

Abstract
A murine macrophage culture system that is both easy to employ and amenable to manipulation has been developed to study the cellular processes involved in AA amyloid formation. Amyloid deposition, as identified by Congo red-positive, green birefringent material, is achieved by providing cultures with recombinant serum amyloid A2 (rSAA2), a defined, readily produced, and highly amyloidogenic protein. In contrast to fibril formation, which can occur in vitro with very high concentrations of SAA and low pH, amyloid deposition in culture is dependent on metabolically active macrophages maintained in neutral pH medium containing rSAA2 at a concentration typical of that seen in acute phase serum. Although amyloid-enhancing factor is not required, its addition to culture medium results in larger and more numerous amyloid deposits. Amyloid formation in culture is accompanied by C-terminal processing of SAA and the generation of an 8.5-kd fragment analogous to amyloid A protein produced in vivo. Consistent with the possibility that impaired catabolism of SAA plays a role in AA amyloid pathogenesis, treatment of macrophages with pepstatin, an aspartic protease inhibitor, results in increased amyloid deposition. Finally, the amyloidogenicity exhibited by SAA proteins in macrophage cultures parallels that seen in vivo, eg, SAA2 is highly amyloidogenic, whereas CE/J SAA is nonamyloidogenic. The macrophage culture model presented here offers a new approach to the study of AA amyloid pathogenesis.
Reactive (secondary) amyloidosis is characterized by the extracellular deposition of protein fibrils (amyloid) containing predominantly amyloid A protein (AA), a proteolytically derived fragment of serum amyloid A (SAA). 1-4 SAA, an apolipoprotein synthesized by the liver, circulates in association with high-density lipoprotein particles and is normally present at very low levels. 5-7 As a major component of the acute phase response to injury, SAA is subject to hyperinduction, with plasma levels increasing as much as 1000-fold. 8-10 It is believed that persistent overproduction of SAA in response to chronic inflammatory disease coupled with impaired catabolism of SAA leads to assembly of the partially degraded N-terminal fragment (AA) into protease-resistant fibrils. Pathology results from displacement of normal tissue structure by the fibrillar deposits.
AA amyloidosis is the prototype of all forms of amyloidosis, a group of disparate diseases having in common the production of fibrillar protein aggregates, which in histological tissue sections bind Congo red and exhibit green birefringence under polarized light. 11 Although all amyloid fibrils have the physical characteristics of AA fibrils, they may be composed of any one of a number of plasma proteins that have, or can acquire, the requisite β-structure of amyloid. 12 Thus the essence of amyloidosis, rather than residing in a particular molecule, lies in the process of transforming a soluble, functional protein into insoluble, biologically inert fibrils. The steps or factors involved in this process have been the subject of intense investigation because, as a whole, the various forms of amyloidosis (eg, immunoglobulin light chain, reactive, hereditary, Alzheimers, prionoses) have a very significant impact on human health. 13,14
Although reactive amyloid is one of the rarer forms of amyloidosis, it is the form that has been studied most extensively and has given the greatest insight into the process termed amyloidogenesis. These advances have been possible principally because AA amyloidosis, unlike other forms of amyloidosis, occurs in most mammalian species, and, therefore, numerous animal models of this disease have been available for study. 15-17 In addition, AA amyloidosis involves most of the biological mechanisms deemed important to a greater or lesser extent in the various types of amyloidosis. These mechanisms include overproduction of the fibril precursor protein, incomplete catabolism and/or proteolytic processing of the protein, acquisition of increased β-structure, and interaction with other substances involved in fibril stability. While many of these factors have been clarified by the use of animal models of AA amyloid, new questions leading to a more thorough understanding of the amyloidogenic pathway have been raised. For instance, the amino acid sequence of SAA predicts little if any β-structure, which implies that SAA must undergo significant structural alteration to produce amyloid. 18 The physiological details of this transition, including the microenvironment in which SAA gains β-structure, have yet to be identified. Similar data concerning the carboxyl-terminal cleavage of SAA to AA are still lacking. It remains to be determined whether proteolytic processing occurs before or after fibril formation and whether this step contributes to fibril stability. 19
Another factor made obvious in studies employing the murine model of AA amyloidosis is the major impact that relatively minor changes in primary structure can have on the amyloidogenicity of proteins. Although most strains of mice, as well as most mammalian species, produce two nearly identical acute phase SAA proteins, 20-23 only one of them (mouse SAA2) is incorporated into amyloid fibrils. 24,25 Mice of the CE/J strain are unusual in that they produce only a single SAA whose primary structure is a hybrid of SAA1 and SAA2. 26 Although CE/J SAA differs from highly amyloidogenic SAA2 at only six of 103 positions, it does not form amyloid. 27
Studies of AA amyloid formation have also led to the concept that amyloidogenesis takes place as a biphasic process. 28 A slow preamyloid phase is believed to involve accumulation and nucleation of precursor protein, whereas fibril deposition occurs during the second phase. The duration of the first phase in experimental AA amyloid formation, and possibly in other forms as well, can be tremendously shortened by the administration of amyloid-enhancing factor (AEF). 29,30 This material, generally prepared as a glycerol extract of spleen from amyloidotic animals, has eluded characterization in terms of both its biochemical identity and its mechanism of action.
To address the various aforementioned issues at a cellular level and on the basis of numerous data implicating macrophages in the amyloidogenic pathway, 30-33 we have developed a murine peritoneal macrophage culture system that reproducibly demonstrates amyloid deposition in the presence of recombinantly expressed mouse SAA2 (rSAA2). This system is quite similar to the mesangial cell culture model previously developed for the study of AL amyloid formation. 34,35 In the latter model, AL amyloid deposition is achieved in cultures of mesangial cells isolated from human kidneys and incubated with amyloidogenic immunoglobulin light chains. Both systems represent models in which cellular activities can be manipulated and analyzed, and thereby introduce new approaches to the study of amyloid pathogenesis.
Materials and Methods
Collection and Culture of Resident Peritoneal Cells
Cells were collected by lavage from the peritoneal cavity of normal C57BL/6 mice. Briefly, 8 ml of collection medium (RPMI 1640 (Life Technologies, Grand Island, NY), 25 mmol/L HEPES, pH 7.0, and 1× antibiotic-antimycotic (Life Technologies)) was injected intraperitoneally, the abdomen was gently massaged, and fluid and cells were withdrawn. Cells were collected by centrifugation, resuspended at a concentration of 5 × 10 6 cells/ml, and plated at a density of 1.7 × 10 6 cells/well in 8-well chamber slides. Culture medium contained RPMI 1640, 2 mmol/L l-glutamine, 1× antibiotic-antimycotic, and 15% fetal calf serum (Hyclone Laboratories, Logan, VT). Cells were allowed to attach for 3 hours, after which wells were rinsed thoroughly to remove nonadherent cells. Adherent cells had a uniform, round morphology typical of freshly plated, nonactivated macrophages. Cells were maintained in 350 μl of culture medium per well at 37°C in an atmosphere of 5% CO2.
Experimental media were supplemented with 1) mouse rSAA2 at a final concentration of 140 μg/ml, 2) AEF at a final concentration of 12 μg/ml, or 3) mouse rSAA2 plus AEF. SAA (7 μl) was added directly to medium (350 μl) from a 7–10 mg/ml stock solution prepared by dissolving purified, lyophilized rSAA2 in 6 mol/L urea, 25 mmol/L HEPES, pH 7.2. Concentrations of stock solutions were adjusted after analysis by SDS-PAGE and densitometric quantitation of Coomassie blue-stained SAA bands. AEF stock solutions (2 mg/ml) were thoroughly mixed to resuspend precipitated material before withdrawal of an aliquot (2 μl) for addition to culture medium (350 μl). In experiments employing pepstatin, treatment of cells with this inhibitor was initiated at the same time as treatment with rSAA2. Pepstatin (Sigma Chemical Co., St. Louis, MO) was added to the culture medium to achieve a final concentrations of 5 μg/ml; stock solutions (100×) were prepared by dissolving pepstatin in dimethyl sulfoxide. All culture media were replaced every 2–3 days.
In some experiments, cells were fixed with formalin before they received rSAA2 and AEF. These cells were first rinsed four times with serum-free RPMI and then covered with 350 μl of phosphate-buffered 10% formalin for 10 minutes. After four rinses to remove the formalin, rSAA2 and AEF were added as described above. The same procedure was used to fix cells that had already initiated amyloid formation by a 5-day treatment with rSAA2 and AEF.
Culture of L-Cells and IC-21 Cells
Frozen stocks were obtained from the American Type Culture Collection (Manassas, VA) and maintained according to the supplier’s recommendations. Both lines grew as adherent monolayers and were passaged when cells reached confluence. For subculturing, L-cells were released from flasks by trypsinization, and IC-21 cells were released by incubation in calcium/magnesium-free phosphate-buffered saline (PBS) and then striking flasks against the palm of the hand. To test each line for amyloid formation, cells were cultured in 350 μl of medium in 8-well chamber slides, grown to 80% confluence, and then treated with rSAA2 (140 μg/ml) or with rSAA2 (140 μg/ml) and AEF (12 μg/ml).
Production and Purification of rSAA
Mouse rSAA2, rSAA1, and rSAA corresponding to the SAA in mice of the CE/J strain (CE/J SAA) were produced in Escherichia coli BL834 cells, using the pET-21a vector expression system (Novagen, Madison, WI) as previously described. 36 Purification from E. coli lysates was accomplished by Sepharose CL-6B chromatography in 4 mol/L guanidine, 0.05 mol/L Tris-HCl, pH 8.2, followed by chromatofocusing over a range of pH 8 to pH 5 in 6 mol/L urea. 36 Final preparations were precipitated in ammonium sulfate (80% saturated), extensively dialyzed against water, and lyophilized.
Preparation of AEF
AEF was prepared from the spleens of amyloidotic mice that had received daily subcutaneous injections of casein (0.5 ml of a 10% solution) for 25–40 days. AEF was extracted by homogenizing spleens in 8 volumes of 4 mol/L glycerol, 0.01 mol/L Tris-HCl, pH 7.8. Spleen extracts were incubated at 4°C with shaking and then centrifuged at 250,000 × g for 3 hours. 29 The supernatant was dialyzed against PBS and stored frozen at −20°C until use. When thawed, these preparations contained insoluble material.
Latex Bead Uptake
The uptake of blue-dyed polystyrene latex beads, 0.8 μm in diameter, (Sigma) was examined to assess phagocytic capability. Beads were suspended in serum-free RPMI at a concentration of 7.2 × 108/ml. Cells were rinsed three times with serum-free RPMI and then incubated for 30 minutes with 300 μl of the bead suspension. During the incubation, chamber slides were tightly wrapped in Parafilm and shaken gently in a water bath at 37°C. The bead suspension was removed, and cells were rinsed a minimum of 10 times with RPMI. After immersing the slides in two changes of water, cells were fixed with formalin and stained with hematoxylin.
Lysozyme Assay
Secreted lysozyme activity was measured as a marker of macrophage function. Adherent peritoneal cells were treated with rSAA2, AEF, or both for 48 hours as described above. Medium was then collected from cells and centrifuged at 14,000 × g for 5 minutes at 4°C; the supernatant was assayed for lysozyme activity. Micrococcus lysodeikticus (Sigma) was used as the substrate; the lyophilized bacteria were suspended in complete culture medium at a concentration of 1.5 mg/ml. The reaction was initiated by mixing 300 μl of medium collected from cells with 300 μl of M. lysodeikticus suspension and immediately determining the A450 of the mixture (0 time). Clearance of the suspension was followed by determining the A450 at 5-minute intervals over the course of 30–60 minutes. A unit of lysozyme was defined as a change in the A450 of 0.001 per minute at pH 6.25 at 22°C. The cells from which medium was collected were lysed in a solution of 50 mmol/L Tris, pH 8.0, 10 mmol/L EDTA, and 0.2% (w/v) sodium dodecyl sulfate (SDS). DNA was isolated from cell lysates by phenol-chloroform extraction and ethanol precipitation and quantified spectrophotometrically. Units of lysozyme were normalized to μg of DNA per well. Values obtained from two separate cultures were averaged; the two values were within 15% of each other.
Congo Red Staining
Cells were fixed in ice-cold 100% methanol and then stained for 45 minutes with Congo red prepared in alkaline 80% ethanol. 37 After several quick dips in water, slides were immersed in hematoxylin for 2 minutes. They were then dipped once in acidified 70% ethanol, several times in water, and once in a 1% solution of NaOH. Dehydration was accomplished by washing sequentially in 95% ethanol and 100% ethanol; slides were cleared in xylene, and coverslips were applied using Permount. The extent of Congo red staining was scored by visual examination (+1 to +5). Medium collected from amyloid-forming cultures was applied to microscope slides either by cytospinning or by direct application of whole medium or medium after centrifugation into soluble and pelletable fractions. After air-drying and gentle heat fixation to promote adherence to the glass slide, medium samples were fixed and stained with Congo red as described above.
In Situ Staining with Trypan Blue
Exclusion of trypan blue was used as an indication of cell viability. Before staining, cells were rinsed three times with serum-free RPMI. They were then covered for 1 minute with a solution of 2% (w/v) trypan blue in PBS. 38 Immediately after the trypan blue was removed, cells were fixed with 4% (w/v) paraformaldehyde, pH 7.5, for 10 minutes at room temperature, rinsed four times with PBS with gentle shaking, and examined microscopically before staining with Congo red.
SDS-PAGE, Immunoblotting, and N-Terminal Sequence Analysis
To analyze the SAA components of amyloid produced in cell cultures, cell layers were rinsed three times with serum-free RPMI, scraped into PBS, pelleted by centrifugation, and solubilized in SDS sample buffer. For analysis of SAA in culture medium collected from cells, 15 μl aliquots of medium were added directly to SDS sample buffer. Samples were subjected to tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis (tricine-SDS-PAGE) as previously described. 39 Separating, spacing, and stacking layers contained 16.5%, 10%, and 4% polyacrylamide, respectively. Immunoblotting was performed as described previously. 40 Rabbit anti-SAA antiserum (diluted 1:1000) generated against purified mouse rSAA2 produced in insect Sf9 cells from a recombinant baculovirus served as the primary antibody. 40 Two detection systems were employed. One system utilized goat anti-rabbit immunoglobulin conjugated to alkaline phosphatase (diluted 1:1000) (Biosource International, Camarillo, CA) as the second antibody. Color was developed by incubation with the alkaline phosphatase substrate 5-bromo-4-chloro-3-indolyl phosphatenitroblue tetrazolium (BCIP)/NZT) (Bio-Rad Laboratories, Richmond, CA). In the other system, ECL Western blotting detection reagents (Amersham Life Science, Buckinghamshire, England) were used according to the manufacturer’s recommendations. Horseradish peroxidase-labeled donkey anti-rabbit immunoglobulin (diluted 1:1000) served as the second antibody. For N-terminal sequence analysis, proteins were transferred electrophoretically after SDS-PAGE onto polyvinylidene difluoride (PVDF) membranes (ProBlott, Applied Biosystems, Foster City, CA). Amino-terminal sequencing was carried out using an Applied Biosystems model 473A protein sequencer.
Immunocytochemistry
Cells were rinsed four times with PBS, fixed in 10% formalin, and permeabilized by immersion in 0.1% Triton X-100 in PBS for 4 minutes. Endogenous peroxidase was quenched by a 20-minute incubation in 1% (v/v) hydrogen peroxide in methanol. Immunostaining was performed using a Vectastain ABC Kit (Vector Laboratories, Burlingame, CA). Cells were washed with PBS and blocked with diluted goat serum in PBS for 20 minutes at room temperature. Incubation with the primary antibody (rabbit anti-mouse rSAA2, 1:5000 dilution) was carried out for 1 hour at room temperature. Cells were then washed with PBS, incubated with the second antibody (biotinylated goat anti-rabbit antiserum) for 30 minutes at room temperature, washed with PBS, and incubated with ABC reagent for 45 minutes and then with substrate for 6 minutes. The substrate for horseradish peroxidase was prepared using Fast DAB and urea H2O2 tablets (Sigma). Cells were counterstained with hematoxylin.
Results
General Description of Amyloid Deposition
SAA-derived amyloid deposition has been characterized in cultures of adherent murine peritoneal cells maintained in the presence of mouse rSAA2 (140 μg/ml). Cultures developed foci of amyloid, which enlarged and became increasingly Congophilic and birefringent with time. The earliest deposition occurred in cultures incubated with AEF in addition to rSAA2 and was observed as early as 24–48 hours after treatment. The amyloid masses in AEF-treated cultures were larger and more numerous than those in cultures treated with SAA only (Figure 1, A–D) ▶ . Most, if not all, of the Congo red-positive material remained attached to chamber slides throughout the culture period (2–24 days). Medium collected from amyloid-forming cultures was routinely negative for the presence of Congo red positive-material. Amyloid appeared to adhere via contact with cells rather than the slide surface. Sequential staining first in situ with trypan blue and then with Congo red after fixation revealed that the vast majority of cells, including those associated with amyloid masses, were viable. Congo red staining was never observed in cultures maintained in the absence of rSAA2, with or without AEF (data not shown). In addition, wells that contained culture medium and rSAA2 with or without AEF, but lacked cells, did not develop Congo red-positive material either on the surface of the slide or in the medium.
Figure 1.

Amyloid deposition in cultures of adherent peritoneal cells. Panels A, C, E and G show Congo red and hematoxylin staining. Panels B, D, F and H show green birefringence in A, C, E and G respectively, under polarized light. Magnification, ×200. A and B: Cells derived from C57BL/6 mice and incubated with rSAA2 and AEF. C and D: Cells derived from C57BL/6 mice and incubated with rSAA2 without AEF. E and F: Cells derived from nude Balb/c mice and incubated with rSAA2 and AEF. G and H: Cells derived from amyloidotic C57BL/6 mice and incubated with rSAA2 and pepstatin (5 μg/ml).
The morphology of adherent peritoneal cells in cultures undergoing amyloid deposition varied among experiments. In some cultures, adherent cells in the absence of rSAA2 remained fairly uniform in shape and evenly distributed for as long as 24 days (Figure 2A) ▶ . Identical-looking cultures established from the same collection of peritoneal cells exhibited remarkable cell clustering after the addition of rSAA2. Clusters eventually evolved into aggregates of cells coated with or embedded in Congo red-positive material (Figure 2B) ▶ . In other experiments, adherent peritoneal cells developed over time into a pleomorphic population that included multinucleated giant cells, elongated cells, and nodular masses of cells, as well as round, typical macrophage-looking cells. When provided with rSAA2, these mixed cell cultures also exhibited amyloid deposition.
Figure 2.
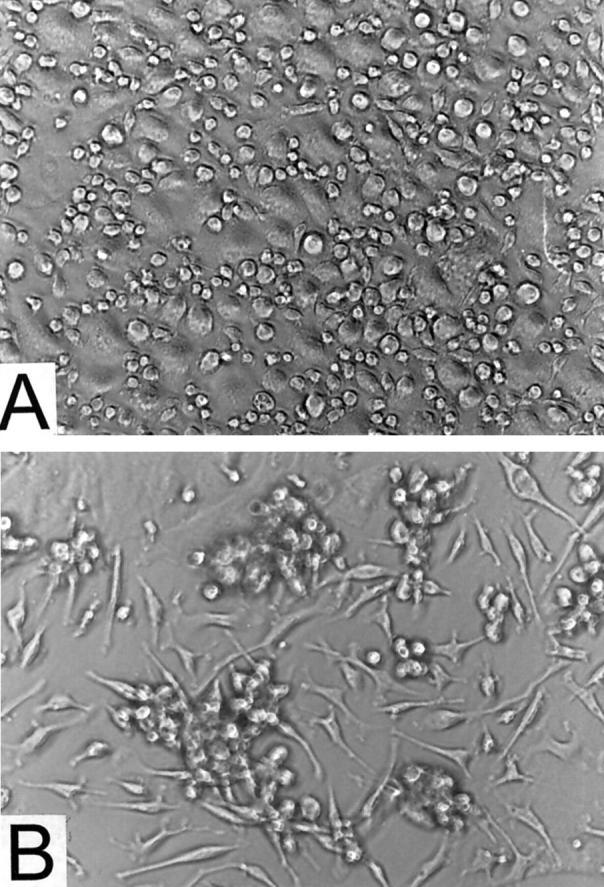
Cultures of adherent peritoneal cells viewed by phase-contrast microscopy. A: Cells maintained in the absence of rSAA2 and AEF. B: Cells incubated with rSAA2 and AEF; amyloid is seen at sites of clustered cells. Magnification, ×200.
Evidence Supporting a Key Role for Macrophages in Amyloid Formation
Although adherence-selected peritoneal cells are generally assumed to represent a macrophage population, cells undergoing amyloid deposition were examined to confirm that they exhibited properties characteristic of macrophages. Lysozyme activity was present in medium collected from adherent peritoneal cell cultures. After a 48-hour incubation, the level of secreted lysozyme activity was 7.4, 4.6, 3.3, and 2.8 units per μg DNA in untreated, rSAA2-treated, AEF-treated, and rSAA2/AEF-treated cultures, respectively. In addition, cells in amyloid-forming cultures demonstrated uptake of latex beads, indicative of phagocytic activity (Figure 3) ▶ .
Figure 3.

Adherent peritoneal cells that have phagocytosed latex beads. Cells were incubated with a suspension of latex beads (0.8-μm diameter) for 30 minutes at 37°C, rinsed, formalin-fixed, and stained with hematoxylin. Magnification, ×400.
Although it appeared that the vast majority of cells in amyloid-forming cultures were macrophages, a limited number of T cells or T-cell products also may have been present. To investigate the possibility that T cells or T-cell products were required for amyloid formation, adherent peritoneal cell cultures were established from nude BALB/c mice known to be immunosuppressed because of a lack of T cells. When provided with rSAA2 or with rSAA2 plus AEF, these cultures developed amyloid in the same manner as those established from immunocompetent mice (Figure 1, E and F) ▶ .
Further data supporting the specific involvement of macrophages in amyloid formation were obtained by testing established cell lines of different origin (fibroblast versus macrophage) for amyloid-forming capability. L-cells, a murine line that is derived from connective tissue and which grows as an adherent monolayer, were cultured under amyloid-forming conditions for 15 days (ie, confluent cultures maintained in the presence of 140 μg/ml rSAA2 and 12 μg/ml AEF) and then stained with Congo red. No Congo red-positive material was present either on the slide or in the medium. In contrast to these results, IC-21 cells, an SV-40-transformed line derived from C57BL/6 mouse peritoneal macrophages, demonstrated amyloid deposition after a 5-day treatment with rSAA2 and AEF.
Experiments were then performed to determine whether amyloid formation in culture occurred by a mechanism requiring metabolically active macrophages or by a process in which cells served merely as an anchor or substratum for fibril assembly. Cells were fixed in situ with formalin and then maintained under amyloid-forming conditions for 14 days. These cultures failed to develop Congo red-positive masses, whereas unfixed cultures treated in parallel with the same preparations of rSAA2 and AEF showed extensive amyloid deposition. Another set of cultures was incubated with rSAA2 and AEF for 5 days to achieve modest amyloid development, as judged by phase-contrast microscopy, and then fixed with formalin. Incubation with rSAA2 and AEF was resumed and continued until day 14. Although these cultures contained Congo red-positive foci, amyloid deposition had not progressed beyond that observed on day 5. All of the deposits in cultures fixed on day 5 were much smaller than those in parallel cultures maintained for 14 days without fixation, consistent with the hypothesis that metabolically active macrophages play an essential role in amyloid fibril formation.
Effect of Exogenously Added AEF on Amyloid Formation
To begin to probe the process of amyloid deposition and gain clues about the mechanism of AEF action, SAA was tracked immunochemically in culture medium and in the adherent cell layer during amyloid formation. Medium was collected from cultures after incubation with rSAA2 in the presence or absence of AEF and then examined by SDS-PAGE and Western analysis. As seen in samples collected on days 3 and 6, intact SAA persisted in medium lacking AEF (Figure 4 ▶ , lanes 2 and 4), whereas no immunodetectable SAA, neither intact nor fragments thereof, remained in AEF-supplemented medium (Figure 4 ▶ , lanes 3 and 5). Thus, in the presence of AEF, cellular adherence, uptake, and/or degradation of SAA was greatly enhanced.
Figure 4.

SDS-PAGE immunoblot comparing the fate of SAA in medium conditioned by cells in the absence of AEF versus medium conditioned by cells in the presence of AEF. Lane 1: Purified rSAA2. Lane 2: Medium supplemented with rSAA2, conditioned by cells during days 1–3. Lane 3: Medium supplemented with rSAA2 and AEF, conditioned by cells during days 1–3. Lane 4: Same as lane 2, conditioned during days 4–6. Lane 5: Same as lane 3, conditioned during days 4–6.
Immunocytochemistry was performed to compare the amount of SAA associated with cells in the presence and absence of AEF. Cultures treated with rSAA2 and AEF for 2 days contained numerous immunostained masses (Figure 5A) ▶ . In contrast, parallel cultures treated solely with SAA contained few such areas (Figure 5B) ▶ . Higher magnification revealed that all immunostaining was associated with cells rather than randomly distributed on the slide and was present both intra- and extracellularly (Figure 5C) ▶ . Small immunopositive masses were seen within 2 hours of the first addition of rSAA2 and AEF (Figure 5D) ▶ ; their prominence well in advance of the appearance of Congo red-positive material may reflect the high sensitivity of immunodetection. It is more likely that these masses lack the β-sheet conformation of amyloid fibrils required for Congo red binding. No immunostaining was detected in rSAA2/AEF-treated cultures when immunocytochemistry was performed using nonimmune serum in place of anti-SAA antiserum. Cultures maintained in the absence of rSAA2 and AEF showed no immunostaining, suggesting a lack of endogenous SAA production.
Figure 5.
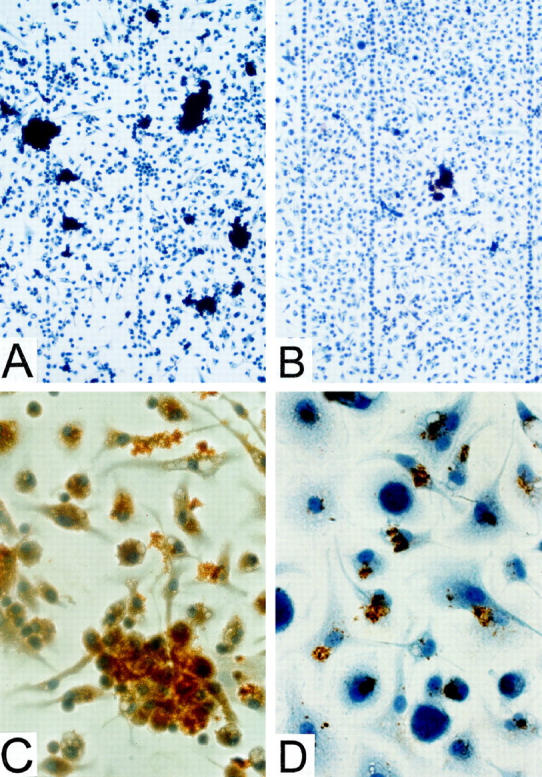
Immunochemical detection of SAA/AA in cultures of adherent peritoneal cells after incubation with rSAA2 +/− AEF. A: Cells cultured with rSAA2 and AEF for 48 hours; relatively high density of immunostained (dark brown) areas seen under low magnification, ×100. B: Cells cultured with rSAA2 for 48 hours in the absence of AEF; relatively low density of immunostained areas seen under low magnification, ×100. C: Cells cultured with rSAA2 and AEF for 48 hours; magnification, ×400. All immunostaining is in contact with cells. D: Cells cultured with rSAA2 and AEF for 2 hours; magnification, ×600.
Amyloid Deposition in Cultures of Adherent Peritoneal Cells from Amyloidotic Mice
Peritoneal macrophages collected from mice that had received a single injection of AEF 8 days earlier followed by seven daily injections of casein were cultured in the presence of rSAA2 without AEF supplementation. The rate of amyloid formation in these cultures, as well as the size and number of Congo red-positive foci, paralleled that seen in AEF-supplemented cultures established from nonamyloidotic mice, supporting the hypothesis that macrophages acquire AEF activity in vivo during amyloid formation.
Carboxyl-Terminal Cleavage of SAA
To determine whether SAA is C-terminally cleaved in culture as it is in vivo during amyloid formation, cultures were incubated with rSAA2 and AEF until they exhibited extensive amyloid deposition. The adherent material consisting of amyloid and cells was scraped from the wells and analyzed by tricine-SDS-PAGE. Coomassie blue staining of cell layer proteins revealed a relatively prominent band at nearly the same position as a human AA standard (∼8.4 kd) (Figure 6, A and C) ▶ . This band, as well as several bands at higher molecular mass positions (9, 10, and 12 kd), were shown by Western blot analysis to represent SAA peptides (Figure 6B) ▶ . Immunoreactivity was also detected in bands corresponding in position to dimeric forms of SAA and AA. The SAA identity of 8-, 9-, 10-, and 12-kd peptides was confirmed by N-terminal sequence analysis after transfer to PVDF membranes (Figure 6C ▶ , thin lines at right indicate sequenced peptides). All four bands gave the N-terminal sequence of murine SAA2, starting with glycine at residue 1, indicating that the parent SAA had undergone C-terminal cleavage.
Figure 6.

SDS-PAGE of cell layers harvested from amyloid-laden cultures showing C-terminal cleavage of SAA. Cultures were rinsed with RPMI, scraped into PBS, pelleted by centrifugation, and solubilized in SDS gel-loading buffer. A: Coomassie blue-stained PVDF membrane. Lane 1: human AA protein isolated from spleen. Lane 2: Cell layer with amyloid (14-day culture). Lane 3: purified rSAA2. B: Immunoblot of A, probed with anti-mouse SAA antiserum. Lane 1: cell layer containing SAA and AA peptides (dimers of SAA and AA are also present). Lane 2: Purified rSAA2. C: Coomassie blue-stained PVDF membrane. Lane 1: Human AA protein from spleen. Lane 2: Cell layer with amyloid (19-day culture). Thin lines at right indicate peptides subjected to N-terminal sequencing.
Enhanced Amyloid Deposition in Peritoneal Cell Cultures Incubated with Pepstatin
In previous studies, mice given pepstatin in conjunction with casein exhibited increased amyloid deposition relative to mice treated with casein only. 41 To determine if pepstatin, an inhibitor of aspartic proteases such as cathepsin D and cathepsin E, also enhanced AA deposition in macrophage cultures, medium was supplemented with pepstatin (5 μg/ml) in addition to rSAA2. Although a complete dose-response curve has not been established, cultures treated with pepstatin at a concentration of 5 μg/ml exhibited a more extensive pattern of Congo red staining than cultures not treated with pepstatin and, at the same time, maintained viability equivalent to that of untreated cultures, even after as long as 24 days. Pepstatin-treated cultures developed large areas of diffuse, pinkish-red staining in addition to dense, localized masses of amyloid associated with cell clusters (Figure 1, G and H) ▶ . In general, these amorphous amyloid deposits produced intense yellow-green birefringence and occurred in areas where the cells were evenly dispersed rather than clustered. A graded evaluation of the relative amount of amyloid deposition in three independent experiments, each employing a different preparation of rSAA2, yielded scores of (+4 versus +2), (+3 versus +2), and (+3 versus +1) for pepstatin-treated cultures versus cultures lacking the inhibitor.
Inability of Other Murine rSAAs to Form Amyloid
It is generally accepted that in vivo murine amyloid deposits are derived exclusively from SAA2, whereas SAA1 and CE/J SAA do not appear to be capable of fibril formation. To determine if this is also true for amyloid deposition in cell culture, peritoneal macrophages were treated with purified mouse rSAA1 and AEF under conditions identical to those used with rSAA2 and AEF. These cultures exhibited a relatively modest degree of cell clustering and limited SAA/AA deposition, as judged by immunocytochemical staining, but did not develop Congo red-positive material. Cultures treated in the same way with CE/J SAA or a mutant of CE/J SAA containing glycine in place of histidine at position 7 (His7Gly) also failed to produce Congo red-positive deposits. However, peritoneal macrophages derived from either C57BL/6 or CBA/J mice and treated with equal amounts of rSAA2 and CE/J SAA (140 μg/ml of each) developed extensive Congo red-positive masses and were in no way protected against amyloid deposition. N-terminal sequence analysis of the amyloid harvested from cultures maintained in the presence of the two SAAs revealed that it was exclusively composed of SAA2-derived peptides.
Discussion
A cell culture system that is both easy to employ and amenable to manipulation has been developed for studying AA amyloid pathogenesis and, in particular, elucidating the cellular processes involved in amyloid fibril formation. Murine peritoneal macrophages cultured in fetal calf serum-supplemented RPMI medium serve as the cellular component of this system, while murine rSAA2, readily produced with the pET–E. coli expression system, is provided as the amyloidogenic precursor protein. Macrophages treated with rSAA2 at a concentration typical of that present in acute-phase serum (140 μg/ml) exhibit amyloid deposition within 5 days of SAA treatment.
The mechanism by which amyloid fibril formation takes place is an area of intense investigation. The findings presented here indicate that macrophages play an essential role in this process in the case of AA amyloidosis. Adherent peritoneal cells before or in the process of amyloid production were shown to secrete lysozyme and take up latex beads, verifying that these cells indeed behave like macrophages. The demonstration that amyloid deposition occurred in cultures of peritoneal cells obtained from nude mice indicates that T lymphocytes are not essential for fibril formation. Evidence thus far obtained supports a specific role for macrophages as opposed to other cell types. L cells, an established line of connective tissue origin, were incapable of amyloid formation, whereas IC-21 cells, derived from peritoneal macrophages, demonstrated amyloid deposition analogous to that occurring in primary cultures of peritoneal macrophages. No amyloid deposition occurred in the absence of macrophages or with cells formalin-fixed before incubation with rSAA2 and AEF. Furthermore, cultures that had developed small amyloid masses did not show further deposition after they were fixed with formalin, suggesting an ongoing requirement for viable macrophages during amyloid progression.
Although the exact function served by macrophages in AA amyloid formation remains to be determined, roles in concentrating SAA and inducing structural alterations in SAA seem likely based on current models of amyloid fibrillogenesis. It has been proposed that the polymerization of amyloidogenic proteins, including SAA, into fibrils is a nucleation-dependent process that can only occur above a critical protein concentration. 42-44 According to this mechanism, formation of amyloid fibrils under physiological conditions requires that either the effective concentration is increased, possibly through entrapment in a subcellular compartment or through adsorption, or that the critical concentration is decreased through interaction of the amyloidogenic protein with intra- or extracellular components. In the studies described here, the concentration of SAA in medium was 140 μg/ml, similar to that found in serum during an acute-phase response and well below that used previously to achieve amyloid fibrils in vitro in the absence of cells (10 mg/ml). 45 Based on this study and on recently reported findings, 46 it is reasonable to propose that macrophages concentrate SAA through binding and uptake in clathrin-coated vesicles. Consistent with this hypothesis, SAA has been shown previously to bind to macrophages in a specific, saturable fashion. 47 The immunochemical analyses described here provide further evidence for intracellular uptake of SAA. Peritoneal macrophages treated with rSAA2 demonstrated intracellular immunostaining for SAA, whereas cultures lacking exogenous rSAA2 were not immunostained.
Internalized SAA is likely to be trafficked through cells via the endosomal network. 33,46 While some or even most of the SAA is expected to be completely degraded, a portion may not progress to catabolic lysosomes, but instead may undergo low pH-induced conformational changes. Such changes are presumed to be necessary for amyloid fibril formation, based on the prediction that SAA contains little or no β-sheet structure. 18 The idea that acidic conditions induce β-sheet structure stems from the observation that in vitro fibril formation from SAA can be achieved by using low pH conditions (pH 2.5). 45 In striking contrast, amyloid formation in peritoneal cell cultures occurs when rSAA2 is added to neutral pH culture medium, leading us to hypothesize that endosomes provide the acidic environment in which β-sheet structure is acquired. Immunochemical evidence for endosomal localization of SAA/AA, possibly in the form of nascent fibrils, has been reported previously. 31,33,48 The fact that amyloid in vivo, as well as in macrophage cultures, exists as extracellular deposits suggests that there must be a mechanism by which SAA/AA exits the cell. Double-staining studies using trypan blue followed by Congo red indicate that the vast majority of cells maintain viability in the midst of amyloid deposition and imply that active cellular processes, rather than cell death, serve to translocate SAA/AA to the extracellular space. Immunochemical analyses suggest that amyloid deposits in macrophage cultures originate at cell surfaces. This observation is in agreement with what has been described previously in an immunoelectron microscopic study of amyloid formation in murine spleen. 32 In that analysis, SAA reactivity was detected in granular form along cell membranes and in fibrillar bundles radiating from the surface of mononuclear cells.
Whether all SAA molecules must be internalized, processed, and exported before being deposited, or whether some can be added directly to extracellular amyloid remains to be determined. In vitro studies with amyloidogenic Alzheimer Aβ peptide suggest that the slow nucleation phase preceding fibril formation can be bypassed in the presence of preformed amyloid. 49,50 Solutions of Aβ seeded with amyloid fibrils polymerize rapidly according to first-order kinetics. 49 Possibly acting as “seeds” in vivo, AA fibrils extracted from amyloid deposits, as well as short synthetic peptides corresponding to various amyloidogenic proteins, have been shown to accelerate AA amyloid deposition when injected into experimental animals in conjunction with an inflammatory stimulus. 51,52 AEF, although undefined biochemically, is also known to accelerate amyloid deposition in mice and has been proposed to work by seeding amyloid formation. 29,53 Similar mechanisms may be operating in macrophage cultures, which demonstrate greatly enhanced amyloid formation when treated with AEF. The addition of AEF to cell cultures is accompanied by very rapid appearance of SAA deposits that are immunoreactive but not Congo red positive. From the appearance of these deposits in immunochemically stained cultures, we speculate that SAA may have collected at these sites via an adsorptive mechanism or possibly through specific interaction with “seeds” of β-sheet present in the AEF preparation. Further studies will address whether the early SAA deposits evolve into amyloid as defined by Congo red positivity or whether they serve as localized, concentrated sources of SAA that must be taken up and processed by cells before forming amyloid.
A mechanism of amyloid formation that bypasses the necessity for intracellular processing of SAA must take into account our observation that viable cells are required for growth and not just initiation of fibrils (ie, progression from small to large Congo red deposits was arrested by fixation of cells). We propose that macrophages not only facilitate structural transitions in SAA, but also produce and possibly secrete the protease(s) responsible for generating AA protein, most commonly consisting of the N-terminal 76 residues of SAA. We further speculate that C-terminal processing of SAA may be required to form stable β-sheet structures. It follows that if this protease, as well as its source, were inactivated by fixation, amyloid growth would terminate. Evidence for production of a C-terminal-specific protease was provided by SDS-PAGE and N-terminal sequence analysis of proteins harvested from amyloid-laden cultures. These studies revealed that although intact SAA (12 kd) had been added to the cultures, AA protein (8.5 kd) was a prominent component of amyloid-laden cultures.
The N-terminal portion of SAA has been shown to contain determinants critical for fibrillogenesis. 45 As demonstrated in vitro, removal of this region precludes amyloid fibril formation. 54,55 In this context, we have shown previously that pepstatin inhibits cleavage of SAA at specific sites in the N-terminal region, exactly the region required for amyloid fibril formation. 41 We have also shown that mice injected concurrently with pepstatin and casein exhibit enhanced amyloid deposition relative to mice treated with casein alone. 41 It is possible that increased in vivo deposition results from an increased pool of N-terminally intact SAA. By inhibiting SAA degradation, pepstatin could, in effect, increase the concentration of molecules which retain a capacity for amyloid fibril formation. The increased amount of amyloid deposition observed in macrophage cultures treated with pepstatin is consistent with this hypothesis. Using the macrophage culture system described here, we will be able to further investigate the role played by pepstatin-inhibitable cathepsins in normal SAA catabolism and determine whether modulation of their activity is a contributing factor in AA pathogenesis.
Footnotes
Address reprint requests to Dr. Barbara Kluve-Beckerman, Indiana University School of Medicine, Department of Medical and Molecular Genetics, 975 West Walnut Street, IB130, Indianapolis, IN 46202-5251. E-mail: bkluvebe@iupui.edu.
Supported by grants from the United States Public Health Service (DK-42657) and Veterans Affairs Medical Research (MRIS 583-0888).
References
- 1.Benditt EP, Eriksen N, Hermodsen MA, Ericsson LH: The major proteins of human and monkey amyloid substance: common properties including unusual N-terminal amino acid sequences. FEBS Lett 1971, 19:169-173 [DOI] [PubMed] [Google Scholar]
- 2.Levin M, Franklin EC, Frangione B, Pras M: The amino acid sequence of a major nonimmunoglobulin component of some amyloid fibrils. J Clin Invest 1972, 51:2773-2776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Husebekk A, Skogen B, Husby G, Marhaug G: Transformation of amyloid precursor SAA to protein AA and incorporation in amyloid fibrils in vivo. Scand J Immunol 1985, 21:283-287 [DOI] [PubMed] [Google Scholar]
- 4.Husby G, Marhaug G, Dowton B, Sletten K, Sipe JD: Serum amyloid A (SAA): biochemistry, genetics and the pathogenesis of AA amyloidosis. Amyloid Int J Exp Clin Invest 1994, 1:119-137 [Google Scholar]
- 5.Benson MD, Kleiner E: Synthesis and secretion of serum amyloid A (SAA) by hepatocytes in mice treated with casein. J Immunol 1980, 124:495-499 [PubMed] [Google Scholar]
- 6.Hoffman JS, Benditt EP: Secretion of serum amyloid protein and assembly of serum amyloid protein-rich high density lipoprotein in primary mouse hepatocyte culture. J Biol Chem 1982, 257:10518-10522 [PubMed] [Google Scholar]
- 7.Benditt EP, Eriksen N: Amyloid protein SAA is associated with high density lipoprotein from human serum. Proc Natl Acad Sci USA 1977, 74:4025-4028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McAdam KPW, Elin RJ, Sipe JD, Wolff SM: Changes in human serum amyloid A and C-reactive protein following etiocholanlolone-induced inflammation. J Clin Invest 1978, 61:390-394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lowell CA, Stearman RS, Morrow JF: Transcriptional regulation of serum amyloid A gene expression. J Biol Chem 1986, 261:8453-8461 [PubMed] [Google Scholar]
- 10.Sipe JD, Rokita H, de Beer FC: Cytokine regulation of the mouse SAA gene family. Mackiewicz A Kushner I Baumann H eds. Acute Phase Proteins: Molecular Biology, Biochemistry, and Clinical Applications. 1993, :pp 511-526 FL, CRC Press, Boca Raton [Google Scholar]
- 11.Cooper JH: Selective amyloid staining as a function of amyloid composition and structure. Histochemical analysis of the alkaline Congo red, standardized toluidine blue, and iodine methods. Lab Invest 1974, 31:232-238 [PubMed] [Google Scholar]
- 12.Glenner GG: Amyloid deposits and amyloidois. The β-fibrilloses. N Engl J Med 1980, 302:1283–1292, 1333–1343 [DOI] [PubMed]
- 13.Husby G: Amyloidosis and rheumatoid arthritis. Clin Exp Rheumatol 1985, 3:173-180 [PubMed] [Google Scholar]
- 14.Benson MD: Amyloidosis. ed 7 Scriver CR Beaudet AL Sly WS Valle D eds. The Metabolic and Molecular Bases of Inherited Disease, 1995, :pp 4159-4191 McGraw-Hill, New York [Google Scholar]
- 15.Skinner M, Shirahama JT, Benson MD, Cohen AS: Murine amyloid protein AA in casein-induced experimental amyloidosis. Lab Invest 1977, 36:420-427 [PubMed] [Google Scholar]
- 16.Kisilevsky R, Benson MD, Axelrad MA, Boudreau L: The effect of a liver protein synthesis inhibitor on plasma SAA levels in a model of accelerated amyloid deposition. Lab Invest 1979, 41:206-210 [PubMed] [Google Scholar]
- 17.Ali-Khan Z, Li W, Chan SL: Animal model for the pathogenesis of reactive amyloidosis. Parasitol Today 1996, 12:297-302 [DOI] [PubMed] [Google Scholar]
- 18.Turnell W, Sarra R, Glover ID, Baum JO, Caspi D, Baltz ML, Pepys MB: Secondary structure prediction of human SAA1. Presumptive identification of calcium and lipid binding sites. Mol Biol Med 1986, 3:387-407 [PubMed] [Google Scholar]
- 19.Kisilevsky R, Narindrasorasak S, Tape C, Tan R, Boudreau L: During AA amyloidogenesis is proteolytic attack on serum amyloid A a pre- or post-fibrillogenic event? Amyloid Int J Exp Clin Invest 1994, 1:174-183 [Google Scholar]
- 20.Yamamoto K-I, Migita S: Complete primary structures of two major murine serum amyloid A proteins deduced from cDNA sequences. Proc Natl Acad Sci USA 1985, 82:2915-2919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dwulet FE, Wallace DK, Benson MD: Amino acid structures of multiple forms of amyloid-related serum protein SAA from a single individual. Biochemistry 1988, 27:1677-1682 [DOI] [PubMed] [Google Scholar]
- 22.Sletten K, Husebekk A, Husby G: The primary structure of equine serum amyloid A (SAA) protein. Scand J Immunol 1989, 30:117-122 [DOI] [PubMed] [Google Scholar]
- 23.Marhaug G, Husby G, Dowton SB: Mink serum amyloid A protein. Expression and primary sequence based on cDNA sequences. J Biol Chem 1990, 265:10049-10054 [PubMed] [Google Scholar]
- 24.Hoffman JS, Ericsson LH, Eriksen N, Walsh KA, Benditt EP: Murine tissue amyloid protein AA. NH2-terminal sequence identity with only one of two serum amyloid protein (ApoSAA) gene products. J Exp Med 1984, 159:641-646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shiroo M, Kawahara E, Nakanishi I, Migita S: Specific deposition of serum amyloid A protein 2 in the mouse. Scand J Immunol 1987, 26:709-716 [DOI] [PubMed] [Google Scholar]
- 26.de Beer MC, de Beer FC, McCubbin WD, Kay CM, Kindy MS: Structural prerequisites for serum amyloid A fibril formation. J Biol Chem 1993, 268:20606-20612 [PubMed] [Google Scholar]
- 27.Sipe JD, Carreras I, Gonnerman WA, Cathcart ES, de Beer MC, de Beer FC: Characterization of the inbred CE/J mouse strain as amyloid resistant. Am J Pathol 1993, 143:1480-1485 [PMC free article] [PubMed] [Google Scholar]
- 28.Sipe JD, McAdam KPWJ, Uchino F: Biochemical evidence for the biphasic development of experimental amyloidosis. Lab Invest 1978, 38:110-114 [PubMed] [Google Scholar]
- 29.Axelrad MA, Kisilevsky R, Willmer J, Chen SJ, Skinner M: Further characterization of amyloid-enhancing factor. Lab Invest 1982, 47:139-146 [PubMed] [Google Scholar]
- 30.Shirahama T, Miura K, Ju ST, Kisilevsky R, Gruys E, Cohen AS: Amyloid-enhancing factor-loaded macrophages in amyloid formation. Lab Invest 1990, 62:61-68 [PubMed] [Google Scholar]
- 31.Takahashi M, Yokota T, Kawano H, Gondo T, Ishihara T, Uchino F: Ultrastructural evidence for intracellular formation of amyloid fibrils in macrophages. Virchows Arch [A] 1989, 415:411-419 [DOI] [PubMed] [Google Scholar]
- 32.Arai K, Miura K, Baba S, Shirasawa H: Transformation from SAA2-fibrils to AA-fibrils in amyloid fibrillogenesis: in vivo observations in murine spleen using anti-SAA and anti-AA antibodies. J Pathol (Lond) 1994, 173:127-134 [DOI] [PubMed] [Google Scholar]
- 33.Chan SL, Chronopoulos S, Murray J, Laird DW, Ali-Khan Z: Selective localization of murine ApoSAA1/SAA2 in endosomes-lysosomes in activated macrophages and their degradation products. Amyloid Int J Exp Clin Invest 1997, 4:40-48 [Google Scholar]
- 34.Tagouri YM, Sanders PW, Picken MM, Siegal GP, Kerby JD, Herrera GA: In vitro AL-amyloid formation by rat and human mesangial cells. Lab Invest 1996, 74:290-302 [PubMed] [Google Scholar]
- 35.Isaac J, Kerby JD, Russell WJ, Dempsey SC, Sanders PW, Herrera GA: In vitro modulation of AL-amyloid formation by human mesangial cells exposed to amyloidogenic light chains. Amyloid Int J Exp Clin Invest 1998, 5:238-246 [DOI] [PubMed] [Google Scholar]
- 36.Kluve-Beckerman B, Yamada T, Hardwick J, Liepnieks JJ, Benson MD: Differential plasma clearance of murine acute phase serum amyloid A proteins SAA1 and SAA2. Biochem J 1997, 322:663-669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Puchtler H, Sweat F, Levine M: On the binding of Congo red by amyloid. J Histochem Cytochem 1962, 10:355-364 [Google Scholar]
- 38.Perry SW, Epstein LG, Gelbard HA: In situ trypan blue staining of monolayer cell cultures for permanent fixation and mounting. BioTechniques 1997, 22:1020-1024 [DOI] [PubMed] [Google Scholar]
- 39.Schagger H, von Jagow G: Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range of 1 to 100 kDa. Anal Biochem 1987, 166:368-379 [DOI] [PubMed] [Google Scholar]
- 40.Kluve-Beckerman B, Song M, Benson MD, Liepnieks JJ: Recombinant murine serum amyloid A from baculovirus-infected insect cells: purification and characterization. Biochim Biophys Acta 1993, 1182:303-310 [DOI] [PubMed] [Google Scholar]
- 41.Yamada T, Liepnieks JJ, Benson MD, Kluve-Beckerman B: Accelerated amyloid deposition in mice treated with the aspartic protease inhibitor, pepstatin. J Immunol 1996, 157:901-907 [PubMed] [Google Scholar]
- 42.Harper JD, Lansbury PT: Models of amyloid seeding in Alzheimer’s disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu Rev Biochem 1997, 66:385-407 [DOI] [PubMed] [Google Scholar]
- 43.Lomakin A, Chung DS, Benedek GB, Kirschner DA, Teplow DB: On the nucleation and growth of amyloid β-protein fibrils: detection of nuclei and quantitation of rate constants. Proc Natl Acad Sci USA 1996, 93:1125-1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kisilevsky R, Fraser PE: Aβ amyloidogenesis: unique, or variation on a systemic theme? Crit Rev Biochem Mol Biol 1997, 32:361-404 [DOI] [PubMed] [Google Scholar]
- 45.Westermark GT, Engstrom U, Westermark P: The N-terminal segment of protein AA determines its fibrillogenic property. Biochem Biophys Res Commun 1992, 182:27-33 [DOI] [PubMed] [Google Scholar]
- 46.Rocken C, Kisilevsky R: Comparison of the binding and endocytosis of high-density lipoprotein from healthy (HDL) and inflamed (HDLSAA) donors by murine macrophages of four different mouse strains. Virchows Arch 1998, 432:547-555 [DOI] [PubMed] [Google Scholar]
- 47.Kisilevsky R, Subrahmanyan L: Serum amyloid A changes high density lipoprotein’s cellular affinity. A clue to serum amyloid A’s principal function. Lab Invest 1992, 66:778-785 [PubMed] [Google Scholar]
- 48.Shirahama TS, Cohen AS: Intralysosomal formation of amyloid fibrils. Am J Pathol 1975, 81:101-116 [PMC free article] [PubMed] [Google Scholar]
- 49.Jarrett JT, Berger EP, Lansbury PT: The carboxy terminus of the β amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer’s disease. Biochemistry 1993, 32:4693-4697 [DOI] [PubMed] [Google Scholar]
- 50.Naiki H, Nakakuki K: First-order kinetic model of Alzheimer’s β-amyloid fibril extension in vitro. Lab Invest 1996, 74:374-383 [PubMed] [Google Scholar]
- 51.Niewold TA, Gruys E, Kisilevsky R, Shirahama TS: Fibril amyloid enhancing factor (FAEF)-accelerated amyloidosis in the hamster is not dependent on serine esterase activity and mononuclear phagocytosis. Scand J Immunol 1991, 34:101-107 [DOI] [PubMed] [Google Scholar]
- 52.Johan K, Westermark G, Engstrom U, Gustavsson A, Hultman P, Westermark P: Acceleration of amyloid protein A amyloidosis by amyloid-like synthetic fibrils. Proc Natl Acad Sci USA 1998, 95:2558-2563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kisilevsky R, Gruys E, Shirahama T: Does amyloid enhancing factor (AEF) exist? Is AEF a single biological entity? Amyloid Int J Exp Clin Invest 1995, 2:128-134 [Google Scholar]
- 54.Yamada T, Kluve-Beckerman B, Liepnieks JJ, Benson MD: In vitro degradation of serum amyloid A by cathepsin D and other acid proteases: possible protection against amyloid fibril formation. Scand J Immunol 1995, 41:570-574 [DOI] [PubMed] [Google Scholar]
- 55.Patel H, Bramall J, Waters H, de Beer M, Woo P: Expression of recombinant human serum amyloid A in mammalian cells and demonstration of the region necessary for high-density lipoprotein binding and amyloid fibril formation by site-directed mutagenesis. Biochem J 1996, 318:1041-1049 [DOI] [PMC free article] [PubMed] [Google Scholar]