemically in DS brain as diffuse plaques at 12 years. 5 In addition, other proteins have been found to associate with Aβ in AD plaques. 5, 8-14 The deposition of some Aβ-associated proteins may be indicative of a local inflammatory response to the amyloid, and the accrual of others may stabilize the Aβ or promote its aggregation.
Recently, abundant Aβ-negative AMY plaques were described immunohistochemically in AD brain. 15 A monoclonal antibody (MAb) used in that study, AMY 117, was raised against an as-yet-unidentified 100-kd protein present in paired-helical-filaments-tau-rich AD brain extracts. An accompanying commentary to this report raised the possibility that AMY plaques are a non-amyloid precursor to Aβ-bearing senile plaques. 16 In collaboration with the authors of the original report, we sought to determine the temporal sequence of deposition of the AMY 117 antigen relative to that of Aβ. The immunoreactivity (IR) of the AMY 117 MAb was compared with that of each of three Aβ antibodies in brains obtained from three temporal models of AD pathogenesis: DS patients (aged 12 to 73 years), monkeys (aged 17 to 34 years), and PD-APP transgenic mice (aged 8 to 20 months). The immunohistochemical protocols were first optimized for each antibody in AD brain sections for each of the fixation and embedding conditions used in these three temporal models of AD.
Materials and Methods
Subject Groups
Autopsied brains from 22 AD patients (aged 64 to 96 years; mean, 83 years) and 10 aged human controls (aged 60 to 87 years; mean, 78 years) were used to optimize immunostaining protocols for each tissue preparation and to characterize the spatial patterns of immunoreactivity (IR) of antibodies to Aβ or AMY. A series of 29 brains from clinically diagnosed DS patients (aged 12 to 73 years; mean, 38 years), the neuropathology of most of which has been previously described, 5 was examined to determine the temporal sequence of deposition of Aβ and AMY relative to each other. Within this series, brain tissues from 10 young DS cases (aged 12 to 29 years) were generously provided by Dr. K. Wisniewski (Institute for Basic Research in Developmental Disabilities, Staten Island, NY). In addition, brain sections from three young DS patients lacking AD pathology were kindly provided by Dr. D. Anthony (Department of Pathology, Harvard Medical School, Boston, MA). Brain tissues from the remaining 16 DS patients (aged 36 to 73 years) were collected at autopsy by us at Brigham and Women’s Hospital. Brain tissues from two animal models of AD pathogenesis were also examined: eight monkeys (aged 17 to 34 years) and 30 PD-APP transgenic mice 17 aged 8 to 20 months (kindly provided by Athena Neurosciences, South San Francisco, CA).
Tissue Preparation
Blocks of human and monkey brain tissues from cerebral cortex, hippocampus, and cerebellum were fixed in 10% neutral buffered formalin for three time intervals ranging from 1) 1 to 2 hours (AD, aged human controls, older DS (>29 years), and monkey brains; brief fixation) to 2) an unknown period longer than 1 week (duplicate blocks from AD, older DS, and monkey brains; routine fixation) to 3) several years (12- to 29-year-old DS brains; long-term fixation). For several AD, DS, and aged control brains, additional blocks were fixed in 70% ethanol in 125 mmol/L NaCl at 4°C for 2 days. PD-APP transgenic mice were saline perfused and their hemibrains immersion fixed in 70% ethanol in 125 mmol/L NaCl at 4°C for 1 to 2 days. Fixed brain tissue was dehydrated and embedded in paraffin. Eight-micron sections were baked at 58°C for 1 hour. In addition, fresh-frozen 6-μm cryostat sections were prepared for several subjects in each group (except the monkeys). Frozen sections were fixed with cold acetone before immunolabeling.
Immunohistochemistry
Serial sections were immunostained with the avidin-biotin horseradish peroxidase/diaminobenzidine (DAB) method (rabbit or mouse ABC Elite kit, Vector Laboratories, Burlingame, CA), using the antibodies detailed below. Details of the immunostaining protocol have been previously described. 5 Double labeling was accomplished using the horseradish peroxidase/DAB kit to detect the first primary antibody and the alkaline phosphatase ABC kit with Vector Red substrate (Vector Laboratories) to detect the second primary antibody. All antibodies were tested on long-term, routinely and briefly fixed AD and DS paraffin sections as well as on frozen sections to determine optimal staining conditions. Our sensitive general Aβ antibody R1282 (1:1000), 18 which detects multiple Aβ forms, was used as a reference antibody for Aβ plaque distribution. A highly sensitive Aβ42-endspecific MAb, 21F12 (1:1000), 19 was used to detect early, diffuse plaques as well as more mature plaques (gift of Athena Neurosciences, South San Francisco, CA). In long-term and routinely formalin-fixed tissues, both Aβ antibodies required formic acid pretreatment (88% formic acid for 8 minutes at room temperature (RT)) to optimize visualization of Aβ deposits. AMY 117 hybridoma supernatant MAb (AMY 117; gift of Lee and Trojanowski Laboratories, The Center for Neurodegenerative Research, University of Pennsylvania School of Medicine, Philadelphia, PA) 15 was used neat on all tissues. AMY 117 ascites MAb (AMY 117asc; 1:5000; gift of Lee and Trojanowski Laboratories) was used in absorption experiments with Aβ1–40 and Aβ1–42 peptides (20 μg of Aβ peptide/1 μl of antibody) as well as to confirm AMY 117 staining. In long-term and routinely formalin-fixed tissues, the AMY 117 staining required a double pretreatment: antigen retrieval by microwaving the sections for 10 minutes in citrate buffer solution (BioGenex, San Ramon, CA) followed by proteinase K digestion (Dako Corp., Carpinteria, CA) for 6 minutes at RT. No pretreatment was required for AMY 117 immunostaining of briefly formalin-fixed or ethanol-fixed paraffin sections or on cryosections. The HistoMouse-SP kit (Zymed Laboratories, South San Francisco, CA), an immunohistochemistry kit designed to use MAbs on mouse sections, was used in conjunction with MAb AMY 117 on the PD-APP transgenic mouse brain sections so as to avoid cross-reactivity with endogenous mouse IgG. Selected sections from all subject groups were stained for amyloid deposits with thioflavin S.
Microscopy
For light microscopy, photomicrographs were generated using an Olympus BX50 microscope. For the confocal image shown in Figure 1 ▶ , kindly provided by Dr. M. L. Schmidt (The Center for Neurodegenerative Diseases, University of Pennsylvania School of Medicine, Philadelphia, PA), a polyclonal antibody raised against native Aβ purified from AD brain and made in our laboratory, Angela (1:250), 20 was used for double-immunofluorescent labeling of Aβ and AMY 117 on a 40-μm ethanol-fixed, cryoprotected AD brain section. Here, Aβ IR was visualized using a Texas-Red-conjugated donkey anti-rabbit secondary antibody (1:400; Jackson ImmunoResearch Laboratories, West Grove, PA) whereas AMY 117 IR was visualized using a fluorescein-isothiocyanate-conjugated donkey anti-mouse secondary antibody (1:100; Jackson ImmunoResearch Laboratories). A series of six confocal images were obtained through the section at 1-μm intervals using a Leica confocal laser scanning microscope, as previously described. 15
Figure 1.
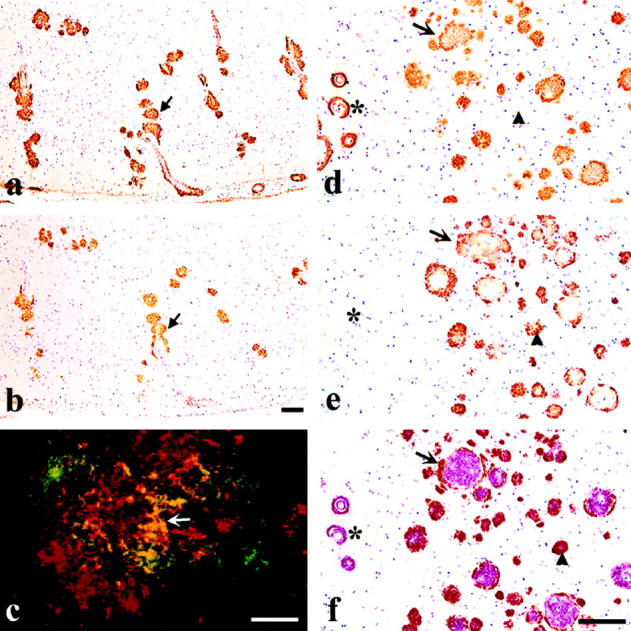
Immunostaining with antibodies to Aβ and AMY 117 demonstrated the co-occurrence of the two antigens in the vast majority of plaques in AD brain. a and b: Adjacent 8-μm ethanol-fixed, paraffin sections of occipital cortex from a 90-year-old AD patient show overlapping immunoreactivities between Aβ labeled with R1282 (a) and AMY 117 (b); arrows mark one example. c: Double-immunofluorescent labeling with AMY 117 (ATC green) and the Ap antibody, Angela (Texas Red) on a 40-μm ethanol-fixed AD brain cryosection shows partial overlap in yellow (arrow) of the two antigens within an individual plaque lesion. The confocal image shown in c, kindly provided by Dr. M. L. Schmidt (The Center for Neurodegenerative Diseases, University of Pennsylvania School of Medicine, Philadelphia, PA), is the compilation of six images taken at 1-μm intervals. This image reflects the same yellow-labeled co-localization between AMY 117 and Aβ antibody R1282 we have observed in 8-μm AD brain sections by non-confocal fluorescent microscopy (data not shown). d to f: Three adjacent 8-μm briefly formalin-fixed, paraffin sections of frontal cortex from a 69-year-old AD patient illustrate overlapping Aβ and AMY 117 immunoreactivities. d shows plaques immunostained with Aβ antibody R1282, e shows plaques immunostained with AMY 117, and f shows double labeling with both antibodies (AMY 117 visualized in brown with DAB and Aβ (R1282) visualized in red with alkaline phosphate). The arrow indicates a large Aβ plaque surrounded by AMY 117 IR. The arrowheads indicate a single plaque that is negative for Aβ in the first section (d), shows AMY 117 IR in the second (e), and shows the presence of both antigens (red and brown reaction products) in the third section f.Note that the Aβ IR blood vessels (asterisk) shown in d and f are AMY 117 negative in e. Scale bars, 100 μm a, b, d to f and 10 μm (c).
Results
AD and Older DS Brains
In both single-labeled adjacent sections and in double-labeled sections, the vast majority of AMY 117 immunoreactive plaques were co-localized to some degree with Aβ immunoreactivity (IR) detected by antibodies R1282 (a general Aβ polyclonal antibody) and 21F12 (Aβ42-specific MAb) in both AD and older (≥29 years) DS brains. AMY 117 IR was always restricted to those cortical and hippocampal regions that contained Aβ deposits. Many, but not all, AMY IR plaques overlapped with thioflavin-S-labeled amyloid plaques. As exemplified in Figure 1 ▶ , AMY 117 IR frequently co-localized (eg, overlapped) with Aβ IR (Figure 1, a–c) ▶ , was interspersed with it (Figure 1c) ▶ , or, in the more pathologically severe brains, surrounded it (Figure 1, d–f) ▶ within an individual plaque lesion. In the latter case, the two antigens were found to partially overlap or to segregate but abut each other. Subpial Aβ deposits, large diffuse Aβ42 IR bands, cerebellar Aβ deposits, and vessel wall Aβ were all devoid of any AMY 117 IR (see asterisks in Figure 1, d–f ▶ ). Occasionally, small punctate AMY 117 immunoreactive deposits that did not appear to overlap with Aβ IR were observed; such deposits occurred only in brain regions bearing abundant Aβ IR plaques (see arrowheads in Figure 1, d and e ▶ ). However, the presence of both antigens could often be detected in these same lesions in sections just above or below the plane of the initially stained section (see arrowhead in Figure 1f ▶ ). Absorption of antibodies R1282 (Aβ) and AMY 117asc with synthetic Aβ1–40 and Aβ1–42 peptides caused ablation of plaque staining by R1282 (absorption with Aβ1–40 peptide shown in Figure 2, a and b ▶ ) but did not diminish AMY 117 IR (Figure 2, c and d) ▶ . Figure 2 ▶ further illustrates the close co-occurrence of Aβ and AMY 117 within plaques, at both high and low magnification, in the brain of a 65-year-old DS patient. Regions of compacted Aβ42 IR plaques in hippocampus, parahippocampal gyrus, and temporal cortex were also AMY 117 IR, as shown at low magnification in Figure 2 ▶ (eg, small arrowheads in e and f). However, AMY 117 IR was absent in Aβ42 IR diffuse plaques in the parahippocampal gyrus (arrows in Figure 2, e and f ▶ ), in Aβ42 IR plaques in the subpial layers of temporal cortex (asterisks in Figure 2, e and f ▶ ), and in Aβ42 IR plaques in deep cortical layers and white matter (large arrowheads in Figure 2, e and f ▶ ) as judged in immediately adjacent sections. In AD and DS brains in general, Aβ IR, especially as detected with the Aβ42 MAb 21F12, was more abundant than AMY 117 IR.
Figure 2.

AMY 117 IR was not diminished by pre-absorption with Aβ peptide and was less abundant than Aβ42 in old DS brain. a and b: Adjacent 8-μm briefly formalin-fixed, paraffin sections of hippocampus from a 65-year-old DS patient with clinical and neuropathological AD show AβIR (a) was ablated by absorption of Aβ antibody (R1282) with synthetic Aβ1-40 peptide (b). c and d: AMY 117ascIR (c) in serial sections adjacent to those shown in a and b was not diminished by pre-absorption of the AMY 117 antibody with Aβ1-40 peptide (d), indicating that anti-AMY 117 is not an Aβ antibody. Note the close co-occurrence of Aβ and the AMY 117 antigen in virtually all plaques, exemplified by the arrowhead. e and f: Immunoreactivities of Aβ42 (using antibody 21F12) and AMY 117 in adjacent sections approximately 200 μm deeper into the same tissue block shown in a to d indicate that AMY 117 IR occurs only in regions bearing Aβ. Many compacted plaques in the 65-year-old DS brain were labeled by Aβ42 (e) and AMY 117 (f) in hippocampus (DG, dentate gyrus; small arrowheads), parahippocampus (PHC), and temporal cortex (TC). However, AMY 117 IR was absent from Aβ42-rich plaque regions in the subiculum (arrow in e and f) and parahippocampus, in the deep cortical layers and adjacent white matter in temporal cortex (large arrowheads in e and f), and in the subpial layer of cortex (alongside asterisks in e and f). Scale bar, 100 μm (a to d) and 1 mm (e and f).
Aged Human Control Brains
Of the 10 nondemented aged control brains examined, 2 had no Aβ or AMY 117 IR. In the remaining eight cases, varying amounts of Aβ deposition were detected in the cortex in each brain. Only three of the eight Aβ-bearing brains had any AMY 117 IR; Aβ IR was always much more abundant than that of AMY 117. In general, AMY 117 IR was associated with spherical, compacted plaques and plaques apparently undergoing compaction, but not with diffuse, thioflavin-negative plaques in these aged control brains (data not shown). As in AD brains, no vessel wall AMY 117 IR was detected, even when Aβ IR was present in the blood vessel.
Down Syndrome Brains
To determine the relative temporal sequence of deposition of Aβ and the AMY 117 antigen, we immunolabeled adjacent sections of DS brains from patients ranging from 12 to 73 years old using the highly sensitive Aβ42 MAb 21F12 and the AMY 117 MAb. Frontal cortex sections from 13 young DS patients (aged 12 to 29 years) were examined; in addition, large sections containing both temporal cortex and hippocampus were available for three of these young DS cases and were immunostained with both antibodies. Because the young DS brains had been subjected to long-term fixation in formalin, various pretreatments were tested and then employed to allow visualization of the AMY 117 antigen. A combination pretreatment involving antigen retrieval by microwaving the section in a citrate buffer solution followed by a brief proteinase K digestion allowed the unmasking of the AMY 117 antigen in the long-term fixed tissues. Aβ42 plaque IR was observed in 7 of the 13 young DS brains (aged 12, 15, 16, 17, 21, 27, and 29 years) (exemplified in Figure 3, a, c, e, and g ▶ ). Quantitative analyses of Aβ deposition and other neuropathological characterization of these brains has been previously reported. 5 AMY 117 IR was detected in three of the seven young DS brains that had Aβ deposits (and in none of those that did not). Specifically, the frontal and temporal cortices and hippocampus of a 15-year-old DS patient having thioflavin-positive amyloid plaques and shown previously to have compacted and cored Aβ IR plaques, gliosis, and some neuritic changes had some AMY 117 IR plaques (not shown); the hippocampus of a 16-year-old DS patient that showed compacted Aβ IR plaques had some AMY 117 IR (Figure 3, e and f) ▶ , whereas the frontal and temporal cortices having only diffuse Aβ42 IR plaques did not (Figure 3, c and d) ▶ ; and the frontal cortex of a 29-year-old DS patient (Figure 3, g and h) ▶ previously shown to have compacted and cored Aβ IR plaques, gliosis, and some neuritic changes had many plaques positive for both Aβ and AMY 117. In the brains of the four young DS patients (aged 12, 17, 21, and 27 years) that had almost exclusively diffuse Aβ42 IR plaques, AMY 117 IR was not detected (eg, Figure 3, a and b ▶ ). Brains from middle-aged and older DS patients showed AMY 117 IR very similar to that described above for AD, as demonstrated in the brain of a 65-year-old DS patient in Figure 2 ▶ . As in AD cases, no vascular or cerebellar AMY 117 IR was detected in DS brains at any age, even though abundant Aβ deposition occurred in each structure in these older DS brains.
Figure 3.
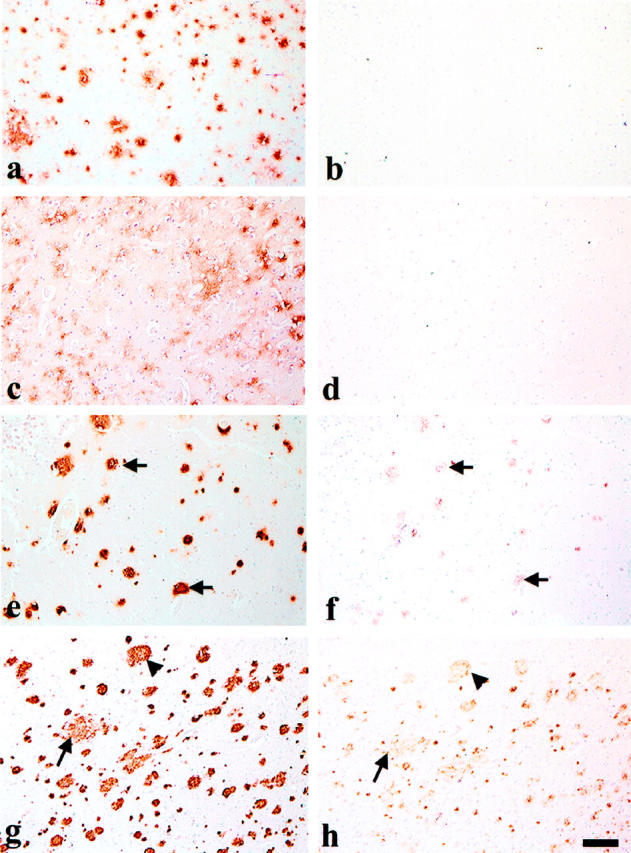
Aβ42 deposition precedes that of the AMY 117 antigen in young DS brain. Eight-micron, long-term formalin-fixed, paraffin sections from young DS brain were immunostained with Aβ42 antibody 21F12 and are shown in the left column (a, c, e, and g). Adjacent sections were immunostained with AMY 117 after double pretreatment of the tissue (microwaving and proteinase K) and are shown in the right column (b, d, f, and h). a and b: The frontal cortex of a 12-year-old DS patient shows many Aβ42 IR diffuse plaques (a) but no AMY 117 IR (b). c to f: Adjacent sections of a large block of tissue containing both temporal cortex and hippocampus from a 16-year-old DS patient show Aβ42 IR in many diffuse plaques in temporal cortex (c) and in more compacted plaques in hippocampus (e). AMY 117 IR is not seen in the temporal cortex (d) but is visible in some compacted plaques in the hippocampus (f). Arrows in e and f indicate examples of overlapping Aβ and AMY 117 immunoreactivities. g and h: Adjacent sections of frontal cortex from a 29-year-old DS patient show abundant IR for both Aβ (g) and AMY 117 (h). Arrowheads indicate relatively compacted plaques immunoreactive for both antigens, whereas arrows show a relatively less compated Aβ42 IR plaque that has less abundant, finely punctate AMY 117 IR. Scale bar, 100 μm for all images.
Monkey Brains and PD-APP Transgenic Mouse Brains
To further characterize the temporal accrual of AMY 117 in Aβ plaque lesions, two animal models of AD pathogenesis were examined. Cortical sections bearing Aβ (R1282 and 21F12) immunoreactive plaques from the brains of eight monkeys ranging in age from 17 to 34 years were immunostained with the AMY 117 MAb. In addition, hemibrain sections from 30 Aβ (R1282) immunoreactive plaque-bearing PD-APP transgenic mice, aged 8 to 20 months, were examined for AMY 117 IR (using the HistoMouse kit (Zymed Laboratories, South San Francisco, CA) to avoid mouse IgG cross-reactivity). No AMY 117 IR was detected in any of these monkey or transgenic mouse brains, regardless of Aβ plaque deposition (data not shown). Even in the 18- and 20-month-old PD-APP transgenic mice that have very abundant compacted and cored Aβ IR and thioflavin-positive plaques, AMY 117 IR was not observed.
Discussion
By optimizing the immunostaining protocols for the AMY 117 MAb and the Aβ antibodies on AD brain sections, we were able to document the clear co-occurrence of the AMY 117 and Aβ antigens within the vast majority of nondiffuse plaque lesions in AD cortex and hippocampus. The degree of AMY 117 IR within Aβ plaques varied from plaque to plaque, but importantly, the two antigens very frequently existed in the same lesion. AMY 117 IR was primarily associated with more rounded, compacted Aβ plaques, whereas its co-localization with large diffuse Aβ deposits was rare. The images in Figure 1 ▶ indicate that AMY 117 and Aβ can both co-mingle and specifically co-localize (eg, overlap) within an individual plaque. In a subset of more mature plaques, AMY 117 IR surrounds that of Aβ; in such cases, the two antigens may partially overlap or may segregate but abut each other. AMY 117 IR was not diminished by absorption of the antibody with Aβ peptide, suggesting that AMY 117 is a non-Aβ antigen. In general, the Aβ antigen was more abundant than the AMY 117 antigen in AD, aged human, and DS brains.
The discrepancy between our results and those reported earlier, 15 in which limited or sometimes no co-localization was described between AMY 117 and Aβ IR in AD brain sections, is probably due, in part, to differences in staining conditions. First, the pretreatments used in our study enhanced the Aβ IR, sometimes even in the ethanol-fixed sections, implying that some Aβ deposits may have been missed in the study by Schmidt et al. 15 Second, the Aβ42 MAb 21F12 and the Aβ polyclonal antibody R1282 are extremely sensitive at detecting multiple forms of Aβ deposits and, as such, allowed greatly increased detection of Aβ deposits relative to those detected by the polyclonal Aβ antibody 2332 used in the previous study. 15 Indeed, we have performed side-by-side comparisons of the three antibodies under the optimal staining conditions for each and have consistently found Aβ42 MAb 21F12 to detect the greatest number of Aβ deposits (C. A. Lemere and T. J. Grenfell, unpublished data). Our Aβ polyclonal antibody R1282 also detected more Aβ deposits than polyclonal antibody 2332 under optimal conditions but, in some cases, labeled fewer Aβ deposits relative to those stained by Aβ42 MAb 21F12. Third, in our study, we always single-labeled sections adjacent to the double-labeled sections so as to characterize the staining pattern for each antibody on its own to avoid the potential for steric competition between the antibodies for their respective antigens within lesions. Both in our hands, and recently in those of M. L. Schmidt (personal communication), a competition between Aβ and AMY 117 antibodies for their respective antigens in individual plaque lesions was observed and may explain why some AMY-positive plaques appeared to be Aβ negative in their study.
In full agreement with the earlier report, no AMY 117 IR was observed in Aβ-bearing blood vessels or in cerebellum, implying that there is a regional and cellular specificity for this protein to associate with Aβ. In our study, AMY 117 IR occurred only in those brain areas having Aβ immunoreactive plaques and never in regions devoid of Aβ. In contrast, low-magnification photomicrographs previously published by Schmidt and colleagues 15 (as shown in Figure 3 ▶ of their paper) illustrate an example of an AD case in which AMY 117 IR was detected in a region of parahippocampal gyrus devoid of Aβ using polyclonal antibody 2332. Upon request, the authors kindly provided us with several adjacent sections from the same block of tissue depicted in the figure. In our hands, the region previously described as being devoid of Aβ was Aβ42 plaque-rich using a different Aβ antibody (Aβ42 MAb 21F12) and pretreatment of the tissue with formic acid. Pretreatment of tissue and improved Aβ antibody sensitivity may account for the increased detection of Aβ deposits and, as such, the visualization of a much greater co-occurrence of Aβ with AMY 117 in the current study. Our conclusion is that regions of AMY IR are Aβ-rich. Careful inspection of Table 1 in the aforementioned paper confirms that in AD brain regions where both antigens were examined (positive staining listed as present (Y) or absent (N)), Aβ IR alone or Aβ and AMY 117 IR together were described, but not AMY 117 IR alone. 15 In the current study, small, punctate AMY 117 immunoreactive deposits that were not directly associated with Aβ IR were occasionally observed, but only in areas of abundant Aβ immunoreactive plaques. In general, these small, punctate AMY 117 deposits were located between and close to plaques and, as such, may represent the outer edge of a plaque that exists above or below the plane of the AMY-117-stained section, so that Aβ may be surrounded by the AMY 117 antigen (as we indeed observed in mature plaques (Figure 1, d–f ▶ , arrows)) or the two antigens partially overlap (exemplified by Figure 1, d–f ▶ , arrowheads).
By performing immunohistochemical studies of plaque development in a unique temporal series of 29 DS brains from patients between the ages of 12 and 73 years, we were able to determine that Aβ deposition clearly precedes that of the AMY 117 antigen. Early, diffuse Aβ42 plaques in young DS brains were AMY 117 negative; it was only after the appearance of more mature, compacted Aβ plaques that AMY 117 IR was observed. This point was best exemplified by the immunostaining of adjacent sections from a single block of brain tissue containing both temporal cortex and hippocampus from a 16-year-old DS patient (see Figure 3, c–f ▶ ). Diffuse Aβ42 IR was detected throughout the temporal cortex, whereas more compacted Aβ42 IR plaques were seen in the hippocampus in the same section. In the adjacent section, AMY 117 IR was observed only in the more compacted plaques in the hippocampus; the temporal cortex in the same section was entirely AMY 117 negative. We cannot exclude the possibility that the AMY 117 antigen within diffuse plaques (but not compacted plaques) was destroyed by the harsh (long-term) fixation conditions or by the double pretreatment to expose antigens in the young DS brains. However, the lack of AMY 117 IR in the long-term-fixed young DS brains bearing exclusively diffuse Aβ42 IR is consistent with the lack of AMY 117 IR in diffuse Aβ42 IR deposits observed in briefly fixed tissues from middle-age and older DS patients (see Figure 2, e and f ▶ ) and in AD cases. Furthermore, the staining protocol for each antibody under each of the fixation conditions was optimized before use. Therefore, we believe it is very unlikely that technical factors could explain the lack of AMY 117 IR in the aforementioned young DS brains.
The pattern of AMY 117 IR seen in the young DS brains was also observed in briefly fixed aged human control brains in which only three of eight Aβ-bearing brains showed any AMY 117 IR, and then only in a small portion of all Aβ plaques. Again, it was the Aβ plaques that appeared to be in the process of compaction that showed AMY 117 IR. Neither AMY 117 nor Aβ IR were observed in two aged human control brains and in six young DS brains, lending further support to the conclusion that the AMY 117 antigen is not detectable in brain lesions before the appearance of Aβ.
Brains from two animal models of AD pathogenesis, aged monkey (17 to 24 years) and PD-APP transgenic mice (aged 8 to 20 months), were examined for Aβ and AMY 117 IR. Aβ IR deposits were observed in all of the monkey and mouse brains, and the number of Aβ deposits increased strikingly with age. AMY 117 IR was not detected in either species, regardless of Aβ plaque burden. The lack of AMY 117 IR in monkey and PD-APP transgenic mouse brain suggests two possibilities. First, a species difference in the AMY 117 antigen may make it unrecognizable by the human MAb. Second, insufficient maturation of the plaques in monkeys and transgenic mice, compared with that in AD brain, may be responsible for the lack of AMY 117 detection in the plaques of these animals.
In summary, we conclude that the AMY 117 antigen is a non-Aβ amyloid-associated protein that accrues in AD plaques after Aβ deposition, rather than existing as the subunit of a novel, Aβ-negative lesion in Alzheimer’s disease. Because AMY 117 appears at the time of compaction of Aβ plaques, it may turn out to play a significant role in the evolution of the plaque. This new information is critical to the interpretation of AMY IR plaques in AD brain and to the further search for the AMY antigen.
Acknowledgments
We are indebted to Dr. Marie Luise Schmidt (The Center for Neurodegenerative Disease Research, University of Pennsylvania School of Medicine, Philadelphia, PA) for providing the confocal image shown in Figure 1 ▶ and to Drs. Schmidt and John Trojanowski for many interesting and helpful discussions. In addition, we are grateful to Drs. Krystyna Wisniewski (Institute for Basic Research in Developmental Disabilities, Staten Island, NY) and Douglas Anthony (Department of Pathology, Harvard Medical School, Boston, MA) for generously providing brain tissues from young DS patients. Lastly, we thank Dr. Dora Games and Karen Khan for kindly providing fixed PD-APP transgenic mouse brains.
Footnotes
Address reprint requests to: Dr. Cynthia A. Lemere, Center for Neurologic Diseases, Harvard Institutes of Medicine Room 622, 77 Avenue Louis Pasteur, Boston, MA 02115. E-mail: lemere@cnd.bwh.harvard.edu.
Supported by NIH grant (AG06173).
References
- 1.Selkoe DJ: Amyloid β-protein and the genetics of Alzheimer’s disease. J Biol Chem 1996, 271:18295-18298 [DOI] [PubMed] [Google Scholar]
- 2.Wisniewski K, Wisniewski H, Wen G: Occurrence of neuropathological changes and dementia of Alzheimer’s disease in Down’s syndrome. Ann Neurol 1985, 17:278-282 [DOI] [PubMed] [Google Scholar]
- 3.Rumble B, Retallack R, Hilbich C, Simms G, Multhaup G, Martins R, Hockey A, Montgomery P, Beyreuther K, Masters CL: Amyloid A4 protein and its precursor in Down’s syndrome and Alzheimer’s disease. N Engl J Med 1989, 320:1446-1452 [DOI] [PubMed] [Google Scholar]
- 4.Mann DMA, Brown A, Prinja D, Davies CA, Landon M, Masters CL, Beyreuther K: An analysis of the morphology of senile plaques in Down’s syndrome patients of different ages using immunocytochemical and lectin histochemical techniques. Neuropathol Appl Neurobiol 1989, 15:317-329 [DOI] [PubMed] [Google Scholar]
- 5.Lemere CA, Blustzjan JK, Yamaguchi H, Wisniewski T, Saido TC, Selkoe DJ: Sequence of deposition of heterogeneous amyloid β-peptides and Apo E in Down syndrome: implications for initial events in amyloid plaque formation. Neurobiol Dis 1996, 3:16-32 [DOI] [PubMed] [Google Scholar]
- 6.Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina H, Ihara Y: Visualization of Aβ42(43) and Aβ40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is Aβ42(43). Neuron 1994, 13:45-53 [DOI] [PubMed] [Google Scholar]
- 7.Iwatsubo T, Mann DM, Odaka A, Suzuki N, Ihara Y: Amyloid β protein (Aβ) deposition: Aβ42(43) precedes Aβ40 in Down syndrome. Ann Neurol 1995, 37:294-299 [DOI] [PubMed] [Google Scholar]
- 8.Dickson DW: The pathogenesis of senile plaques. J Neuropathol Exp Neurol 1997, 56:321-339 [DOI] [PubMed] [Google Scholar]
- 9.Abraham CR, Selkoe DJ, Potter H: Immunochemical identification of the serine protease inhibitor, α1-antichymotrypsin in the brain amyloid deposits of Alzheimer’s disease. Cell 1988, 52:487-501 [DOI] [PubMed] [Google Scholar]
- 10.Ma J, Yee A, Brewer HB, Jr, Das S, Potter H: Amyloid-associated proteins α1-antichymotrypsin and apolipoprotein E promote the assembly of the Alzheimer β-protein into filaments. Nature 1994, 372:92-94 [DOI] [PubMed] [Google Scholar]
- 11.Eikelenboom P, Hack CE, Rozemuller JM, Stam FC: Complement activation in amyloid plaques in Alzheimer’s dementia. Virchows Arch B Cell Pathol 1989, 56:259-262 [DOI] [PubMed] [Google Scholar]
- 12.Eikelenboom P, Stam FC: Immunoglobulins and complement factors in senile plaques: an immunoperoxidase study. Acta Neuropathol 1982, 57:239-242 [DOI] [PubMed] [Google Scholar]
- 13.Rogers J, Webster S, Lue L-F, Brachova L, Civin WH, Emmerling M, Shivers B, Walker D, McGeer P: Inflammation and Alzheimer’s disease pathogenesis. Neurobiol Aging 1996, 17:681-686 [DOI] [PubMed] [Google Scholar]
- 14.Zhan S-S, Veerhuis R, Kamphorst W, Eikelenboom P: Distribution of beta amyloid associated proteins in plaques in Alzheimer’s disease and in the non-demented elderly. Neurodegeneration 1995, 4:291-297 [DOI] [PubMed] [Google Scholar]
- 15.Schmidt ML, Lee VM-Y, Forman M, Chiu T-S, Trojanowski JQ: Monoclonal antibodies to a 100-kd protein reveal abundant Aβ-negative plaques throughout gray matter of Alzheimer’s disease brains. Am J Pathol 1997, 151:69-80 [PMC free article] [PubMed] [Google Scholar]
- 16.Dickson DW: Discovery of new lesions in neurodegenerative diseases with monoclonal antibody techniques. Is there a non-amyloid precursor to senile plaques? Am J Pathol 1997, 151:7-11 [PMC free article] [PubMed] [Google Scholar]
- 17.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagopian S, Johnson-Wood K, Khan K, Lee M, Leibowitz P, Lieberburg I, Little S, Masliah E, McConlogue L, Montoya-Zavala M, Mucke L, Paganini L, Penniman E, Power M, Schenk D, Seubert P, Snyder B, Soriano F, Tan H, Vitale J, Wadsworth S, Wolozin B, Zhai J: Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature 1995, 373:523-527 [DOI] [PubMed] [Google Scholar]
- 18.Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL, Lieberburg I, Koo EH, Schenk D, Teplow DB, Selkoe DJ: Amyloid β-peptide is produced by cultured cells during normal metabolism. Nature 1992b, 359:322-325 [DOI] [PubMed] [Google Scholar]
- 19.Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, Khan K, Gordon M, Tan H, Games D, Lieberburg I, Schenk D, Seubert P, McConlogue L: Amyloid precursor protein processing and Aβ42 deposition in a transgenic mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA 1997, 94:1550-1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Selkoe DJ, Abraham CR, Podlisny MB, Duffy LK: Isolation of low-molecular-weight proteins from amyloid plaque fibers in Alzheimer’s disease. J Neurochem 1986, 146:1820-1834 [DOI] [PubMed] [Google Scholar]