

Abstract
During the past several years, a panel of human tumor cell lines (predominantly ovarian) with acquired resistance to cisplatin, the orally bioavailable analogue JM216, and the structurally hindered analogue AMD473, has been established and characterized for underlying mechanisms of resistance. We have examined these resistant cell lines for gains and losses of DNA associated with the acquisition of resistance using the molecular cytogenetic technique of comparative genomic hybridization. Our comparison of three analogues has shown the most frequently observed changes to include amplification of 4q (5/7) and 6q (5/7), followed by amplification of 5q (3/7). We have defined four minimal common overrepresented regions, two each on 4q and 6q, which are potential loci of genes associated with platinum analogue resistance. Additional consistent abnormalities appear to be associated with cell lines sharing specific resistance mechanisms. For example, amplification of 12q was observed in the CH1 lines made respectively resistant to JM216 and AMD473 in which increased DNA repair appears to be a major mechanism of resistance for both agents. Hence, these comparative genomic hybridization studies have identified distinct chromosomal aberrations which may correlate with defined mechanisms of resistance and contain hitherto unrecognized genes that may provide targets for future therapeutic intervention.
The major thrust in anticancer therapeutic development is the identification of selective therapies against molecular targets. 1,2 The identification of molecular mechanisms of drug resistance has been expedited by the examination of cell lines with acquired resistance, using modern molecular techniques. These techniques include classical cytogenetics, differential display, fluorescent in situ hybridization (FISH), and the more modern approaches of comparative genomic hybridization (CGH) and spectral karyotyping (SKY).
CGH is a new technique used to examine an entire genome for variations in DNA sequence copy number 3 (amplifications and deletions). It does not require replicating cells and therefore produces results which are representative of the tumor as a whole and not just the dividing population. In contrast to FISH, it does not require a previous knowledge of genetic aberrations. It can be employed with DNA extracted from fresh tumor material or material that has been frozen, formalin-fixed, or paraffin embedded. Finally, in contrast to differential display, CGH provides information on the chromosomal location of the amplified or deleted region. We have used DNA from corresponding pairs of resistant and sensitive cell lines labeled with fluorochromes of different colors, eg, green and red. These two DNAs are hybridized simultaneously to metaphase spreads from control (normal) cells. Comparison of the ratio of red:green signal along each chromosome axis reveals regions of gain and loss between the sensitive and resistant cell lines.
CGH is currently being used to determine aberrations in solid tumors and hematological malignancies in order to identify changes common to particular sub-types of tumor. 4,5 For example, high-level amplifications of regions of chromosomes 17q and 20q have been identified in breast carcinoma. 6 In a few instances, these findings have correlated with prognostic significance, eg, the identification of amplification of the REL proto-oncogene in diffuse large cell lymphoma has been associated with progression of disease. 7
We have confirmed that genetic loci associated with known mechanisms of resistance show the corresponding chromosomal imbalance with CGH; for example, we have demonstrated that the CH1 cell line with acquired resistance to doxorubicin shows significant amplification of the P-glycoprotein (MDR1) gene (Figure 1) ▶ . Other authors have also shown amplification of this region of 7q21 in cell lines with classical multi-drug resistance using CGH 8 and reverse in situ hybridization. 9 Moreover, in a cell line with acquired resistance to etoposide known to be mediated through topoisomerase II, we have demonstrated deletions of both the topoIIα gene on chromosome 17q21–22 and the topoIIβ gene on chromosome 3p24–25.
Figure 1.
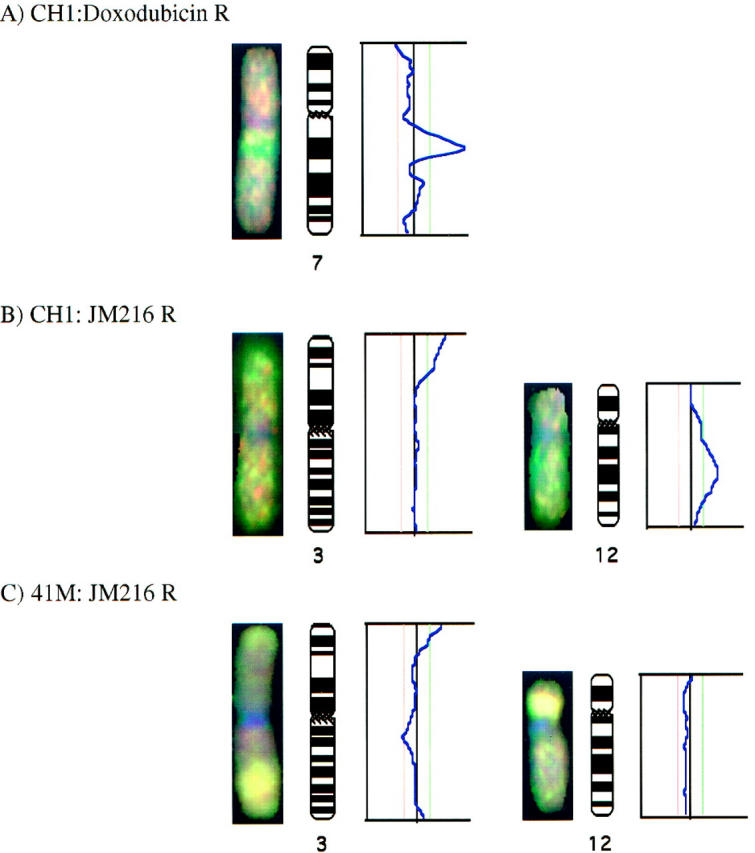
Individual chromosomes and their corresponding mean ratio profiles are shown for chromosomes with significant imbalances associated with acquired resistance to Doxorubicin in the CH1 cell line (A), JM216 in the CH1 cell line (B), and JM216 in the 41M cell line (C). The average of the red:green ratios along the axis for a number of copies of each chromosome is indicated by the blue line in each graph. The red and green dotted lines indicate the significance level (confidence limits of 99%) for 30% of the cells having the imbalance in a diploid population.
A panel of acquired cisplatin-resistant human tumor cell lines (predominantly ovarian) has previously been established and characterized for the underlying mechanisms of resistance to cisplatin in order to support our mechanism-directed approach to the circumvention of resistance. 10,11 Previous studies have indicated that cisplatin is able to circumvent acquired resistance due to reduced drug transport (eg, in the 41M:cisR line 12 ). Orr et al have also reported recently that JM216 (and other ammine/amine platinum (IV) dicarboxylates) are able to circumvent acquired resistance to cisplatin, carboplatin, and tetraplatin in murine L1210 leukemia sublines. 13 A study has been undertaken to determine whether cisplatin resistance mechanisms are also involved in acquired resistance to JM216 using two cell lines (41M and CH1) with derived resistance to JM216. Interestingly, and in contrast to its acquired cisplatin-resistant counterpart, JM216 resistance in the 41M cell line appeared to be due to enhanced glutathione levels, rather than drug transport. 14 In addition, following 2 years of exposure to JM216, only a relatively low level of resistance (1.9-fold) was achievable. These data suggest that acquired resistance to JM216 is less likely to occur through reduced drug accumulation, a common mechanism of resistance in acquired cisplatin-resistant cell lines. In the CH1:JM216-resistant cell line, in common with its cisplatin-resistant counterpart, acquired resistance appeared to be due to the increased DNA repair of JM216-induced adducts. Half-times for the removal of total platinum bound to DNA after JM216 exposure were 20 hours for the parental CH1 line, in contrast to 11 hours for the CH1:JM216R line. The broad-spectrum third-generation platinum drug AMD473 was designed to circumvent tumor resistance to cisplatin and carboplatin by being less susceptible than cisplatin to thiol binding. AMD473 was evaluated against our established panel of human ovarian carcinoma cell lines containing examples of acquired cisplatin resistance mediated through defined mechanisms. 10-12 AMD473 has shown promising circumvention of acquired cisplatin resistance in many of these in vitro human ovarian cell lines, which included specific models of acquired cisplatin resistance. 15
We report here for the first time the examination of these resistant cell line models for genomic aberrations associated with the acquisition of resistance using the molecular cytogenetic CGH technique. The profiles of three different platinum analogues (cisplatin, JM216, AMD473) have been compared in the same cell line, and the same members of the platinum family have also been studied in three different cell lines (CH1, 41M, and A2780).
Materials and Methods
Cell Lines
Ovarian carcinoma cell lines 41M and A2780 were established from previously untreated patients. CH1 was established from an ovarian carcinoma patient previously treated with and resistant to cisplatin and carboplatin. The resistant cell lines were established by serial passage in the presence of increasing concentrations of drug. 16,17 The resistance was stable for at least 6 months and did not require any maintenance dosing with cisplatin or JM216. All cell lines were maintained as monolayers in Dulbecco’s minimum essential medium supplemented with 10% fetal calf serum, 50 μg/ml gentamicin, 0.5 μg/ml hydrocortisone, and 2 mmol/L L-glutamine in a 10% CO2 atmosphere.
Drugs
Cisplatin
Cis-diamminedichloroplatinum(II) was first shown to have antitumor activity in 1969 18 and has subsequently become a pivotal component of many therapeutic regimens against a wide variety of solid tumors. However, its clinical use is severely limited by both nephrotoxicity and neurotoxicity. Hence, numerous attempts have been made to synthesize derivatives with an improved therapeutic index.
JM216
Bis-acetato-amminedichloro(cyclohexylamine) platinum(IV) was developed to be orally bioavailable. It has activity in several cisplatin-resistant human ovarian cancer cell lines in vitro.
AMD473
Cis-amminedichloro-(2-methylpyridine)-platinum(II) was designed to introduce steric hindrance close to the platinum center through the 2-methylpyridine ligand, thereby reducing the drug’s proclivity for deactivation by endogenous thiol-containing species.
CGH
One microgram of DNA from each sensitive and resistant cell line pair was labeled with digoxigenin or biotin respectively, using High-Prime (Boehringer Mannheim, Lewes, UK). Unincorporated nucleotides were removed using Nick columns (Pharmacia, St. Albans, UK). The labeled DNAs were ethanol precipitated together with a 50-fold excess of COT1 DNA (Gibco/BRL, Paisley, UK), resuspended in 10 μl Hybrisol VIII (Oncor, Gaithersburg, MD) and denatured at 75°C for 8 minutes. This mixture was then used to probe slides containing normal male lymphocyte metaphases (Vysis, Downers Grove, IL) as previously described. 19 Following stringent washing, the hybridized signals were detected using fluorescin-avidin and rhodamine-antidigoxigenin, and counterstained with DAPI, again under conditions previously described. 19
Computer-Assisted Image Analysis
Slides were examined using an Axioskop microscope (Carl Zeiss, Welwyn Garden City, UK) equipped with appropriate filters. Separate images were collected for red, green, and blue fluorescence, using a CCD camera (Photometrics, Tuscon, AZ) and Quips software (Vysis). At least 10 metaphases were captured for each hybridization, the chromosomes were karyotyped, and the axis defined based on the DAPI banding pattern. The DAPI image was used as a mask for the red and green images to exclude background fluorescence. The total image intensity for the masked red and green images was then independently normalized to accommodate differences in the image capture, so that the average red:green ratio for each cell was 1.0. The red:green ratios for sections of each chromosome were measured perpendicular to the axis. Chromosomal imbalances affecting more than 30% of the cell population, were identified where the ratio was greater than 1.15–1.20 for gains and less than 0.85–0.80 for losses (all imbalances were within 99% confidence limits). Chromosomes 1p, 16, 19, and 22 have previously been reported as having an unreliable CGH profile due to a variable number of interspersed repetitive elements between individuals. 20 We have therefore considered only imbalances affecting more than 50% of the cells in these regions to be significant.
Results
Regions of chromosomal imbalance associated with acquired resistance were seen in all the cell lines examined, varying in number from 3 to 10. The specific chromosome band locations of all the imbalances observed are listed in Table 1 ▶ . All the amplifications observed were low level (less than 5 extra copies). This contrasts with the high level amplification of the MDR1 gene observed in the CH1:doxorubicin resistant cell line (see Figure 1A ▶ ): however, this CH1:doxorubicin cell line is approximately 80-fold resistant to doxorubicin whereas the highest level of resistance of any of the platinum analogue resistant cell lines in this study is 11-fold.
Table 1.
Resistance Levels and Chromosome Aberrations Associated with Acquired Resistance to Platinum Analogues
| Cell line: Selection agent | Resistance | Sites of chromosomal imbalance |
|---|---|---|
| CH1: cisplatin R. | 6.4-fold | +Xq24–28, +4q13–27,+7. |
| 41M: cisplatin R. | 4.7-fold | −3q24–29, +4q23–28,−5p12–15.3,+5q11.2–31,+6q16–27,+7p11.2–22, −7q32–36,−9p21,+10q11.2–24,−12q22–24.3. |
| A2780: cisplatin R. | 16-fold | +1q21–23,−1q32–44,+6q21–27,−13q14–34, +17q22–25. |
| CH1: JM216 R. | 6.2-fold | +3p21–26,+12q13–24.1, −18q21–23. |
| 41M: JM216 R. | 1.9-fold | +3p23–26, −3q13.2–13.3,+4q21–23,+5p13–q23,+6p12–q12,+6q25–27,−8q13, −9p12–21,+9q22–32,+14q12–13,−14q24–32,+17q21–25. |
| CHI: AMD473 R. | 3- to 4-fold | −Xp21–22.3,+2p14,+4q26–33,+5p14, +6q15–23,+9p21–24,+11q22,+12q13–23,−17p11–13,+20p11.2–q13.3. |
| A2780: AMD473 R. | 3- to 4-fold | −Xp21–22.1,+Xq21, +4q13–31.3,+5q21–23,+6,−7q11.2–21,−10p11.2–13,−12q23–24.1, −17p11–13,+18p11.1–11.3,−20. |
It is clearly critically important to dissociate genes associated with drug resistance from those associated with growth advantage. We have currently reported the extreme variability developed in four different sub-cultures of the MCF7 breast cancer cell line. 21 Hence, we were scrupulous in every case to use the exact parental sensitive cell line from which the resistant line had been developed as the appropriate control in each CGH analysis.
Cisplatin resistance was examined in the cell lines CH1, 41M, and A2780. The CGH ratio profiles for these cell lines with acquired resistance to cisplatin, compared with their sensitive ‘parental’ cell lines, are shown in Figure 2 ▶ . Each of these cell lines shows changes in copy number of discrete regions of DNA during the acquisition of drug resistance. However, few of the changes are common between these cell lines, with the notable exception of the gain of 6q in 41M and A2780 and the gain of 4q and 7p in CH1 and 41M.
Figure 2.

CGH karyotype for the cell line CH1 (A) and mean ratio profiles, associated with acquired resistance to cisplatin, are shown for the cell lines CH1 (B), 41M (C), and A2780 (D). Visual examination of the karyotype (A) confirms that both the sensitive and resistant DNAs have labeled and hybridized successfully, and also provides an indication of areas of imbalance; the amplification of chromosome 7 is clearly significant on this examination. The areas of imbalance are indicated next to the chromosome ideograms, red for deletion and green for amplification, so that the chromosome location can be identified.
For the cell line CH1 (B), significant amplifications of 4q, 7, and Xq are apparent. For the 41M cell line (C), significant amplifications of 4q, 5q, 6q, 7p, and 10q and deletions of 3q, 5p, 7q, 9p, and 12q are demonstrated. For the cell line A2780 (D), significant amplifications of 1q, 6q, and 17q and deletions of 1q and 13q are shown.
Overall for the three different platinum analogues in three different parent cell lines, we have shown the most frequently observed changes to include gain of 4q (5/7) and 6q (5/7), followed by gain of 5q21–23 (3/7). The minimal common overrepresented region (MCOR) on chromosome 4 was q23 in 4/7 lines and q26–27 also in 4/7 cell lines. Likewise, the MCOR on chromosome 6 was q21–23 in 4/7 lines and q25–27 also in 4/7 cell lines. Thus, two different sites on 4q and 6q may be important loci for genes involved in platinum analogue resistance. A summary of amplified and deleted regions, acquired with resistance to the three different analogues, is illustrated with chromosome ideograms in Figure 3 ▶ .
Figure 3.

Amplifications (right) and deletions (left) of genomic sequences associated with acquired drug resistance to platinum analogues.
In terms of chromosomal aberrations associated with specific analogues, for the more lipophilic analogues, gain of 3p occurs only in the two cell lines resistant to JM216 (Figure 1, B and C) ▶ , and deletions of 17p and Xp occur only in the two cell lines resistant to AMD473. Gain of 12p was seen only in the CH1 cell lines with acquired resistance (Figure 1B) ▶ . The 41M-sensitive cell line showed high level amplifications of 3q, 9p, and 12p when compared with normal DNA. With the acquisition of resistance to cisplatin, some of these extra copies were lost from chromosomes 3q and 9p; the acquisition of resistance to JM216 was accompanied by loss of extra copies from 9p only. However, the high-level amplification of 12q was retained in both the JM216 and AMD473 resistant cell lines; this phenomenon of equal amplification in sensitive and resistant cell lines is illustrated in Figure 1C ▶ as a bright yellow region on the chromosome, with no alteration in the ratio profile.
Conclusions
A steadily increasing number of specific chromosomal abnormalities are found to be associated with particular tumor subtypes. In leukemias, these usually involve specific translocations, whereas in solid tumors they frequently include extensive rearrangements. These cytogenetic abnormalities define the sites of specific genes whose alteration is implicated in the neoplastic process. High level amplifications occur frequently in solid tumors and are often manifested cytogenetically in the form of homogeneously staining regions and double minute chromosomes.
The true incidence and significance of solid tumor chromosome abnormalities are limited in at least three different ways. First, these investigations are often performed late in the disease on samples from effusions or metastases, when the karyotype may be dominated by secondary abnormalities acquired during progression. Second, the chromosome preparations obtained from solid tumors are usually of poorer quality than those obtained from bone marrow or peripheral blood. Third, the majority of karyotypes obtained from solid tumors contain marker chromosomes in which the contributing chromosomes cannot be identified. The advent of CGH has enabled investigators to overcome most of these limitations.
The use of CGH and molecular cytogenetic techniques in the study of drug resistance has received little attention to date. Hoare et al used reverse in situ hybridization to detect a new amplicon in the doxorubicin-resistant lung cancer cell line, GLC4-ADR, which mapped to chromosome 1. They concluded that these techniques were ideally suited to characterizing genetic changes specific to drug resistance within a background of genetic anomalies associated with tumor progression. 9 This work builds upon prior publications by Kallioniemi and Tanner, who used CGH to screen 15 breast cancer cell lines and 33 primary tumors to identify and map regions of the genome with increased DNA-sequence copy number. 6 The majority of 26 chromosomal subregions involved did not correspond to the loci of currently known amplified genes in breast cancer with the highest frequency observed in 17q22–24 and 20q13. They subsequently studied the area of 20q13 by interphase FISH with anonymous cosmid probes and gene-specific P1 clones. They narrowed the critical region of interest to approximately 1.5 mb at 20q13.2. Previously known genes were excluded as candidates, 22 implying that this chromosomal region harbors a novel oncogene that contributes to the malignant progression of breast cancer. This demonstrates that CGH can be used to search directly for novel oncogenes associated with tumor progression and should be equally applicable to the search for novel oncogenes associated with specific resistance pathways.
In 1997, Wasenius et al 23 used CGH to look at cell lines with acquired resistance to cisplatin and acquired resistance to antimony and arsenite cross-resistant to cisplatin. In agreement with our work, they observed only low-level amplifications. They reported regions of 9 chromosomes to be frequently associated with resistance to cisplatin, of which the most frequently observed change was loss of 2p and gain of 2q (3/4 cell lines resistant to cisplatin). In this study we benefited from the extensive bank of ICR cell lines in which we were able to examine 7 cell lines resistant to 3 platinum analogues. Our results have demonstrated that gains of material on chromosome arms 4q and 6q are the most consistently observed changes and potentially the most significant (these were among the changes noted by Wasenius et al). The MCORs that we have identified on 4q and 6q are broadly consistent with the areas identified by Wasenius et al.
We also report the following chromosomal aberrations associated with specific analogues in the context of their biochemical mechanism of action.
Cisplatin
It is interesting to note that for cisplatin, the chromosomal aberrations observed in the three resistant cell lines differ substantially from each other. Previous biochemical studies confirm that the mechanisms of resistance differ significantly in these three cell lines. Kelland, Harrap, and others have shown that resistance in the 41M cell line is predominantly mediated by a transport mechanism, 12 whereas increased DNA repair and/or tolerance of DNA-platinum adducts is the major mechanism of resistance in the CH1 cell line. 10 Reduced drug accumulation, elevated glutathione levels, enhanced capacity to remove platinum adducts, and failure to engage the appropriate apoptotic response all play a role in the acquired resistance of the A2780 cell line. 11
JM216
In contrast, the platinum analogue JM216 is sufficiently lipophilic that transport does not play any significant role in the development of acquired resistance in the 41M cell line. Hence, one would presume that the CH1 and 41M cell lines share similar mechanisms of resistance to JM216. Our CGH results show overrepresentation of the same region of chromosome 3p for both CH1 and 41M cell lines made resistant to JM216. It is interesting that this region encompasses the sites of the MLH-1 DNA repair gene and topoIIβ.
AMD473
The sterically hindered platinum analogue AMD473 has a lipophilicity between that of cisplatin and JM216. The mechanisms of resistance to AMD473 have been shown to include reduced accumulation, reduced DNA platination, and increased capacity to forgo apoptosis in both the CH1 and A2780 cell lines. 24 Our CGH results show a unique loss of copies of the regions Xp and 17p associated only with AMD473 resistance.
Although these associations of chromosome abnormalities with potential mechanisms are thought-provoking, they should not be overinterpreted in light of the small number of cell lines. There is a degree of cross-resistance between cell lines with acquired resistance to JM216 and AMD473, as well as to cisplatin itself. Thus, changes in common among cell lines with acquired resistance to the different analogues would be expected, corresponding to the sites of genes involving shared mechanisms of resistance, such as the changes on 4q, 5q, and 6q.
The classically quoted biochemical pathways associated with cisplatin resistance (decreased intracellular accumulation, elevated glutathione, elevated metallothioneins, enhanced DNA repair, altered mitochondrial membrane potential, increased oncogene expression, and signal transduction) were summarized by Marshall and Andrews 25 ; multiple publications have confirmed either protein and/or mRNA changes in these parameters. 26,27 In the majority of cell lines, cisplatin resistance appears to be multifactoral. A summary of specific genes which have been associated with resistance to cisplatin and their chromosomal location is presented in Table 2 ▶ . It is unclear whether these specific genetic changes are primary or secondary. A recent paper by Anthoney and Brown 56 has demonstrated the appearance of microsatellite alleles at multiple loci in resistant lines, suggesting an association between selection for cisplatin resistance and the development of genomic instability.
Table 2.
Chromosome Localization of Genes Implicated in Platinum Resistance
| Genes involved in signal transduction pathways | Chromosome location |
|---|---|
| EGF 28 (epidermal growth factor) | 4q25 |
| CyclinB p34 29 | 5q12 |
| PKCθ 30,31 (protein kinase C) | 10p15 |
| CALM1 32 (calmodulin) | 14q24–31 |
| PKCβ 30,31 | 16p11 |
| HER2/NEU 33 | 17q21.2 |
| PKCα 30,31 | 17q22–23 |
| Genes involved in the recognition of DNA damage and repair | |
| TopoIIβ 26,34 | 3p24–25 |
| HMG2 35 | 4q31 |
| POLB 36,37 (DNA polymerase β) | 8p11.2 |
| DDB1 38 (DNA damage binding protein) (xeroderma pigmentosum E) | 11q12–13 |
| HMG1 39 (high mobility group protein 1) | 13q12 |
| TopoIIα 26,34 | 17q21–22 |
| ERCC1 40 (excision repair complementing defective in chinese hamster) | 19q13 |
| POLA 37 (DNA polymerase α) | Xp21–22 |
| HSSB 41 (human single-stranded DNA binding protein) | n/a |
| SSRP1 42 (structure specific recognition protein) | n/a |
| Other genes implicated in modulating cisplatin resistance | |
| JUN 30 | 1p31–32 |
| MLH1 43 | 3p21–23 |
| WAF1 44 (wildtype p53 activated fragment) | 6p21 |
| PMS2 43 (post meiotic segregation) | 7p22 |
| MDR1 26 (multidrug resistance) | 7q21 |
| MYC 45 | 8q24 |
| H-RAS 46 | 11p15 |
| GSTπ 47 | 11q13 |
| MDM2 48 | 12q14–15 |
| FOS 49 | 14q24 |
| MRP 26,34 (multidrug resistance protein) | 16p13 |
| LRP 34 (lung resistance protein) | 16p11 |
| Metallothioneins 50 | 16q13 |
| p53 44,48 | 17p13 |
| ERBB2 51 | 17q21 |
| BCL2 52 | 18q21.3 |
| PLS3 53 (T-plastin) | n/a |
| HSP-60 54 (heat shock protein) | n/a |
| BCLX 55 | n/a |
In summary, our results comparing seven cell lines and three agents have shown the most frequently observed changes to involve amplification of 4q(5/7) and 6q (5/7), followed by amplification of 5q (3/7). These common areas of amplification and deletion across different cell lines in response to the acquisition of resistance to the same drug or analogues indicate potential sites of genes involved in common mechanisms of drug resistance. These aberrations include the sites of EGF, HMG2, and cyclin B, all of which have been previously implicated in cisplatin resistance. Other candidate genes in these regions include MSH3 (5q11–12), APC (5q21–22), and DHFR (5q11–13), which are all involved in DNA repair; the genes cyclin C (6q21), CD24 (6q21), and PDCD2 (programmed cell death 2) (6q27), involved in signal transduction; and GST2 (4q28–31), the glutathione s-transferase gene.
Some aberrations were induced only in response to a specific analogue. These included the amplification of 3p (site of both the topoIIβ and MLH1 genes) seen in response to acquired resistance to JM216; deletions of 17p (site of p53) and Xp (site of CLCN4 and POLA) seen only in response to acquired resistance to AMD473. These areas may include genes involved with a more selective mechanism of resistance. The regions that we have identified are currently being investigated by FISH using specific gene probes to narrow the critical region and identify candidate genes.
These cell line data have provided insight into the association of defined chromosomal abnormalities with specific mechanisms of resistance. It is now critical to extend this work into tumor biopsies obtained from patients. Hence, we are now beginning a major study to analyze samples from patients with intrinsic and acquired resistance to the three platinum analogues that we have described.
Acknowledgments
We thank Johnson Matthey Company and AnorMED for drug synthesis and supply.
Footnotes
Address reprint requests to Brian Leyland-Jones, Department of Oncology, McGill University, 546 Pine Ave. W, Montreal, QC H2W 1S6 Canada. E-mail: leylandj@med.mcgill.ca.
Supported by the Haddow Research Fellowship (B.L.J.), Cancer Research Campaign (L.R.K. and K.R.H.), and the Leukaemia Research Fund (L.R.H.).
References
- 1.: British Association for Cancer Research: New targets in cancer chemotherapy. Br J Cancer 1997, 75 (suppl 1):1-54 [PMC free article] [PubMed] [Google Scholar]
- 2.New Molecular Targets. Proceedings of the EORTC winter meeting. Nancy, France. Jan. 22, 1998.
- 3.Kallioniemi A, Kallioniemi O-P, Sudar D, Rutovitz D, Gray JW, Waldman F, Pinkel D: Comparative genomic hybridisation for molecular cytogenetic analysis of solid tumours. Science 1992, 258:818-821 [DOI] [PubMed] [Google Scholar]
- 4.Visakorpi T, Kallioniemi AH, Syvanen AC, Hyytinen ER, Karhu R, Tammela T, Isola JJ, Kallioniemi O-P: Genetic changes in primary recurrent prostate cancer by comparative genomic hybridisation. Cancer Res 1995, 55:342-347 [PubMed] [Google Scholar]
- 5.Bentz M, Huck K, du-Manoir S, Joos S, Werner CA, Fischer K, Dohner H, Lichter P: Comparative genomic hybridisation in chronic B-cell leukemias shows a high incidence of chromosomal gains and losses. Blood 1995, 85:3610-3618 [PubMed] [Google Scholar]
- 6.Kallioniemi A, Kallioniemi O-P, Piper J, Tanner M, Stokke T, Chen L, Smith HS, Pinkel D, Gray JW, Waldman FM: Detection and mapping of amplified DNA sequences in breast cancer by comparative genomic hybridization. Proc Natl Acad Sci USA 1994, 91:2156-2160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Houldsworth J, Mathew S, Rao PH, Dyomina K, Louie DC, Parsa N, Offit K, Chaganti RS: REL proto-oncogene is frequently amplified in extranodal diffuse large cell lymphoma. Blood 1996, 87:25-29 [PubMed] [Google Scholar]
- 8.Schwendel A, Petersen I, Hartmann T, Ried T, Dietel M, Lage H: Mapping of DNA gains and losses in parental and multidrug-resistant cancer cell lines. European Conference on Comparative Genomic Hybridization. Semmering, Austria, 1996, p 42a
- 9.Hoare SF, Freeman CA, Coutts JC, Varley JM, James L, Keith WN: Identification of genetic changes associated with drug resistance by reverse in situ hybridization. Br J Cancer 1997, 75:275-282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kelland LR, Mistry P, Abel G, Loh SY, O’Neill CF, Murrer BA, Harrap KR: Mechanism-related circumvention of acquired cis-diamminedichloroplatinum(II) resistance using two pairs of human ovarian carcinoma cell lines by ammine/amine platinum(IV) dicarboxylates. Cancer Res 1992, 52:3857-3864 [PubMed] [Google Scholar]
- 11.O’Neill CF, Orr RM, Kelland LR, Harrap KR: Comparison of platinum(Pt) binding to DNA, and removal of total Pt adducts and interstrand crosslinks in three human ovarian carcinoma cell lines sensitive and resistant to cisplatin. Cellular Pharmacol 1995, 2:1-7 [Google Scholar]
- 12.Loh SY, Mistry P, Kelland LR, Abel G, Harrap KR: Reduced drug accumulation as a major mechanism of acquired resistance to cisplatin in a human ovarian carcinoma cell line: circumvention studies using novel platinum(II) and (IV) ammine/amine complexes. Br J Cancer 1992, 66:1109-1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orr RM, O’Neill CF, Nicolson MC, Barnard CFJ, Murrer BA, Giandomenico CM, Vollano JF, Harrap KR: Evaluation of novel ammine/amine platinum(IV) dicarboxylates in L1210 murine leukaemia cells sensitive and resistant to cusplatin, tetraplatin or carboplatin. Br J Cancer 1994, 70:415-420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mellish KJ, Kelland LR: Mechanisms of acquired resistance to the orally active platinum-based anticancer drug, Bis-acetato-ammine-dichloro-cyclohexylamine platinum(IV) (JM216) in vivo. Cancer Res 1994, 54:4118-4122 [PubMed] [Google Scholar]
- 15.Holford J, Sharp SY, Murrer BA, Abrams M, Kelland LR: In vitro circumvention of cisplatin resistance by the novel sterically hindered platinum complex AMD473. Br J Cancer 1998, 77:366-373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hills CA, Kelland LR, Abel G, Siracky J, Wilson AP, Harrap KR: Biological properties of ten human ovarian carcinoma cell lines: calibration in vitro against four platinum complexes. Br J Cancer 1989, 59:527-534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Behrens BC, Hamilton TC, Masuda H, Grotzinger KR, Whang-Peng J, Louie KG, Knutsen T, McKoy WM, Young RC, Ozols RF: Characterization of a cis-diamminedichloroplatinum(II)-resistant human ovarian cancer cell line and its use in evaluation of platinum analogues. Cancer Res 1987, 47:414-418 [PubMed] [Google Scholar]
- 18.Rosenberg B, VanCamp L, Trosko JE, Mansour VH: Platinum compounds: a new class of potent antitumour agents. Nature 1969, 222:385-386 [DOI] [PubMed] [Google Scholar]
- 19.Hiorns LR: Comparative genomic hybridisation. Cytogenetic Methods and Protocols: Methods in Molecular Biology. Edited by GJ Swansbury. Humana Press, 1999, in press.
- 20.Kirchhoff M, Gerdes T, Maahr J, Rose H, Lundsteen C: Automatic correction of the interfering effect of unsuppressed interspersed repetitive sequences in comparative genomic hybridization analysis. Cytometry 1997, 28:130-134 [PubMed] [Google Scholar]
- 21.Hiorns LR, Bradshaw TD, Kelland LR, Leyland-Jones B: CGH identification of genomic alterations acquired during cell culture. Proc Am Assoc Cancer Res 1998, 2354a
- 22.Tanner MM, Tirkkonen M, Kallioniemi A, Collins C, Stokke T, Karhu R, Kowbel D, Shadravan F, Hintz M, Kuo W-L, Waldman FM, Isola JJ, Gray JW, Kallioniemi O-P: Increased copy number at 20q13 in breast cancer: defining the critical region and exclusion of candidate genes. Cancer Res 1994, 54:4257-4260 [PubMed] [Google Scholar]
- 23.Wasenius VM, Jekunen A, Monni O, Joensuu H, Aebi S, Howell SB, Knuutila S: Comparative genomic hybridization analysis of chromosomal changes occurring during development of acquired resistance to cisplatin in human ovarian carcinoma cells. Genes Chromosomes Cancer 1997, 18:286-291 [PubMed] [Google Scholar]
- 24.Holford J: Thesis, University of London, 1997, pp 142–158
- 25.Marshall JL, Andrews PA: Preclinical and clinical experience with cisplatin resistance. Haematol Oncol Clin N Am 1995, 9:415-429 [PubMed] [Google Scholar]
- 26.Hasegawa S, Naito S, Kotoh S, Koga H, Yokomizo A, Yamasaki T, Noma H, Kumuzawa J: The expression of drug-resistant genes and the acquirement of drug resistance in human bladder cancer cell lines. Nishinihon J Urol 1996, 58:331-336 [Google Scholar]
- 27.Yang YY, Robbins PD, Lazo JS: Elevated Sp1 and AP-2 DNA binding activity in cisplatin resistant cells. Proc Annu Meet Am Asso Cancer Res 1995, 36:1945a [Google Scholar]
- 28.Christen RD, Hom DK, Porter DC, Andrews PA, MacLeod CL, Hafstrom L, Howell SB: Epidermal growth factor regulates the in vitro sensitivity of human ovarian carcinoma cells to cisplatin. J Clin Invest 1990, 86:1632-1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McClay EF, Albright KD, Jones JA, Eastman A, Christen R, Howell SB: Modulation of cisplatin resistance in human malignant melanoma cells. Cancer Res 1992, 52:6790-6796 [PubMed] [Google Scholar]
- 30.Rubin E, Kharbanda S, Gunji H, Weichselbaum R, Kufe D: Cis-diamminodichloroplatinum(II) induces c-jun expression in human myeloid leukemia cells: potential involvement of a protein kinase C-dependent signaling pathway. Cancer Res 1992, 52:878-882 [PubMed] [Google Scholar]
- 31.Basu A, Teicher BA, Lazo JS: Involvement of protein kinase C in phorbolester induced sensitization of HeLa cells to cisdiamminedichloroplatinum(II). J Biol Chem 1990, 265:8451-8457 [PubMed] [Google Scholar]
- 32.Kikuchi Y, Iwano I, Miyauchi M, Sasa H, Nagata I, Kuki E: Restorative effects of calmodulin antagonists on reduced cisplatin uptake by cisplatin-resistant human ovarian carcinoma cells. Gynecol Oncol 1990, 39:199-203 [DOI] [PubMed] [Google Scholar]
- 33.Tsai C-M, Chang K-T, Perng R-P, Mitsudomi T, Chen M-H, Kadoyama C, Gazdar AF: Correlation of intrinsic chemoresistance of non-small-cell lung cancer cell lines with Her-2/neu gene expression but not with ras gene mutations. J Natl Cancer Inst 1993, 85:897-901 [DOI] [PubMed] [Google Scholar]
- 34.Leegte B, vanEchten J, van-derVeen AY, Withoff S, Timmer-Bosscha H, deVries EG, deJong B: A cytogenetic search for genes involved in the multidrug resistance of different GLC4 cell lines. Proc Annu Meet Am Assoc Cancer Res 1996, 37:3806a [Google Scholar]
- 35.Billings PC, Davis RJ, Engelsberg BN, Skov KA, Hughes EN: Characterization of high mobility group protein binding to cisplatin-damaged DNA. Biochem Biophys Res Commun 1992, 188:1286-1294 [DOI] [PubMed] [Google Scholar]
- 36.Scanlon KJ, Jiao L, Funato T, Wang W, Tone T, Rossi JJ, Kashani-Sabet M: Ribozyme-mediated cleavage of c-fos mRNA reduces gene expression of DNA synthesis enzymes and metallothionein. Proc Natl Acad Sci USA 1991, 88:10591-10595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scanlon KJ, Kashani-Sabet M, Sowers LC: Overexpression of DNA replication and repair enzymes in cisplatin-resistant human colon carcinoma HCT8 cells and circumvention by azidothymidine. Cancer Commun 1989, 1:269-275 [PubMed] [Google Scholar]
- 38.Chu G, Chang E: Cisplatin-resistant cells express increased levels of a factor that recognizes damaged DNA. Proc Natl Acad Sci USA 1990, 87:3324-3327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pil PM, Lippard SJ: Specific binding of chromosomal protein HMG1 to DNA damaged by the anticancer drug cisplatin. Science 1992, 256:234-237 [DOI] [PubMed] [Google Scholar]
- 40.Dabholkar M, Bostick-Bruton F, Weber C, Bohr VA, Egwuagu C, Reed E: ERCC1 and ERCC2 expression in malignant tissue from ovarian cancer patients. J Natl Cancer Inst 1992, 84:1512-1517 [DOI] [PubMed] [Google Scholar]
- 41.Clugston CK, McLaughlin K, Kenny MK, Brown R: Binding of human single stranded DNA binding protein to DNA damage by the anticancer drug cis-diamminedichloroplatinum(II). Cancer Res 1992, 52:6375-6379 [PubMed] [Google Scholar]
- 42.Bruhn SL, Pil PM, Essigmann JM, Housman DE, Lippard SJ: Isolation and characterization of human cDNA clones encoding a high mobility group box that recognize structural distortions to DNA caused by binding of the antitumor agent cisplatin. Proc Natl Acad Sci USA 1992, 89:2307-2311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Drummond JT, Anthoney A, Brown R, Modrich P: Cisplatin and adriamycin resistance are associated with MutL alpha and mismatch repair deficiency in an ovarian tumor cell line. J Biol Chem 1996, 271:19645-19648 [DOI] [PubMed] [Google Scholar]
- 44.Perego P, Giarola M, Righetti SC, Supino R, Caserini C, Delia D, Pierotti MA, Miyashita T, Reed JC, Zunino F: Association between cisplatin resistance and mutation of p53 gene and reduced bax expression in ovarian carcinoma cell systems. Cancer Res 1996, 56:556-562 [PubMed] [Google Scholar]
- 45.Sklar MD, Prochownik EV: Modulation of cis-platinum resistance in friend erythroleukemia cells by c-myc. Cancer Res 1991, 51:2118-2123 [PubMed] [Google Scholar]
- 46.Kashani-Sabet M, Lu Y, Leong L, Haedicke K, Scanlon KJ: Differential oncogene amplification in tumor cells from a patient treated with cisplatin and 5-fluorouracil. Eur J Cancer 1990, 26:383-390 [DOI] [PubMed] [Google Scholar]
- 47.Ozols RF, O’Dwyer PJ, Hamilton TC, Young RC: The role of glutathione in drug resistance. Cancer Treat Rev 1990, 17a:45-50 [DOI] [PubMed] [Google Scholar]
- 48.Fajac A, DaSilva J, Ahomadegbe JC, Ratear JG, Bernaudin JF, Riou G, Benard J: Decreased susceptibility to cisplatin-induced apoptosis is associated with increased p53 expression in a cisplatin-resistant human ovarian carcinoma cell line, IGR-OV1/DDP. Proc Annu Meet Am Asso Cancer Res 1996, 37:2814a [Google Scholar]
- 49.Funato T, Yoshida E, Jiao L, Tone T, Kashani-Sabet M, Scanlon KJ: The utility of an anti-fos ribozyme in reversing cisplatin resistance in human carcinomas. Adv Enzyme Regul 1992, 32:195-209 [DOI] [PubMed] [Google Scholar]
- 50.Kelley SL, Basu A, Leicher BA, Hacker MP, Hamer DH, Lazo JS: Overexpression of metallothionein confers resistance to anticancer drugs. Science 1988, 241:1815-1818 [DOI] [PubMed] [Google Scholar]
- 51.Wu L, Wu A, Jiang K: Effect of antisense c-erbB2 on biologic behaviour and chemotherapeutic drug sensitivity in human ovarian cancer cells. Chung Hua Fu Chan Ko Tsa Chih 1996, 31:169-172 [PubMed] [Google Scholar]
- 52.Eliopoulos AG, Kerr DJ, Herod J, Hodgkins L, Krajewski S, Reed JC, Young LS: The control of apoptosis and drug resistance in ovarian cancer: influence of p53 and bcl-2. Oncogene 1995, 11:1217-1228 [PubMed] [Google Scholar]
- 53.Hisano T, Ono M, Nakayama M, Naito S, Kuwano M, Wada M: Increased expression of T-plastin gene in cisplatin-resistant human cancer cells: Identification by mRNA differential display. FEBS Lett 1996, 397:101-107 [DOI] [PubMed] [Google Scholar]
- 54.Kimura E, Enns RE, Alcaraz JE, Arboleda J, Slamon DJ, Howell SB: Correlation of the survival of ovarian cancer patients with mRNA expression of 60kD heat shock protein HSP-60. J Clin Oncol 1993, 11:891-898 [DOI] [PubMed] [Google Scholar]
- 55.Minn AJ, Rudin CM, Boise LH, Thompso CB: Expression of bcl-X1 can confer a multidrug resistance phenotype. Blood 1995, 86:1903-1910 [PubMed] [Google Scholar]
- 56.Anthony DA, McIlwrath AJ, Gallagher WM, Edlin ARM, Brown R: Microsatellite instability, apoptosis, and loss of p53 function in drug resistant tumor cells. Cancer Res 1996, 56:1374-1381 [PubMed] [Google Scholar]