

Abstract
Endostatin is a carboxyl-terminal proteolytic fragment of collagen XVIII and a potent inhibitor of angiogenesis. The mechanism of action is unknown, but the crystal structure of endostatin predicts a prominent heparan sulfate binding site, suggesting that endostatin competitively inhibits heparin-binding angiogenic factors, such as basic fibroblast growth factor (FGF-2). The goal of the study was to map endostatin binding sites in intact human tissues and to determine whether this binding is heparan sulfate dependent. In situ binding was performed with recombinant epitope-tagged murine endostatin. Endostatin predominantly binds to blood vessels of different calibers in a saturable fashion. In addition, binding to some epithelial basement membranes is seen. The localization pattern is similar to that reported for collagen XVIII, endostatin’s parent molecule. In breast carcinomas, endostatin co-localizes largely with FGF-2. In a surprising contrast to FGF-2, endostatin binding is resistant to treatment with heparitinase, demonstrating that binding is not mediated by heparan sulfate proteoglycans. Furthermore, FGF-2 and heparin do not compete for endostatin binding, providing additional evidence for the discreteness of endostatin and FGF-binding sites.
The dependence of tumor progression on the formation of new blood vessels is now undisputed. Blood vessel formation, or angiogenesis, is under tight control of both stimulatory and inhibitory factors. 1 Inhibitors are speculated to predominate in normal adult tissues when the vasculature is quiescent. In contrast, the balance is disturbed in favor of angiogenic stimulators when tumors are expanding.
Endostatin was recently identified as a potent endogenous inhibitor of angiogenesis. 2 The molecule induces unprecedented anti-tumor responses in several in vivo models. 2,3 Mouse endostatin consists of 184 amino acids at the noncollagenous carboxy terminus of collagen XVIII and is released through proteolytic cleavage. Human serum and tissue forms of endostatin have also been identified. 4,5 The origin from a larger precursor molecule is shared by angiostatin, another potent suppressor of angiogenesis. 6 Angiostatin is an internal fragment of the plasma protein plasminogen. This mechanism to activate potent suppressors of angiogenesis might provide a rapid response to unwanted angiogenic stimuli, similar to the activation of blood coagulation and fibrinolysis factors.
The mechanism behind endostatin is unknown, but recent physical-chemical data have provided leads. Three-dimensional analysis of the molecule by x-ray crystallography predicts a prominent heparan sulfate binding site. 7 The observation is supported by an affinity of endostatin for heparin, which has been exploited for its purification by affinity chromatography. 2 This property has also led to the hypothesis that endostatin may competitively inhibit the binding of angiogenic stimulators to cell surface heparan sulfate proteoglycans (HSPGs). 7 Interestingly, the strongest promoters of angiogenesis, fibroblast growth factor (FGF)-2 and vascular endothelial growth factor (VEGF), not only bind HSPGs but also require them as cofactors for signaling. 8,9 Alternative mechanisms of endostatin action include binding to its own putative signaling receptor, interference with endothelial cell adhesion/migration, or prevention of proteolytic degradation of the extracellular matrix.
As a first step toward investigating endostatin’s mechanism of action, we have mapped its binding sites in intact human tissues using an in situ assay. This approach allowed us to successfully map HSPG binding sites of different members of the FGF family. 10 Endostatin binds predominantly to its functional target, blood vessels. Specifically, it binds to the subendothelial matrix of the vasculature and co-localizes with a subset of FGF-2 binding sites. Surprisingly, in contrast to FGF-2, binding of endostatin is resistant to treatment of the tissues with heparan sulfate lyases, demonstrating that binding is not mediated through heparan sulfates. Furthermore, endostatin does not compete with FGF-2 for tissue binding. These results show that competitive displacement of FGF-2 by endostatin is an unlikely mechanism of action.
Materials and Methods
Production of Recombinant Epitope-Tagged Endostatin
The vector (pSec) containing the entire murine endostatin cDNA (encoding the 184 carboxyl-terminal amino acids of collagen XVIII α-chain) with an amino-terminal influenza virus hemagglutinin (HA) tag was kindly provided by Dr. Judah Folkman (Harvard University). This expression vector contains an immunoglobulin signal peptide enabling efficient secretion of the recombinant protein. The accuracy of the endostatin insert was confirmed by DNA sequencing. The construct was transfected into COS-7 cells using lipofectamine (Gibco-BRL, Gaithersburg, MD). Conditioned medium was collected both after transient transfection and after selection of populations with zeocin (Invitrogen, Carlsbad, CA). Tissue culture reagents were from Gibco-BRL. HA-endostatin was enriched by heparin-affinity chromatography using heparin-agarose beads (Sigma Chemical Co., St. Louis, MO). The identity of the molecule was verified by SDS-polyacrylamide gel electrophoresis and probing the blots with anti-HA antibody (clone 12CA5, Boehringer-Mannheim, Indianapolis, IN) and with rabbit anti-endostatin serum (gift from Dr. J. Folkman). The concentration of endostatin was determined by Western analysis using MultiTag marker protein (Boehringer-Mannheim) as standards.
In Situ Binding and Labeling Assays
Tissue was obtained fresh from the operating room, embedded in OCT compound, snap frozen, and stored at −70°C. National Institutes of Health guidelines for the use of human material were followed, and institutional review board approval was obtained. The in situ binding assay was performed essentially as described previously for FGF-2 and FGF-7. 10 Briefly, after fixation and treatment steps to reduce autofluorescence, the frozen sections were placed in Sequenza immunohistochemistry staining racks (Shandon, Pittsburgh, PA). After blocking with Tris-buffered saline plus bovine serum albumin (room temperature for 30 minutes), sections were incubated with the ligand (endostatin or FGF-2). HA-endostatin was added at a concentration of 2.6 to 130 nmol/L for 60 minutes at room temperature. We used both unprocessed conditioned medium and material enriched on heparin columns. In addition, in situ binding of unlabeled endostatin (200 nmol/L) expressed in the yeast Piscia pastoris was measured, detecting bound reagent with rabbit anti-endostatin antiserum (both kindly provided by Dr. J. Folkman). These experiments were done to rule out an effect of the epitope tag on in situ binding. FGF-2 was applied at a concentration of 10 nmol/L for 60 minutes. After washing, primary antibody was added for 60 minutes at room temperature. Anti-HA antibody (clone 12CA5, Boehringer-Mannheim) was used at a dilution of 1:400 and anti-FGF-2 (clone DE-6, gift from Dupont, Wilmington, DE) at a dilution of 1:100. Secondary Alexa-546-conjugated donkey anti-mouse antibody (Molecular Probes, Eugene, OR) was used in both cases for visualization. Immunolabeling with antibody to factor-VIII-related antigen (anti-von Willebrand factor antibody, DAKO, Carpinteria, CA) was performed at a dilution of 1:1000 to co-localize endothelial cells. Co-localization of HA-endostatin binding with the basement membrane component perlecan was carried out using anti-perlecan antiserum (gift from Dr. J. Hassell, Tampa, FL) at a dilution of 1:100. Both anti-factor-VIII-related antigen and anti-perlecan antibodies were visualized with Alexa-488-conjugated donkey anti-rabbit antibody (Molecular Probes). Sections were examined using a Nikon Microphot FX microscope equipped for epifluorescence. Images were acquired with a Photometrics CCD camera (Tucson, AZ) and Image-Pro-Plus analysis software (Media Cybernetics, Silver Springs, MD).
Heparitinase Digestion
Tissue sections were exposed to heparitinase enzymes to distinguish heparan sulfate from non-heparan-sulfate binding sites. Heparitinase (mixture of 95% heparitinase I and 5% heparitinase II) and pure heparitinase II (purchased from Seikagaku, Ijamsville, MD, and ICN Pharmaceuticals, Costa Mesa, CA, respectively) were reconstituted and diluted in heparitinase buffer (50 mmol/L Hepes, 50 mmol/L sodium acetate, 150 mmol/L NaCl, 9 mmol/L CaCl2, 0.1% bovine serum albumin, pH 6.5). Both enzymes were incubated at a final concentration of 4 mIU/ml in the Sequenza racks at 37°C for 4 hours, replacing the enzyme after 2 hours.
Competition Experiments and Additional Controls
The specificity and saturability of the in situ binding interaction was tested by comparing binding of HA-endostatin (30 nmol/L) in the presence and absence of unlabeled P. pastoris-derived endostatin (3 μmol/L = 100-fold excess). Reciprocal competition experiments between endostatin and FGF-2 were performed. A 100-fold excess (5 μmol/L) of FGF-2 was used to compete with HA-endostatin (50 nmol/L) binding and 300-fold excess endostatin (1 μmol/L) to compete with FGF-2 binding (3 nmol/L). In a separate experiment, the tissue sections were preincubated with excess competitor for 30 minutes before adding the binding ligand of interest. Heparin was tested as possible competitor of endostatin by preincubating endostatin with 0.1 mg/ml heparin overnight at 4°C. In addition, we examined the possibility that COS-7 cell-derived heparan sulfate might occupy the heparan sulfate binding site on the recombinant endostatin, masking potential interactions with tissue heparan sulfate proteoglycans. For this purpose, equimolar HA-endostatin preparations of heparin column eluates and flow-through with and without preincubation with heparin were used on tissue sections with and without heparitinase pretreatment.
Results and Discussion
Transfection of HA-endostatin into COS-7 cells results in efficient expression of a protein with the correct size of approximately 20 kd (data not shown). The band is detected on the Western blots by either anti-HA monoclonal or rabbit anti-endostatin antibodies. Endostatin elutes from a heparin-agarose column at a sodium chloride concentration of approximately 0.4 mol/L (data not shown). This result agrees with findings by Timpl and co-workers 5 and reflects a lower affinity of endostatin for heparin/heparan sulfate than that originally reported by O’Reilly. 2
On tissue sections, HA-endostatin binds to localized regions in a specific manner, providing a fluorescence signal of relatively high intensity. There is no difference in the location of endostatin binding whether HA-endostatin is used at 13 nmol/L or 130 nmol/L, although the signal increases at the higher concentration. Only a weak fluorescence signal is detected at 2.6 nmol/L. HA-endostatin binding is completely eliminated in the presence of 100-fold unlabeled endostatin, demonstrating specificity/saturability of the in situ binding interaction (not shown). Endostatin binding is predominantly seen on blood vessels in all organs examined. Binding sites are present in blood vessels of all calibers ranging from larger muscular arteries to the smallest capillaries and also including venules and veins. Endostatin binds in a linear pattern consistent with binding to vascular basement membrane. The subendothelial location of endostatin binding sites is confirmed by double labeling with antibody to factor-VIII-related antigen. Endothelial cells show cell surface and granular cytoplasmic labeling with this antibody contrasting with subjacent linear binding of endostatin (Figure 1A) ▶ . Furthermore, the location of endostatin binding sites within basement membranes is demonstrated by co-localization with perlecan, a well characterized basement membrane proteoglycan (Figure 1B) ▶ . 11,12 The possibility that the HA tag might have altered endostatin’s binding characteristics was examined by a binding experiment with recombinant unlabeled P. pastoris endostatin. The binding pattern is identical to that of tagged endostatin (not shown), excluding the possibility that basement membrane binding is an artifact caused by the HA epitope. Neither does this experiment reveal additional binding sites, dismissing concerns that the HA tag might have masked binding interactions at a different tissue site.
Figure 1.
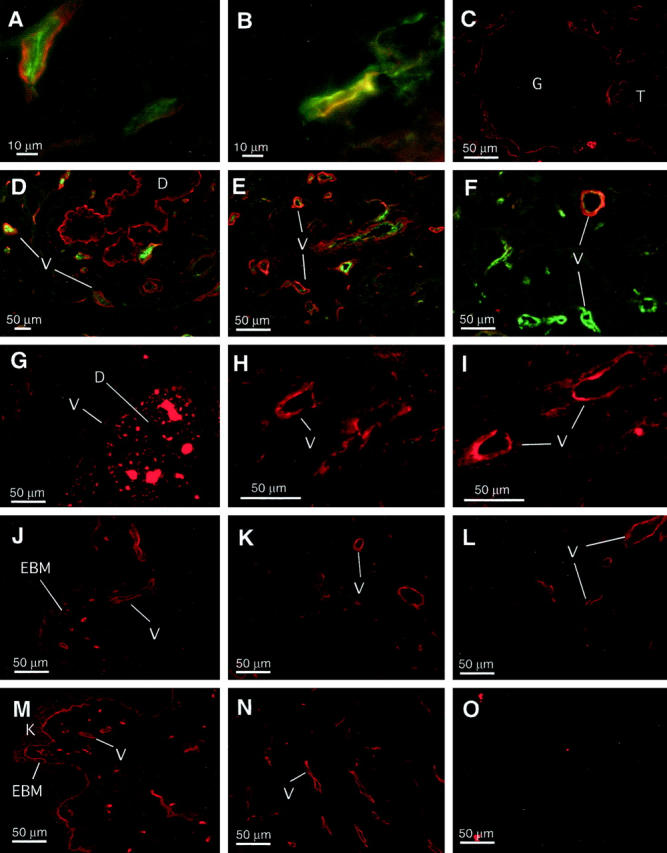
A to F: Endostatin binding in human tissues. Frozen sections of human tissues were incubated with recombinant HA-tagged endostatin (130 nmol/L), and bound endostatin was detected with anti-HA antibodies and Alexa-546-conjugated secondary antiserum (red channel). Double labeling was carried out on some sections (A, B, D, E, and F) with Alexa-488 conjugate (green channel) to determine location of other proteins as indicated. A: Capillaries in normal lung; HA-endostatin binding (red) and factor-VIII-related antigen (green). B: Capillary in breast cancer; co-localization of HA-endostatin binding (red) with perlecan (green). C: Normal renal cortex with glomerulus (G) and renal tubules (T) in the periphery. D: Normal breast tissue with branching duct (D) and stromal capillaries (V); HA-endostatin binding (red) and factor-VIII-related antigen (green). E and F: Infiltrating ductal carcinoma with angiogenic hot spots; HA-endostatin binding (red) and factor-VIII-related antigen (green). G: Ductal carcinoma in situ (DCIS) within ductal lumen (D) and with garland of newly formed capillaries (V). Strong signal within duct lumen is due to autofluorescence of necrotic or calcified material. H and I: Competition between endostatin and FGF-2. Frozen sections of human breast carcinomas were incubated with FGF-2, and bound growth factor was detected with anti-FGF-2 monoclonal antibody and Alexa-546-conjugated secondary antibody. H: FGF-2 (3 nmol/L) binding to blood vessels. I: FGF-2 (3 nmol/L) binding in the presence of 300-fold molar excess (1 μmol/L) of endostatin. The slide was preincubated with the competitor for 30 minutes before adding FGF-2. J to O: Comparison of endostatin and FGF-2 binding. Frozen sections of human skin (J and M) and breast carcinomas (K, L, N, and O) were incubated with HA-tagged endostatin (J, K, and L) or FGF-2 (M, N, and O). Bound protein was detected with anti-HA or anti-FGF-2 monoclonal primary antibodies and visualized by immunofluorescence. Treatment with heparitinase enzyme before the binding reaction was performed on L and O to determine whether the binding sites were heparan sulfates.
However, endostatin binds not only to blood vessels but also to a subset of epithelial basement membranes. For example, in kidney, renal cortical tubular and Bowman’s capsule basement membranes strongly bind endostatin, whereas glomerular basement membrane largely fails to do so (Figure 1C) ▶ . Breast ductal and acinar basement membranes bind endostatin strongly (Figure 1D) ▶ . Binding to epidermal basement membrane in skin is more variable. Some stretches of dermal-epidermal junction showing weak binding alternate with sections lacking any binding interaction (Figure 1J) ▶ . Table 1 ▶ gives a summary description of the findings in different human tissues. Overall, the endostatin binding pattern closely resembles the distribution reported for collagen XVIII, endostatin’s parent molecule. 13 This observation suggests that the same binding partners within basement membranes govern both endostatin and collagen XVIII localization. In addition, this finding also suggests that the primary tissue-targeting domain of collagen XVIII resides in the most carboxyl-terminal endostatin portion of the molecule. No fluorescent signal was observed in negative controls where the HA-endostatin incubation step has been omitted, except for autofluorescence present in several tissues (eg, necrotic debris in intraductal comedo carcinoma (Figure 1G) ▶ , in colon mucosa, and some nonspecific nuclear staining; data not shown).
Table 1.
Endostatin Binding in Normal Human Tissues
| Tissue | Structure | ES binding intensity |
|---|---|---|
| Skin | Dermal blood vessels | ++ |
| Epidermal BM | + | |
| Skin appendage BM (hair follicles, sweat glands etc.) | +++ | |
| Breast | Blood vessels | +++ |
| Epithelial (ducts, acini) BM | +++ | |
| Lung | Blood vessels | +++ |
| Alveolar BM | ++ | |
| Colon | Blood vessels (entire wall thickness) | ++ |
| Mucosal (crypt) BM | − | |
| Uterus | Blood vessels | +++ |
| Myometrial artery intima | +++ | |
| Endometrial arterioles (intima and media) | ++ | |
| Endometrial gland BM | − | |
| Urinary bladder | Blood vessels (entire wall thickness) | +++ |
| Urothelial BM | +++ | |
| Detrusor (smooth) muscle | + | |
| Kidney | Blood vessels (arterioles, extraglomerular capillaries) | ++ |
| Glomerular (capillary) BM | − | |
| Tubular and Bowman’s capsule BM | +++ |
Frozen sections of normal human tissues were incubated with HA-tagged endostatin. Bound endostatin was labeled with anti-HA antibodies and visualized by fluorescence microscopy. Signal intensity in different tissue compartments was evaluated. Signal intensity is graded as negative (−), weak (+), moderate (++), or strong (+++). BM, basement membrane.
Endostatin binding is detected in both normal and neoplastic tissues without obvious differences in amount or affinity. In ductal carcinoma in situ (DCIS), the garland of newly formed capillaries that characteristically surrounds this lesion, 14,15 already contains endostatin binding sites (Figure 1G) ▶ . In angiogenic hot spots 16 of invasive ductal carcinomas, ample endostatin binding is observed (Figure 1E) ▶ . Endostatin binding and factor-VIII-related antigen often do not coincide, as endostatin binding sites can dominate over factor-VIII-related antigen immunoreactivity (Figure 1E) ▶ or vice versa (Figure 1F) ▶ .
As both the angiogenic FGF-2 and the anti-angiogenic endostatin share an affinity for heparan sulfate, we compared their binding on tissues in situ. In skin, FGF-2 localizes strongly to epidermal basement membrane, to basement membranes of the dermal vasculature, and to keratinocyte surfaces (Figure 1M) ▶ . Endostatin binding is similar to that of FGF-2 except that epidermal basement membrane binding is less pronounced and that epidermal keratinocyte binding is absent (Figure 1J) ▶ . In breast cancer tissue, the binding patterns of endostatin and FGF-2 are similar, with blood vessels as conspicuous binding targets (Figure 1, K and N) ▶ . FGF-2 binding sites consist entirely of heparan sulfate glycosaminoglycan chains, proven by their susceptibility to digestion by heparitinase enzyme (Figure 1O) ▶ . In contrast, endostatin binding is not affected by heparitinase treatment (Figure 1L) ▶ . Similarly, chondroitinase digestion has no effect on endostatin binding (not shown). The discreteness of the binding sites is proven further by reciprocal competition experiments between FGF-2 and endostatin. The presence of a 300-fold excess of endostatin does not prevent or reduce binding of FGF-2 to blood vessels within breast carcinoma tissue, even when the tissue sections are preincubated with the competitor for 30 minutes (Figure 1, H and I) ▶ . Similarly, the addition of 100-fold excess FGF-2 does not reduce the binding of HA-endostatin to its target sites (not shown). In addition, preincubating endostatin with high concentrations of heparin diminishes the binding signal only marginally (likely a nonspecific blocking effect), whereas FGF-2 binding is completely eliminated (not shown). A comparison of equimolar preparations of raw conditioned medium, heparin-column eluate, and heparin column flow-through reveals no apparent differences in tissue binding. The latter observation supports the notion that COS-7 cell-derived heparan sulfates play no role in masking potential endostatin-tissue heparan sulfate interactions. These results indicate that endostatin binding in situ is not heparan sulfate or chondroitin sulfate dependent despite the prominent heparan sulfate binding site predicted by three-dimensional modeling. 7 Endostatin has been reported to inhibit FGF-2-induced endothelial cell proliferation. 2 The discreteness of FGF-2 and endostatin binding demonstrated in situ makes it appear unlikely that competitive displacement of FGF-2 by endostatin could be responsible for that effect.
In summary, we show that on tissue sections, recombinant endostatin binds predominantly to its functional target, ie, blood vessels, in a pattern consistent with binding to basement membranes. However, binding to the basal aspect of endothelial cells or additional low levels of binding to endothelial cell surface cannot be excluded. We further show that these binding sites are not heparan sulfates. Efforts to positively identify the nature of the endostatin binding partner(s) are currently ongoing. Other investigators have identified in vitro binding of endostatin and of the entire noncollagenous domain of collagen XVIII to laminin-1, the proteoglycan perlecan, and fibulin-1 and 2. 5 These molecules are present in basement membranes, with laminin-1 being one of the most abundant components of the basement membrane. 17
The biological significance of endostatin binding sites identified in situ is unknown. However, the localization primarily to blood vessels, including angiogenic hot spots, makes an important role in mediating endostatin’s anti-angiogenic effect likely. Angiostatin has a potency similar to endostatin as an inhibitor of angiogenesis. Indeed, these agents show synergism when co-administered to animals. 3 Angiostatin has been shown to induce endothelial cell apoptosis, potentially by hindering the formation of a proper adhesion complex. 18 Despite structural differences between the two molecules, interference with cell adhesion appears to be an attractive theory for endostatin activity also, especially in light of its binding to subendothelial matrix. On the other hand, a recent interpretation of crystallographic data has revealed a zinc binding site, possibly involved in homodimer formation and resembling metal ion binding sites in metalloproteases. 19 This domain occupied by a zinc ion is required for in vivo activity of endostatin. 20
Acknowledgments
We thank Dr. Judah Folkman for generously providing us with reagents and Dr. Thomas Boehm for helpful discussions.
Footnotes
Address reprint requests to Dr. Andreas Friedl, University of Wisconsin-Madison, Department of Pathology and Laboratory Medicine, 5250 Medical Sciences Center, 1300 University Avenue, Madison, WI 53706. E-mail: afriedl@facstaff.wisc.edu.
References
- 1.Hanahan D, Folkman J: Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86:353-364 [DOI] [PubMed] [Google Scholar]
- 2.O’Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, Flynn E, Birkhead JR, Olsen BR, Folkman J: Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell 1997, 88:277-285 [DOI] [PubMed] [Google Scholar]
- 3.Boehm T, Folkman J, Browder T, O’Reilly MS: Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature 1997, 390:404-407 [DOI] [PubMed] [Google Scholar]
- 4.Standker L, Schrader M, Kanse SM, Jurgens M, Forssmann WG, Preissner KT: Isolation and characterization of the circulating form of human endostatin. FEBS Lett 1997, 420:129-133 [DOI] [PubMed] [Google Scholar]
- 5.Sasaki T, Fukai N, Mann K, Goehring W, Olsen BR, Timpl R: Structure, function, and tissue forms of the C-terminal globular domain of collagen XVIII containing the angiogenesis inhibitor endostatin. EMBO J 1998, 17:4249-4256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Reilly MS, Holmgren L, Shing Y, Chen C, Rosenthal RA, Moses M, Lane WS, Cao Y, Sage EH, Folkman J: Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell 1994, 79:315-328 [DOI] [PubMed] [Google Scholar]
- 7.Hohenester E, Sasaki T, Olsen BR, Timpl R: Crystal structure of the angiogenesis inhibitor endostatin at 1.5 angstrom resolution. EMBO J 1998, 17:1656-1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rapraeger AC, Krufka A, Olwin BB: Requirement of heparan sulfate for bFGF-mediated fibroblast growth and myoblast differentiation. Science 1991, 252:1705-1708 [DOI] [PubMed] [Google Scholar]
- 9.Gitay-Goren H, Soker S, Vlodavsky I, Neufeld G: The binding of vascular endothelial growth factor to its receptors is dependent on cell surface-associated heparin-like molecules. J Biol Chem 1992, 267:6093-6098 [PubMed] [Google Scholar]
- 10.Friedl A, Chang Z, Tierney A, Rapraeger AC: Differential binding of FGF-2 and FGF-7 to basement membrane heparan sulfate: comparison of normal and abnormal human tissues. Am J Pathol 1997, 150:1443-1455 [PMC free article] [PubMed] [Google Scholar]
- 11.Noonan DM, Fulle A, Valente P, Cai S, Horigan E, Sasaki M, Yamada Y, Hassell JR: The complete sequence of perlecan, a basement membrane heparan sulfate proteoglycan, reveals extensive similarity with laminin A chain, low density lipoprotein-receptor, and the neural cell adhesion molecule. J Biol Chem 1991, 266:22939-22947 [PubMed] [Google Scholar]
- 12.Murdoch AD, Dodge GR, Cohen I, Tuan RS, Iozzo RV: Primary structure of the human heparan sulfate proteoglycan from basement membrane (HSPG2/perlecan): a chimeric molecule with multiple domains homologous to the low density lipoprotein receptor, laminin, neural cell adhesion molecules, and epidermal growth factor. J Biol Chem 1992, 267:8544-8557 [PubMed] [Google Scholar]
- 13.Rehn M, Pihlajaniemi T: Identification of three N-terminal ends of type XVIII collagen chains and tissue-specific differences in the expression of the corresponding transcripts. The longest form contains a novel motif homologous to rat and Drosophila frizzled proteins. J Biol Chem 1995, 270:4705-4711 [DOI] [PubMed] [Google Scholar]
- 14.Engels K, Fox SB, Whitehouse RM, Gatter KC, Harris AL: Distinct angiogenic patterns are associated with high-grade in situ ductal carcinomas of the breast. J Pathol 1997, 181:207-212 [DOI] [PubMed] [Google Scholar]
- 15.Guidi AJ, Fischer L, Harris JR, Schnitt SJ: Microvessel density and distribution in ductal carcinoma in situ of the breast. J Natl Cancer Inst 1994, 86:614-619 [DOI] [PubMed] [Google Scholar]
- 16.Weidner N, Semple JP, Welch WR, Folkman J: Tumor angiogenesis and metastasis: correlation in invasive breast carcinoma. N Engl J Med 1991, 324:1-8 [DOI] [PubMed] [Google Scholar]
- 17.Timpl R, Brown JC: Supramolecular assembly of basement membranes. Bioessays 1996, 18:123-132 [DOI] [PubMed] [Google Scholar]
- 18.Claesson-Welsh L, Welsh M, Ito N, Anand-Apte B, Soker S, Zetter B, O’Reilly M, Folkman J: Angiostatin induces endothelial cell apoptosis and activation of focal adhesion kinase independently of the integrin-binding motif RGD. Proc Natl Acad Sci USA 1998, 95:5579-5583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding YH, Javaherian K, Lo KM, Chopra R, Boehm T, Lanciotti J, Harris BA, Li Y, Shapiro R, Hohenester E, Timpl R, Folkman J, Wiley DC: Zinc-dependent dimers observed in crystals of human endostatin. Proc Natl Acad Sci USA 1998, 95:10443-10448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boehm T, Oreilly MS, Keough K, Shiloach J, Shapiro R, Folkman J: Zinc-binding of endostatin is essential for its antiangiogenic activity. Biochem Biophys Res Commun 1998, 252:190-194 [DOI] [PubMed] [Google Scholar]