

Abstract
Transforming growth factor-βs (TGF-βs) are multifunctional growth factors that are secreted as inactive (latent) precursors in large protein complexes. These complexes include the latency-associated propeptide (LAP) and a latent transforming growth factor-β binding protein (LTBP). Four isoforms of LTBPs (LTBP-1–LTBP-4) have been cloned and are believed to be structural components of connective tissue microfibrils and local regulators of TGF-β tissue deposition and signaling. By using a gene trap strategy that selects for integrations into genes induced transiently during early mouse development, we have disrupted the mouse homolog of the human LTBP-4 gene. Mice homozygous for the disrupted allele develop severe pulmonary emphysema, cardiomyopathy, and colorectal cancer. These highly tissue-specific abnormalities are associated with profound defects in the elastic fiber structure and with a reduced deposition of TGF-β in the extracellular space. As a consequence, epithelial cells have reduced levels of phosphorylated Smad2 proteins, overexpress c-myc, and undergo uncontrolled proliferation. This phenotype supports the predicted dual role of LTBP-4 as a structural component of the extracellular matrix and as a local regulator of TGF-β tissue deposition and signaling.
Keywords: LTBP, TGF-β, extracellular matrix, colorectal cancer, pulmonary emphysema, cardiomyopathy
Transforming growth factor-βs (TGF-βs) belong to a protein superfamily whose members are necessary for normal development. They also control cell growth and differentiation in a variety of adult tissues and are involved in a wide range of immune responses (Massague and Wotton 2000; Massague et al. 2000). Most cells secrete TGF-β as a functionally inactive, latent cytokine complexed to its latency-associated propeptide (LAP), which interacts with its receptors only after activation. There are two known, structurally different latent complexes of TGF-β. The small latent complex consists of a mature TGF-β dimer bound to the N-terminal portion of the TGF-β–LAP propeptide. The large latent complex contains, in addition to TGF-β–LAP, one of the latent TGF-β-binding proteins (LTBPs). Unless displaced from the LTBPs after secretion, TGF-β is usually deposited in the extracellular matrix (ECM) as a high-molecular-weight complex with LTBP and LAP (Taipale et al. 1998; Koli et al. 2001).
LTBPs belong to the family of fibrillin/LTBP glycoproteins. More than 60% of the protein structure of the LTBP family is composed of repetitive EGF-like domains and 8-cystein repeats, both of which are believed to mediate protein–protein interactions. Like the fibrillins, LTBPs are structural components of the extracellular matrix (ECM) microfibrils, which associate with elastic fibers. Although LTBPs are not involved directly in TGF-β latency, most cells secrete TGF-β in complex with LTBP and, by targeting TGF-β to the ECM, form extracellular deposits of TGF-β. This is believed to provide tissues with a rapidly inducible and highly localized TGF-β signal (Taipale et al. 1998; Koli et al. 2001).
Four isoforms of LTBPs (LTBP-1–LTBP-4) have been cloned thus far (Tsuji et al. 1990; Moren et al. 1994; Yin et al. 1995; Giltay et al. 1997; Saharinen et al. 1998). Each has several splice variants, and at least one (LTBP-1) has two alternative promoters (Koski et al. 1999). The expression of both, isoforms and splice variants, appears tightly controlled in a tissue-specific manner.
Although multiple in vitro studies predicted a dual role for LTBPs as structural components of the extracellular matrix (ECM) and as local regulators of TGF-β signaling (Taipale et al. 1998), their functions at the organismal level are largely unknown. Here we show that severely reduced expression of LTBP-4 (Saharinen et al. 1998), caused by a gene trap integration into the mouse LTBP-4 gene, causes a severe pulmonary emphysema-like condition associated with cardiomyopathy and colorectal cancer in transgenic mice. These abnormalities are correlated with a major fragmentation of elastin fibers and a defective TGF-β signaling.
Results
A stem cell clone recovered from a gene trap screen generates a complex disease phenotype in transgenic mice
From a larger screen for gene trap integrations into genes induced during embryonic stem (ES) cell differentiation (Thorey et al. 1998), we obtained one integration (3C7) that generated a complex disease phenotype when bred to homozygosity in transgenic mice (Table 1).
Table 1.
Summary of abnormalities developing in 3C7 mice
| Age (weeks)
|
Lung
|
Heart
|
Colon
|
Number of processed mice
|
|---|---|---|---|---|
| 4–6 | Mild emphysema | Normal | Mucosal prolapse syndrome of the rectum | 18 |
| 12–14 | Moderate emphysema, rupture of septal walls | Moderate cardiomyopathy with right ventricular dilatation (cardiac body weight index increased >10%) | Colorectal adenoma | 22 |
| 24–32 | Severe emphysema with extensive atelectatic areas | Cardiomyopathy with biventricular dilatation (cardiac body weight index increased >30%) | Invasive colorectal carcinoma | 14 |
All the other inspected organs and tissues were microscopically normal. Analysis included aorta, liver, kidney, intestine, lymph nodes, brain, bone marrow, thymus, adrenals, pituitary, thyroid, pancreas, skin, uterus, and testicles.
By the age of 4 wk, 3C7 mice developed a rectal prolapse induced by an extensive edema in the mucosal layer of the colorectal region (Fig. 1A). Microscopically, the region showed solitary ulcers associated with epithelial hyperplasia and occasional crypts infiltrating the submucosal layer of the colonic wall (Fig. 1C,D). These early lesions are typical for a mucosa prolapse syndrome and most likely reflect some structural abnormalities of the colonic wall (du Boulay et al. 1983). At older ages (12–14 wk) most animals developed adenomas consisting of enlarged and abnormally branched cystic glands with increased numbers of goblet cells (Fig. 1E,F). By the age of 6–8 mo, all mice developed visible tumors in the distal portion of the colorectum with diameters of 0.7–1.5 cm (Fig. 1B). Histologically, these tumors had all features of colorectal adenocarcinomas and were formed by enlarged and dysplastic crypts lined by neoplastic epithelial cells. Crypts showed various degrees of epithelial cell detachment and infiltrated all layers of the colorectal wall (Fig. 1G,H). Interestingly, these lesions are similar if not identical to the colorectal cancers developing in Smad3 knockout mice (Zhu et al. 1998). Because Smad3 is a downstream target of TGF-β, this similarity suggested defective TGF-β signaling in 3C7 mice.
Figure 1.
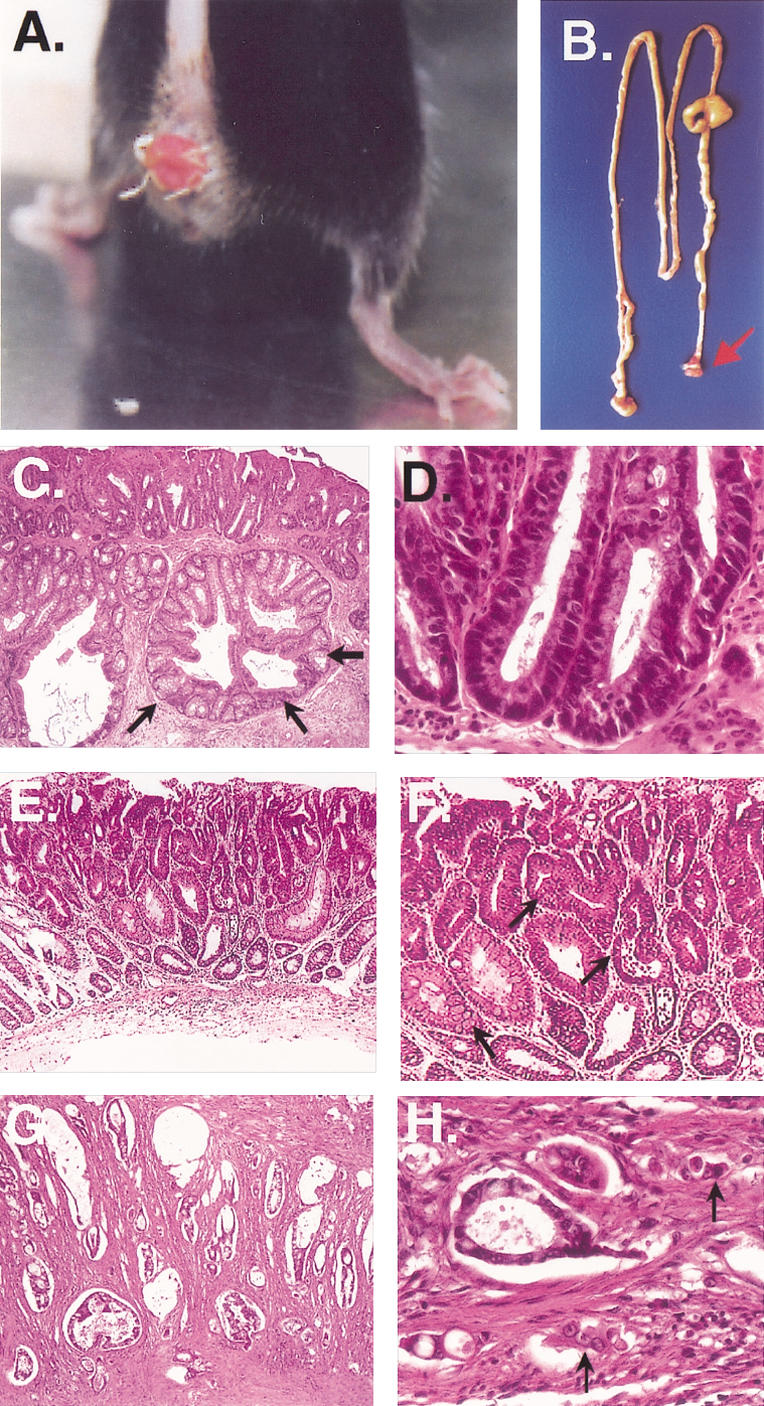
Colorectal cancer in 3C7 mice (−/− = hypomorphic LTBP-4 alleles). (A) Rectal prolapse and (B) digestive tract preparation with colorectal tumor (arrow) of a 4-month-old mouse. (C,D) Mucosal prolapse syndrome of the rectum. Superficial rectal ulceration and well circumscribed pseudoinvasion of crypts into the submucosa (arrows; C, original magnification 100×). Nonneoplastic rectal crypts with regenerative epithelial cells (D, H/E stain, original magnification 600×). (E,F) Colorectal adenoma showing aberrant crypt foci (E, H/E stain, original magnification 100×) with dysplastic crypts (arrows) and increased number of goblet cells (arrow; F, detail of E). (G,H) Invasive rectal adenocarcinoma. Neoplastic tubules invade the muscularis propria (G, H/E stain, original magnification 100×), and show dissociation with small tumor cell clusters (arrows) at the invasive front (H, H/E stain, original magnification 600×).
In addition to colorectal cancer, 3C7 mice developed a pulmonary emphysema-like condition, which, unlike the colonic tumors, was already present at birth. The emphysema worsened with time so that by the age of 6–8 mo the lungs of 3C7 mice exceeded normal size by threefold (Fig. 2A,B). Histologically, alveolar spaces were enlarged, inflated, and significantly reduced in number. They were surrounded by thin, dysplastic, and frequently incomplete septal walls (Fig. 2B). The lobular connective tissue was diminished and replaced by multifocal atelectatic areas. This resulted in an almost complete loss of pulmonary elasticity. The emphysema was associated with a cardiomyopathy that initially involved only the right heart. However, disease progression eventually led to biventricular dilatation and ubiquitous myofibrillar hypertrophy (Table 1; Fig. 2C). By the age of 6–8 mo the majority of the mutant mice had a cardiac/body weight index exceeding normal values by 30%.
Figure 2.

Pulmonary emphysema-like condition and cardiomyopathy in 3C7 mice. (A) Heart/lung preparations from wild-type (+/+) and homozygous 3C7 (−/−, hypomorphic LTBP-4 alleles) mice. (B) Lungs, H/E stain at 200× magnification. (C) Hearts, H/E stain at 200× magnification.
Clinically, the animals were healthy up to 6 mo of age, but thereafter their general condition deteriorated. Most mice had to be killed between 6 and 8 mo of age because of severe dyspnea and rectal prolapse infections that frequently progressed toward ulcerative proctitis. Autopsy revealed massive inflammatory infiltrations of the colonic wall associated with a reactive splenomegaly.
As heterozygous mice were indistinguishable from wild-type mice, and transgene inheritance was consistent with a Mendelian pattern (data not shown), we concluded that the gene trap had disrupted the function of an autosomal recessive gene. Although the described phenotype was stable over 6 generations of back-crossings to C57BL6 mice, abnormalities were more severe in 129/SvJ mice. Accordingly, most homozygous 129/SvJ F1 mice died during the first 10 d of life.
The gene trap integration in 3C7 mice is in the fifth intron of the LTBP-4 gene
To identify the gene responsible for the 3C7 phenotype, we first used 5′ RACE to isolate cellular sequence cotranscribed with the provirus (gene trap sequence tag, GTST; Wiles et al. 2000). Normally, the type of retroviral gene trap used in these experiments selects for integrations into exons (exon trap), and in most cases sequences upstream of the integration site are transcribed as cell–provirus fusion transcripts that can be easily identified by RT-PCR (Wempe et al. 2001). However, 5′ RACE was unsuccessful, as was the detection of cell–provirus fusion transcripts by Northern blotting (Fig. 3B). This suggested that in this case the gene trap, which is normally transcribed from an upstream cellular promoter, was activated instead by a nearby cellular enhancer. Although quite infrequent, we have observed enhancer activations with this type of gene trap, as it still contains a minimal promoter in the 5′-LTR (Fig. 3B; Thorey et al. 1998). Taken together, these results indicated that the gene trap had integrated into an intron and was therefore inaccessible to cloning by RT-PCR. To overcome this problem, we used inverse PCR and genome walking to isolate the genomic sequence spanning the provirus integration site. A 781-nt contig was cloned and sequenced and used to find matches in the public databases. As no matches were found after extensive searching, we assumed that the trapped gene was still unknown. To identify the gene, cDNA and genomic libraries were screened with the genomic contig, thus resulting in the isolation of a 12-kb genomic fragment with two short stretches of homology to the human cDNA encoding the latent transforming growth factor-β binding protein 4 (LTBP-4; GenBank accession nos. AF051344, NM_003573.1, AF051345). The matching segments corresponded to exons 6 and 7 encoding the 5′-end of the first EGF repeat, which is shared among known splice variants (Fig. 3A; Saharinen et al. 1998).
Figure 3.

Gene trap disruption of the LTBP-4 gene locus. (A) Predicted intron/exon structure of the mouse LTBP-4 gene with the gene trap integration site. The exon/intron structure was compiled by using the mouse cDNA sequences for the short and long 5′ splice variants (GenBank accession nos. AF410798 and AF410799), the human cDNA sequences for the various 5′ splice variants (GenBank accession nos. NM_003573, AF054502, AF054501, AF051345, AF051344, and Y13622), and the available genomic sequence of mouse chromosome 7 (GenBank accession no. AC073713). EGF domains and 8Cy repeats are shaded in dark and light gray, respectively. (B) Structure of the U3Cre gene trap provirus (top) and Northern blot analysis of 3C7 ES cells (bottom). Polyadenylated RNAs from ES cells that were grown for 3 d in the absence of LIF and feeder layers were blotted onto nylon filters and hybridized to 32P-labeled Cre- or ribosomal protein L32-specific probes. (C) LTBP-4 expression in wild-type (+/+) and homozygous 3C7 (−/−, hypomorphic LTBP-4 alleles) mice. (Top) Northern blot analysis. Polyadenylated RNAs from selected mouse tissues were blotted onto nylon filters and hybridized to a 32P-labeled LTBP-4 probe. The probe was a 505-nt fragment extending between nucleotides 882 and 1367 of the LTBP-4S cDNA (GenBank accession no. AF410798; for location, see A). This sequence stretch is shared by all known splice variants. (Bottom) Immunoblotting with human anti-LTBP-4 and anti-LTBP-1 antibodies. Proteins from selected tissue digests (see Materials and Methods) were separated on denaturing polyacrylamide gels under reducing conditions, transferred to nitrocellulose membranes, and reacted with polyclonal anti-LTBP-1 or anti-LTBP-4 antibodies raised against the third and fourth 8-Cystein repeats that are shared between known splice variants (Saharinen et al. 1998).
Figure 3A shows that the gene trap integration had occurred into the fifth intron of the mouse LTBP-4 gene in a reverse transcriptional orientation relative to the gene. This inverted orientation explains the absence of a cell–provirus fusion transcript and supports a gene trap activation by a cellular enhancer.
The proviral integration in LTBP-4 severely interrupts gene expression
To investigate whether the proviral intron disruption interfered with gene expression, mRNA from a variety of mouse tissues was hybridized on Northern blots to an LTBP-4-specific probe. As shown in Figure 3C (top), mice homozygous for the gene trap integration expressed <2% of normal LTBP-4 mRNA. Transcript levels in wild-type mice were significantly higher in the lung, heart, and colon than in other tissues, suggesting that tissues that normally express high levels of LTBP-4 are also the most affected by the mutation (Fig. 3C).
To test whether the protein levels were similarly decreased, the same range of tissues was analyzed by immunoblotting using an anti-LTBP-4-specific antibody (Saharinen et al. 1998). As shown in Figure 3C (bottom), the protein was hardly detectable in 3C7 mice, but the levels in various tissues of wild-type mice closely followed the pattern of mRNA expression. Taken together, the results indicate that the gene trap integration into the fifth intron of the LTBP-4 gene resulted in a nearly null allele.
Elastic fibers are fragmented in the lung and colon of 3C7 mice
Together with fibrillins, LTBPs are believed to be components of the sheath of microfibrils that surrounds the elastic fiber's elastin core (Saharinen et al. 1999). Using immuno-electron microscopy (IEM) and immunogold labeling, LTBP-1 and LTBP-2 were shown to associate with the extracellular matrix microfibrils (Taipale et al. 1996; Shipley et al. 2000). Although IEM studies have not yet been performed with the other LTBPs, mainly because of the lack of high-avidity antibodies, in vitro immunofluorescence studies indicate that LTBP-4 has an ECM distribution similar to LTBP-1 and LTBP-3 (K. Koli and J. Keski-Oja, unpubl.). Because some evidence suggests that microfibrils regulate elastic fiber formation by guiding tropoelastin deposition during embryogenesis and early postnatal life (Pereira et al. 1997), we thought that lack of LTBP-4 might alter the ECM's microfibril structure and thus the integrity of elastic fibers, particularly in the lung and colon, which both lost elasticity in 3C7 mice. To test this, we visualized elastic fibers using an elastin-specific histological stain. Figure 4 shows that the lung and colon of 3C7 mice lacked normal elastic fibers and, instead, showed multiple patches of fragmented and condensed elastin. Unexpectedly, elastic fibers in the heart appeared unaltered by the mutation, suggesting that the cardiomyopathy is mostly secondary to the emphysema (Fig. 4). However, because LTBP-4 expression is comparatively high in the heart (Fig. 3C), we cannot exclude a contribution of the mutation to the cardiac phenotype. Taken together, the results suggest that in selected tissues, LTBP-4 interferes with the normal development of elastic fibers in a tissue-specific manner.
Figure 4.

Fragmentation of elastic fibers in 3C7 mice. To visualize elastic fibers (arrows), lung sections of 12-week-old mice were stained with Weigert's resorcin-fuchsin and photographed at 400× magnification (+/+, wild-type mice; −/−, 3C7 mice with hypomorphic LTBP-4 alleles).
Deposition of TGF-β1 to the ECM requires LTBP-4
We found recently that of all three TGF-β isoforms, only TGF-β1 covalently associates with LTBP-4 in vitro (Saharinen and Keski-Oja 2000). Because LTBPs are responsible for the export and deposition of TGF-β in the ECM, this isoform-specific binding suggested that mice lacking LTBP-4 would also lack extracellular TGF-β1. To test this, we used anti-TGF-β1 antibodies that recognize TGF-β1 epitopes selectively displayed inside (LC) or outside (CC) the cell (Flanders et al. 1989; Thompson et al. 1989). Immunohistochemical analysis showed that in 3C7 mice, the lung, colon, and heart contained no detectable extracellular TGF-β1 (Fig. 5). As intracellular levels were essentially normal, the result suggests a defect in export and/or deposition of small latent TGF-β1 (TGF-β1–LAP) in the ECM. This defect appears to be tissue-specific because the kidneys of 3C7 mice had normal levels of extracellular TGF-β1 (Fig. 5).
Figure 5.

Lack of extracellular TGF-β1 in 3C7 mice. To visualize the TGF-β1 deposition in the ECM, tissue sections from 8-week-old mice were stained with polyclonal TGF-β antibodies specific for the intracellular (LC) or extracellular (CC) form of TGF-β1 (see Materials and Methods) and photographed at 200× magnification (+/+, wild-type mice; −/−, 3C7 mice with hypomorphic LTBP-4 alleles). Positive cells and matrix components are reddish-brown (arrows).
To determine whether the tissue-specific reduction in TGF-β1 matrix deposition would correlate with a reduced LTBP-4 transcription, we performed in situ hybridization experiments using oligonucleotide probes. As expected, the 3C7 bronchioalveolar and colonic crypt epithelial cells expressed significantly less LTBP-4 than the corresponding wild-type cells, suggesting a link between TGF-β1 matrix deposition and LTBP-4 expression (data not shown).
To investigate this further, we analyzed ECMs isolated from the colon and the kidneys of 3C7 mice by immunoblotting using an anti-TGF-β1–LAP antibody. As shown in Figure 6, TGF-β1–LAP/LTBP complexes were significantly below normal levels in the colonic ECM but not in the renal ECM, suggesting that TGF-β1 matrix deposition requires LTBP-4 only in the colon. Interestingly, although the anti-β1–LAP antibody recognizes any LTBP/LAPβ1 complex, it produces hardly any signal in the colonic ECM of 3C7 mice. In contrast, it readily visualizes LTBP/LAPβ1 complexes in the kidney ECM, which are similar to, or only slightly below wild-type levels (Fig. 6, bottom). Because only minimal LTBP-4 is transcribed in the 3C7 kidney (Fig. 3), it is very likely LAPβ1 is complexed to another LTBP in this organ. LTBP/LAPβ1 complex intensities are similarly weak in the mutant colonic ECM, regardless of whether they are visualized by anti-LTBP-4 (Fig. 6, upper left panel) or anti-LAPβ1 (Fig. 6, upper right panel) antibodies. Thus, it is quite plausible that in the colon, LTBP-4 is the only isoform responsible for TGF-β1 matrix deposition. Taken together, the results confirm the immunohistochemical analysis and underscore the tissue specificity of LTBP-4.
Figure 6.

Levels of large latent TGF-β1 complexes in 3C7 mice. Proteins from colon and kidney tissue digests (see Materials and Methods) were separated on denaturing polyacrylamide gels under nonreducing conditions, transferred to nitrocellulose membranes, and reacted with anti-LTBP-4 (αLTBP-4) or anti-LAPβ1 (αLAPβ1) antibodies. (A) Free LTBP-4 (left panel) and large latent LTBP complexes (right panel) in the colonic ECM of wild-type (+/+) and mutant 3C7 (−/−, hypomorphic LTBP-4 alleles) mice. (B) Levels of large latent LTBP complexes in the colonic and renal ECMs of 3C7 mice.
Lack of extracellular TGF-β is associated with reduced TGF-β signaling
As TGF-β is a strong inhibitor of epithelial cell proliferation and defective TGF-β signaling has been shown to play a role in the pathogenesis of cancer (Derynck et al. 2001), we hypothesized that the absence of TGF-β1 from the extracellular space might negatively interfere with TGF-β signaling.
To test this, we analyzed the proliferative status of colorectal tumor cells by immunohistochemistry using antibodies against the cell proliferation markers Ki-67 and c-myc. As shown in Figure 7, both markers were highly expressed in the colorectal region of mutant mice, which is consistent with increased proliferation. More importantly, c-myc was already elevated in the normal mucosa adjacent to the prolapse at 4 wk of age (Fig. 7), suggesting a more specific alteration of c-myc expression. As TGF-β is a potent inhibitor of c-myc expression (Alexandrow and Moses 1995; Massague and Wotton 2000), we presumed that its elevation is the result of a defective TGF-β signaling in the mutant mice. To investigate this further, we directly assessed the activation level of the TGF-β signal transduction pathway by estimating the levels of phosphorylated Smad2 proteins (Heldin et al. 1997) in the epithelial cells of various tissues. Immunohistochemical analysis with specific anti-Smad2 antibodies showed that P-Smad levels were decreased significantly in the lung and colon but not in the kidney of mutant mice (Fig. 8A). Because the tissue-specific pattern of TGF-β activation correlated with its deposition in the ECM, we conclude that LTBP-4 is critical for TGF-β signaling in the affected tissues.
Figure 7.

Increased cell proliferation and c-myc induction in the colon of 3C7 mice. Tissue sections were stained with polyclonal rabbit anti-mouse Ki-67 (top) or monoclonal anti-mouse c-myc antibodies (center and bottom) and photographed at 200× magnification (+/+, wild-type mice; −/−, 3C7 mice with hypomorphic LTBP-4 alleles). Positive cells are reddish-brown (arrows).
Figure 8.

Smad2 phosphorylation in 3C7 mice. (A) Decreased Smad2 phosphorylation in the epithelial cells of 3C7 mice. Tissue sections from 8-week-old mice were stained with monoclonal anti-Smad2,3 (Smad) or polyclonal anti-phosphorylated Smad2 (P-Smad) antibodies and photographed at 200× magnification (+/+, wild-type mice; −/−, 3C7 mice with hypomorphic LTBP-4 alleles). Positive cells are reddish-brown (arrows). (B) Induction of Smad2 phosphorylation by TGF-β1 in cultured lung fibroblasts. Fibroblasts were exposed for 1 h to 1 ng/mL TGF-β1. Cell lysates from semiconfluent cultures were separated on denaturing polyacrylamide gels, transferred to nitrocellulose membranes, and reacted with polyclonal anti-P-Smad2 or monoclonal anti-Smad2,3 antibodies.
Finally, to rule out the possibility that an intrinsic defect of the TGF-β signal transduction pathway could have been responsible for the reduced Smad activation, we tested the ability of TGF-β to induce Smad2 phosphorylation in cultured fibroblasts derived from 3C7 lungs. The immunoblot depicted in Figure 8B shows that the mutant fibroblasts responded normally to exogenous TGF-β, indicating that the TGF-β signaling defect is solely the result of its reduced deposition in the extracellular space.
Discussion
A strong hypomorphic allele of the mouse LTBP-4 gene was generated by gene trap mutagenesis in embryonic stem cells. Mice homozygous for the gene trap integration expressed only trace amounts of LTBP-4 and developed severe pulmonary emphysema, cardiomyopathy, and colorectal cancer. The abnormalities were associated with fragmented and disintegrated elastic fibers and with a defective TGF-β deposition in the extracellular space.
Interestingly, the hypomorphic allele was generated by an inverse orientation exon trap insertion into an intron of the LTBP-4 gene, which theoretically should not interfere with gene expression. However, intron insertions have been shown previously to cause mutations. Examples include the ob2J mutation induced by a retrotransposon insertion into the first intron of the mouse leptin gene (Moon and Friedman 1997), the mdr-3 mutation induced by a retroviral LTR insertion into the 22nd intron of the mouse multi-drug resistance gene mdr-3 (Jun et al. 2000), and the Mbp mutation induced by a retrotransposon insertion into the third intron of the rat myelin basic protein gene Mbp (O'Connor et al. 1999). In each case, aberrant splicing precluded the expression of a fully processed, full-length transcript. We found this to be similar in 3C7 cells, in which RT-PCR detected multiple atypical LTBP-4 transcripts (data not shown), indicating that the mutation was caused by aberrant splicing.
The disease model described here is consistent with a dual function of the TGF-β-binding proteins (Taipale et al. 1994). Whereas the absence of intact elastic fibers in the lung and colon underscores the structural requirement of LTBP-4, the lack of extracellular TGF-β implicates LTBP-4 in TGF-β signaling. As TGF-β inhibits epithelial cell proliferation, particularly in the colon, we believe that the lack of extracellular TGF-β is the major oncogenic trigger in a colorectal region already predisposed to cancer by a mucosa prolapse syndrome.
Several studies have associated defects in TGF-β signaling with colorectal cancer. For example, mice with null mutations in the TGF-β signal transducing protein Smad-3, develop tumors that are similar if not identical to the tumors growing in 3C7 mice (Zhu et al. 1998). Although the Smad-3 knockout phenotype could not be replicated by other groups (Datto et al. 1999; Yang et al. 1999), the link between TGF-β signaling defects and colorectal cancer is still compelling because it has been recognized in many other systems, including humans. Accordingly, mutations in the TGF-β signal transducing proteins Smad2 and Smad4 or mutations in the TGF-β II receptors are very common in human colorectal cancers, suggesting that TGF-β and its downstream targets have tumor suppressor functions (Markowitz et al. 1995; Riggins et al. 1997; Zhou et al. 1998, 1999). Whether LTBP-4 itself has such a function remains to be established, but its importance for the integrity of the ECM is likely to prevent cancer cell invasion and metastatic spread.
Whether LTBP-4 has a direct role in elastic fiber formation remains to be established. Because other LTBPs colocalize with fibrillins in the ECM (Taipale et al. 1996; Shipley et al. 2000), it is likely that LTBP-4 is also a component of the microfibril network. In vitro immunofluorescence studies showing similar ECM distribution patterns for all LTBPs, including LTBP-4, support this assumption (K. Koli and J. Keski-Oja, unpubl.). The microfibril network is believed to be crucial for elastic fiber formation because it appears to guide tropoelastin deposition during embryogenesis and early postnatal life (Pereira et al. 1997). Indeed, as has been shown recently, at least two components of the microfibril network, fibrillin 1 and fibrillin 2, are capable of binding tropoelastin, suggesting that the fibrillins (and, by inference, the microfibrils) play an important role in the assembly of elastic fibers (Trask et al. 2000).
One of the most interesting observations made in this study is the tissue selectivity of the LTBP-4 mutation. Only the lung, the colon, and possibly the heart developed disease directly related to the mutation, indicating that LTBP-4 is a highly tissue-specific protein. Such tissue specificity would predict that despite the extensive structural similarities, latent TGF-β-binding proteins have little or no overlapping functions. Accordingly, we found that mutant mice express almost unaltered levels of LTBP-1 and LTBP-2 proteins, suggesting that these isoforms cannot substitute for LTBP-4 (Fig. 3C; data not shown). Moreover, mice expressing null alleles of LTBP-2 die very early in development, most likely because of defective implantation (Shipley et al. 2000), and mice expressing null alleles of LTBP-3 develop skeletal malformations (Dabovic et al. 2002).
In conclusion, we have developed a mouse model of human disease that reveals some crucial functions of LTBP-4. In view of these functions, the LTBP-4 gene seems a likely candidate gene for human connective tissue disorders and cancer.
Materials and methods
ES cell injections, breeding, and genotyping
The 129/SvJ (D3) ES cell-derived chimeras were generated by injecting C57BL/6 blastocysts. The resulting male chimeras were bred to C57BL/6 females, and agouti offspring were tested for transgene transmission by tail blotting (see below). Animals heterozygous for the gene trap insertion were back-crossed to C57BL/6 mice for at least six generations before analyzing the phenotypes in heterozygous and homozygous offspring.
Cloning of provirus flanking sequences and screening of nucleic acid libraries
Inverse PCR from genomic 3C7 ES cell DNA was performed by using the blunt-end restriction enzyme SspI and Cre-specific primers as described previously (Wempe et al. 2001). A 210-nt amplification product was cloned into the p-GEMT-vector (Promega) and sequenced using an ABI 310 Genetic Analyzer (Perkin-Elmer). The upstream provirus flanking sequence was extended by genome walking using the mouse GenomeWalker kit (Clontech) according to the manufacturer's instructions and the specific flanking sequence forward primers 5′-AAGAGG GAGAAGAAACTGGGCTGG-3′ and 5′-TCCTGTAGTGTTT GCCCCTG-3′. A 781-nt genomic contig spanning the proviral integration site was amplified from the kit's EcoRV library and used as a probe to screen the λ-TripleEx 5′-Stretch PLUS mouse kidney cDNA library (Clontech) and the λ EMBL3 mouse liver genomic library (Clontech). Positive plaques were purified by standard methodology, and inserts were sequenced after subcloning into Bluescript vectors.
Nucleic acid hybridization analyses and PCR
Mouse-tail DNA was cleaved with BglII, which does not cut the provirus. The DNA was fractionated on 1% agarose gels, blotted onto Hybond N nylon filters (Amersham/Pharmacia), and hybridized to a 32P-labeled provirus flanking sequence probe. For Northern blots, 2.5 μg of poly(A)+ mRNAs purified with the FastTrack 2.0 mRNA purification kit (Invitrogen) were fractionated in 1% formaldehyde-agarose gels, transferred onto Hybond N nylon filters, and hybridized to a [32P]dCTP-labeled LTBP-4-specific probe. The probe was a 505-nt fragment extending from nucleotides 862 to 1367 of the mouse LTBP4S cDNA (GenBank accession no. AF410798). All probes were prepared by random priming using the Rediprime kit (Amersham/Pharmacia). Blots were exposed to Kodak-Biomax autoradiography film.
Immunoblotting
Equal amounts of selected tissues were homogenized in ice-cold RIPA buffer (150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate in 50 mM Tris-HCl at pH 7.2). Following centrifugation, the deoxycholate insoluble pellet was washed in PBS and digested with plasmin as described previously (Taipale et al. 1994). After adding protease inhibitors (Roche Diagnostics), the supernatants were concentrated in Microcon 30 concentrators (Amicon, Millipore Corp.). Proteins were fractionated by SDS/PAGE (7% acrylamide monomer concentration) and transferred to Protran nitrocellulose membranes (Schleicher & Schuell). Immunodetection after treatment with polyclonal anti-LTBP-1 or anti-LTBP-4 antibodies was performed as described previously (Saharinen et al. 1998).
For Smad2,3 detection, cultured lung fibroblasts were exposed to 1 ng/mL TGF-β1 (Oncogen) for 1 h and then lysed with ice-cold buffer (120 mM NaCl, 0.5% NP-40, 100 mM NaF, 1 mM Na3Vo4 in 20 mM Tris-HCl at pH 8.0; protease inhibitors added as Protease Inhibitor Cocktail, Roche Diagnostics). The lysates were cleared by centrifugation, and equal amounts of protein were separated by polyacrylamide gel (7% Tris-HCl) electrophoresis in the presence of SDS. Proteins were then electrophoretically transferred to Protran nitrocellulose membranes (Schleicher & Schuell). Immunodetection was performed as described (Dooley et al. 2001), using a polyclonal rabbit antibody raised against phosphorylated smad2 (kindly provided by Peter ten Dijke, The Netherlands Cancer Institute). The monoclonal mouse anti-Smad2,3 antibody and the LAP-β1 antibody (AF-246-NA) were purchased from Transduction Laboratories and from R&D Systems, respectively.
Database searches
Cellular sequences flanking the proviral integration site (gene trap sequence tag; GTST) were searched in the NCBI/NIH genomic databases (http://www.ncbi.nlm.nih.gov) using the BlastN algorithm.
Histology, histochemistry, and immunohistochemistry
Paraffin sections of mouse tissues were prepared and stained using standard histology procedures. To visualize elastic fibers, microscopic slides were stained with Weigert's resorcin-fuchsin solution (70% [v/v] ethanol, 1% [v/v] HCl, and 0.2% [v/v] resorcin-fuchsin; Chroma, Münster). For immunostainings, deparaffinized and rehydrated tissue slides were first treated for 30 min with 3% H2O2 to inactivate the endogenous peroxidase. After rinsing in ddH2O and soaking in PBS for 5–10 min, slides were treated with 10% (w/v) BSA in PBS to saturate nonspecific protein binding sites. The slides were then exposed at 4°C overnight to the specific monoclonal or polyclonal antibodies (see below). After removing excess antibody, slides were treated with appropriate biotin-labeled secondary antibodies (Dianova) at 37°C for 30 min and finally with horseradish peroxidase (HRP)-labeled streptavidine (Zymed) at 37°C for 20 min. After washing, slides were incubated in diaminobenzidine (Sigma) at room temperature for 10 min. Slides were examined under a conventional microscope after removing the excess substrate in ddH2O.
For immunohistochemical detection, pretested dilutions of the following antibodies were used: polyclonal mouse anti-TGF-β1-CC (1–30; Flanders et al. 1989), polyclonal mouse anti-TGF-β1-LC (1–30; Flanders et al. 1989), rabbit anti- mouse K-67 (Dianova), monoclonal anti-c-myc (Santa Cruz), monoclonal mouse anti-Smad2,3 (Transduction Laboratories), and rabbit polyclonal anti-P-Smad2 (kindly provided by Peter ten Dijke, The Netherlands Cancer Institute).
Acknowledgments
We thank Marko Hyytiäinen for helpful discussions; C.-H. Heldin, Ludwig Institute for Cancer Research, Uppsala, Sweden, for the anti-LTBP-1 antibody; Peter ten Dijke for the anti-P-Smad2 antibody; and Kathleen Flanders for the LC and CC anti-TGF-β1 antibodies. We acknowledge Armin Reffelmann and Joana Lorenz for excellent technical assistance. This work was supported by grants from the Deutsche Forschungsgemeinschaft and the VW-foundation to H.v.M. and by grants from the Academy of Finland, Finish Cancer Foundation, and Helsinki University Hospital to J.K.-O.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL melchner@em.uni-frankfurt.de; FAX 49-69-63016390.
Article and publication are at http://www.genesdev.org/cgi/doi/10.1101/gad.229102.
References
- Alexandrow MG, Moses HL. Transforming growth factor β and cell cycle regulation. Cancer Res. 1995;55:1452–1457. [PubMed] [Google Scholar]
- Dabovic B, Chen Y, Colarossi C, Obata H, Zambuto L, Perle MA, Rifkin DB. Bone abnormalities in latent TGF-β binding protein (Ltbp)-3-null mice indicate a role for Ltbp-3 in modulating TGF-β bioavailability. J Cell Biol. 2002;156:227–232. doi: 10.1083/jcb.200111080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datto MB, Frederick JP, Pan L, Borton AJ, Zhuang Y, Wang XF. Targeted disruption of Smad3 reveals an essential role in transforming growth factor β-mediated signal transduction. Mol Cell Biol. 1999;19:2495–2504. doi: 10.1128/mcb.19.4.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck R, Akhurst RJ, Balmain A. TGF-β signaling in tumor suppression and cancer progression. Nat Genet. 2001;29:117–129. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- Dooley S, Delvoux B, Streckert M, Bonzel L, Stopa M, ten Dijke P, Gressner AM. Transforming growth factor β signal transduction in hepatic stellate cells via Smad2/3 phosphorylation, a pathway that is abrogated during in vitro progression to myofibroblasts. TGFβ signal transduction during transdifferentiation of hepatic stellate cells. FEBS Lett. 2001;502:4–10. doi: 10.1016/s0014-5793(01)02656-4. [DOI] [PubMed] [Google Scholar]
- du Boulay CE, Fairbrother J, Isaacson PG. Mucosal prolapse syndrome—A unifying concept for solitary ulcer syndrome and related disorders. J Clin Pathol. 1983;36:1264–1268. doi: 10.1136/jcp.36.11.1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanders KC, Thompson NL, Cissel DS, Van Obberghen-Schilling E, Baker CC, Kass ME, Ellingsworth LR, Roberts AB, Sporn MB. Transforming growth factor-β 1: Histochemical localization with antibodies to different epitopes. J Cell Biol. 1989;108:653–660. doi: 10.1083/jcb.108.2.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giltay R, Kostka G, Timpl R. Sequence and expression of a novel member (LTBP-4) of the family of latent transforming growth factor-β binding proteins. FEBS Lett. 1997;411:164–168. doi: 10.1016/s0014-5793(97)00685-6. [DOI] [PubMed] [Google Scholar]
- Heldin CH, Miyazono K, ten Dijke P. TGF-β signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465–471. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- Jun K, Lee SB, Shin HS. Insertion of a retroviral solo long terminal repeat in mdr-3 locus disrupts mRNA splicing in mice. Mamm Genome. 2000;11:843–848. doi: 10.1007/s003350010176. [DOI] [PubMed] [Google Scholar]
- Koli K, Saharinen J, Hyytiainen M, Penttinen C, Keski-Oja J. Latency, activation, and binding proteins of TGF-β. Microsc Res Tech. 2001;52:354–362. doi: 10.1002/1097-0029(20010215)52:4<354::AID-JEMT1020>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Koski C, Saharinen J, Keski-Oja J. Independent promoters regulate the expression of two amino terminally distinct forms of latent transforming growth factor-β binding protein-1 (LTBP-1) in a cell type-specific manner. J Biol Chem. 1999;274:32619–32630. doi: 10.1074/jbc.274.46.32619. [DOI] [PubMed] [Google Scholar]
- Markowitz S, Wang J, Myeroff L, Parsons R, Sun L, Lutterbaugh J, Fan RS, Zborowska E, Kinzler KW, Vogelstein B, et al. Inactivation of the type II TGF-β receptor in colon cancer cells with microsatellite instability. Science. 1995;268:1336–1338. doi: 10.1126/science.7761852. [DOI] [PubMed] [Google Scholar]
- Massague J, Wotton D. Transcriptional control by the TGF-β/Smad signaling system. EMBO J. 2000;19:1745–1754. doi: 10.1093/emboj/19.8.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J, Blain SW, Lo RS. TGFβ signaling in growth control, cancer, and heritable disorders. Cell. 2000;103:295–309. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- Moon BC, Friedman JM. The molecular basis of the obese mutation in ob2J mice. Genomics. 1997;42:152–156. doi: 10.1006/geno.1997.4701. [DOI] [PubMed] [Google Scholar]
- Moren A, Olofsson A, Stenman G, Sahlin P, Kanzaki T, Claesson-Welsh L, ten Dijke P, Miyazono K, Heldin CH. Identification and characterization of LTBP-2, a novel latent transforming growth factor-β-binding protein. J Biol Chem. 1994;269:32469–32478. [PubMed] [Google Scholar]
- O'Connor LT, Goetz BD, Kwiecien JM, Delaney KH, Fletch AL, Duncan ID. Insertion of a retrotransposon in Mbp disrupts mRNA splicing and myelination in a new mutant rat. J Neurosci. 1999;19:3404–3413. doi: 10.1523/JNEUROSCI.19-09-03404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira L, Andrikopoulos K, Tian J, Lee SY, Keene DR, Ono R, Reinhardt DP, Sakai LY, Biery NJ, Bunton T, et al. Targetting of the gene encoding fibrillin-1 recapitulates the vascular aspect of Marfan syndrome. Nat Genet. 1997;17:218–222. doi: 10.1038/ng1097-218. [DOI] [PubMed] [Google Scholar]
- Riggins GJ, Kinzler KW, Vogelstein B, Thiagalingam S. Frequency of Smad gene mutations in human cancers. Cancer Res. 1997;57:2578–2580. [PubMed] [Google Scholar]
- Saharinen J, Keski-Oja J. Specific sequence motif of 8-Cys repeats of TGF-β binding proteins, LTBPs, creates a hydrophobic interaction surface for binding of small latent TGF-β. Mol Biol Cell. 2000;11:2691–2704. doi: 10.1091/mbc.11.8.2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saharinen J, Taipale J, Monni O, Keski-Oja J. Identification and characterization of a new latent transforming growth factor-β-binding protein, LTBP-4. J Biol Chem. 1998;273:18459–18469. doi: 10.1074/jbc.273.29.18459. [DOI] [PubMed] [Google Scholar]
- Saharinen J, Hyytiainen M, Taipale J, Keski-Oja J. Latent transforming growth factor-β binding proteins (LTBPs)—Structural extracellular matrix proteins for targeting TGF-β action. Cytokine Growth Factor Rev. 1999;10:99–117. doi: 10.1016/s1359-6101(99)00010-6. [DOI] [PubMed] [Google Scholar]
- Shipley JM, Mecham RP, Maus E, Bonadio J, Rosenbloom J, McCarthy RT, Baumann ML, Frankfater C, Segade F, Shapiro SD. Developmental expression of latent transforming growth factor β binding protein 2 and its requirement early in mouse development. Mol Cell Biol. 2000;20:4879–4887. doi: 10.1128/mcb.20.13.4879-4887.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale J, Miyazono K, Heldin CH, Keski-Oja J. Latent transforming growth factor-β 1 associates to fibroblast extracellular matrix via latent TGF-β binding protein. J Cell Biol. 1994;124:171–181. doi: 10.1083/jcb.124.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale J, Saharinen J, Hedman K, Keski-Oja J. Latent transforming growth factor-β 1 and its binding protein are components of extracellular matrix microfibrils. J Histochem Cytochem. 1996;44:875–889. doi: 10.1177/44.8.8756760. [DOI] [PubMed] [Google Scholar]
- Taipale J, Saharinen J, Keski-Oja J. Extracellular matrix-associated transforming growth factor-β: Role in cancer cell growth and invasion. Adv Cancer Res. 1998;75:87–134. doi: 10.1016/s0065-230x(08)60740-x. [DOI] [PubMed] [Google Scholar]
- Thompson NL, Flanders KC, Smith JM, Ellingsworth LR, Roberts AB, Sporn MB. Expression of transforming growth factor-β 1 in specific cells and tissues of adult and neonatal mice. J Cell Biol. 1989;108:661–669. doi: 10.1083/jcb.108.2.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorey IS, Muth K, Russ AP, Otte J, Reffelmann A, von Melchner H. Selective disruption of genes transiently induced in differentiating mouse embryonic stem cells by using gene trap mutagenesis and site-specific recombination. Mol Cell Biol. 1998;18:3081–3088. doi: 10.1128/mcb.18.5.3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trask TM, Trask BC, Ritty TM, Abrams WR, Rosenbloom J, Mecham RP. Interaction of tropoelastin with the amino-terminal domains of fibrillin-1 and fibrillin-2 suggests a role for the fibrillins in elastic fiber assembly. J Biol Chem. 2000;275:24400–24406. doi: 10.1074/jbc.M003665200. [DOI] [PubMed] [Google Scholar]
- Tsuji T, Okada F, Yamaguchi K, Nakamura T. Molecular cloning of the large subunit of transforming growth factor type β masking protein and expression of the mRNA in various rat tissues. Proc Natl Acad Sci. 1990;87:8835–8839. doi: 10.1073/pnas.87.22.8835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wempe F, Yang JY, von Melchner H. Gene trapping identifies transiently induced survival genes during programmed cell death. Genome Biol. 2001;2:research0023.1–research0023.10. doi: 10.1186/gb-2001-2-7-research0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiles MV, Vauti F, Otte J, Fuchtbauer EM, Ruiz P, Fuchtbauer A, Arnold HH, Lehrach H, Metz T, von Melchner H, et al. Establishment of a gene-trap sequence tag library to generate mutant mice from embryonic stem cells. Nat Genet. 2000;24:13–14. doi: 10.1038/71622. [DOI] [PubMed] [Google Scholar]
- Yang X, Letterio JJ, Lechleider RJ, Chen L, Hayman R, Gu H, Roberts AB, Deng C. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-β. EMBO J. 1999;18:1280–1291. doi: 10.1093/emboj/18.5.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin W, Smiley E, Germiller J, Mecham RP, Florer JB, Wenstrup RJ, Bonadio J. Isolation of a novel latent transforming growth factor-β binding protein gene (LTBP-3) J Biol Chem. 1995;270:10147–10160. doi: 10.1074/jbc.270.17.10147. [DOI] [PubMed] [Google Scholar]
- Zhou S, Buckhaults P, Zawel L, Bunz F, Riggins G, Dai JL, Kern SE, Kinzler KW, Vogelstein B. Targeted deletion of Smad4 shows it is required for transforming growth factor β and activin signaling in colorectal cancer cells. Proc Natl Acad Sci. 1998;95:2412–2416. doi: 10.1073/pnas.95.5.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S, Kinzler KW, Vogelstein B. Going mad with Smads. N Engl J Med. 1999;341:1144–1146. doi: 10.1056/NEJM199910073411510. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Richardson JA, Parada LF, Graff JM. Smad3 mutant mice develop metastatic colorectal cancer. Cell. 1998;94:703–714. doi: 10.1016/s0092-8674(00)81730-4. [DOI] [PubMed] [Google Scholar]