

Abstract
The plexiform lesions of severe pulmonary hypertension (PH) are complex vascular structures composed primarily of endothelial cells. In this study, we use immunohistochemical markers to identify the various cell layers of pulmonary vessels and to identify different endothelial cell phenotypes in pulmonary arteries affected by severe PH. Our computerized three-dimensional reconstructions of nine vessels in five patients with severe PH demonstrate that plexiform (n = 14) and concentric-obliterative (n = 6) lesions occur distal to branch points of small pulmonary arteries. And, whereas plexiform lesions occur as solitary lesions, concentric-obliterative lesions appear to be only associated with, and proximal to, plexiform structures. The endothelial cells of plexiform lesions express intensely and uniformly the vascular endothelial growth factor (VEGF) receptor KDR and segregate phenotypically into cyclin-kinase inhibitor p27/kip1-negative cells in the central core of the plexiform lesion and p27/kip1-positive cells in peripheral areas adjacent to incipient blood vessel formation. Using immunohistochemistry and three-dimensional reconstruction techniques, we show that plexiform lesions are dynamic vascular structures characterized by at least two endothelial cell phenotypes. Plexiform arteriopathy is not merely an end stage or postthrombotic change—it may represent one stage in an ongoing, angiogenic endothelial cell growth process.
Pulmonary hypertension (PH) with plexiform arteriopathy occurs in a sporadic, idiopathic, or primary form (PPH), as an inherited disease in the familial form, in association with a variety of diseases (eg, scleroderma, mixed connective tissue disorders, HIV infection, hepatic disease with portal hypertension), and with the use of appetite suppressants (including fenfluramine derivatives). 1-7 Structurally, the remodeling of the pulmonary arteries in severe PH involves an increase in vascular smooth muscle cell mass and endothelial cell proliferation, resulting in medial hypertrophy, concentric obliteration of the lumina, and complex vascular structures known as plexiform lesions.
Because of the patchy distribution of plexiform lesions, the vascular changes can be difficult to identify in random two-dimensional histological sections. Using traditional hematoxylin-eosin staining, plexiform vascular remodeling may be detected in only a fraction (10–20%) of the total pulmonary arteries sampled. 8,9 However, the degree of the often fixed pulmonary hypertension of these same patients indicates a significant (greater than 20%) obstruction of the precapillary pulmonary artery system. To address the impact of the vascular changes on pulmonary blood flow, we used three-dimensional computer modeling to reconstruct the cellular layers of the altered blood vessels. Previous three-dimensional studies of the pulmonary vasculature in PH have used casting methods, which permit analysis of regional relationships but cannot address the cellular composition of the vascular lesions. 10,11 Our present study provides the first reconstruction at a cellular level of the complex vascular structures that characterize severe secondary and primary pulmonary hypertensive disorders.
Previously, it has been felt that plexiform lesions were the end-stage result of either postthrombotic or myofibroblastic scarring of the pulmonary arteries. 12,13 Based on more recent observations, however, it is becoming clear that plexiform lesions are driven by endothelial cell growth—in effect, they are dynamic angiogenic lesions. 14,15 In fact, the Executive Summary from the World Symposium—Primary Pulmonary Hypertension 1998 states that the plexiform lesion may represent “endothelial cells that are involved prominently in angiogenesis, perhaps akin to a neoplastic process.” It is also possible that “endothelial markers can be applied to diagnose early lesions.” 16
The endothelial cells of plexiform lesions exhibit several phenotypic alterations that distinguish them from normal pulmonary endothelial cells. 17 For example, the endothelial cells of the plexiform lesions show a reduction or loss of prostacyclin synthase (PGI2S) 18 and nitric oxide synthase 19 and overexpress endothelin-1, 20 5-lipoxygenase, and 5-lipoxygenase activating protein, FLAP. 21 These phenotypical alterations may relate to vasomotor tone abnormalities, in situ thrombosis, or the endothelial cell proliferation observed in patients with severe PH. In addition, the recent finding that the proliferating endothelial cells in the plexiform lesions of PPH—including patients with dexfenfluramine-associated PH—are monoclonal whereas those of secondary PH are polyclonal suggests that a somatic genetic event allows for endothelial cell proliferation in PPH. 22,23
We hypothesize that the growth of the plexiform lesions depends on different developmental stages and phenotypes of pulmonary endothelial cells, such as those described as occurring during angiogenesis and vasculogenesis. 24,25 In the present study, we use immunohistochemical markers of endothelial cells and angiogenesis to identify these different endothelial cell phenotypes. We examine the three-dimensional organization of endothelial and smooth muscle cells, using the immunohistochemical markers factor VIII-related antigen (FVIII-r.ag) and muscle specific actin (MSA), respectively. In addition, we localize three-dimensionally the expression patterns of two “functional” cell markers, the vascular endothelial growth factor (VEGF) receptor KDR, which is expressed in early angiogenesis and vasculogenesis, 25 and p27/kip1, a cell cycle inhibitory protein marker of low growth potential. 26-28
Materials and Methods
Tissue Samples
Lung tissue was obtained from five patients with plexiform pulmonary arteriopathy. Lung tissue from an additional patient with normal pulmonary vessels was used for control studies. Four of the lung specimens were obtained at autopsy—one each from patients with PPH, liver cirrhosis, HIV, and scleroderma. The fifth specimen was the explanted right lung from a patient with PPH; the unused donor’s left lung provided the normal control tissue. Clinical data are summarized in Table 1 ▶ .
Table 1.
Clinical Data
| Patient no. | Diagnosis | Age | Gender | PA S/D/M* | Tissue source |
|---|---|---|---|---|---|
| 1 | PPH† | 49 | F | 93/36/58 | Surgical |
| 2 | PPH | 30 | F | 90/34/50 | Autopsy |
| 3 | Orthotopic liver transplant for hepatitis C | 51 | M | NA‡ | Autopsy |
| 4 | AIDS | 37 | M | NA | Autopsy |
| 5 | Scleroderma | 37 | F | 85/37/59 | Autopsy |
| 6 | Donor lung | Unknown | Unknown | Unknown | Surgical |
*PA S/D/M, Pulmonary artery pressures, systolic/diastolic/mean.
†PPH, Primary pulmonary hypertension.
‡NA, Not available.
When possible, each lung specimen was oriented in the anatomical position. Blocks of tissue were removed from the mid to distal portions of the lobes. The blocks were then cross-sectioned every 1–2 mm and placed in numbered cassettes to maintain the hilar-to-peripheral orientation. In some of the cases, archival tissue blocks were used without a priori knowledge of orientation. Orientation was deduced from the dichotomous branching pattern of the pulmonary vasculature. That is, serial sections were examined to determine the direction in which branching occurred. Branching was assumed to progress in a distal direction. All tissue was fixed in 10% formalin, processed, and embedded in paraffin.
Hematoxylin-eosin-stained sections were screened to select the tissue blocks with plexiform and/or concentric-obliterative lesions. All blocks examined showed plexiform arteriopathy. Five blocks from the two PPH cases, two blocks from the scleroderma case, and one block from each of the remaining non-PPH cases were selected for serial sectioning and computerized three-dimensional reconstruction. The representative blocks (nine total) were sectioned at 5-μm intervals. The total thickness of tissue that was serially sectioned and placed on Superfrost/Plus slides (Fisher Scientific, Pittsburgh, PA) for final reconstruction varied from 145 μm to 540 μm. The depth of cut was labeled on all slides, and planar orientation was maintained.
Immunohistochemistry
The various cell layers of the pulmonary vessels were examined using immunohistochemical staining for endothelial cells, smooth muscle cells, and inflammatory cells. The first slide of each case was stained with H&E, the second with FVIII-related antigen (FVIII-r.ag) (polyclonal, 1:150 dilution; Dako Corp., Carpinteria, CA), the third with muscle-specific actin (MSA) (monoclonal, 1:20 dilution; Enzo, Farmingdale, NY), the fourth with leukocyte common antigen (LCA) (monoclonal, 1:100 dilution; Dako Corp., Carpinteria, CA), and the fifth with CD68 (monoclonal, 1:50 dilution; Dako Corp.), a macrophage marker. This staining rotation was repeated for all remaining slides.
In addition, two tissue blocks from patient 2 (PPH) were serially sectioned every 5 μm (50 sections total) with a staining rotation of FVIII-r.ag, MSA, VEGF receptor KDR (polyclonal antibody, flk-1, 1:50 dilution; Santa Cruz Biotechnologies, Santa Cruz, CA), and p27/kip1 (monoclonal antibody, 1:1000 dilution; Transduction Laboratories, Lexington, KY).
Heat-induced antigen retrieval using pressure cooker heating in a sodium citrate solution was used to optimize immunostaining. After incubation with the primary antibody, immunodetection was performed using biotinylated anti-mouse (for monoclonal) or anti-rabbit (for polyclonal) immunoglobulins. Peroxidase-labeled streptavidin (Vector Laboratories, Burlingame, CA), with diaminobenzidine chromogen as the substrate (Vector Laboratories), completed the immunostaining.
The Ventana ES automatic immunostaining device was employed for the additional immunohistochemical staining of lung tissue from patient 2 (PPH).
Incubations with unrelated antibodies were used as controls for the above methods. Appendix was used as the positive control tissue for MSA, FVIII-r.ag, and KDR. Lymph node was used for the positive control tissue for p27/kip1. In addition, each lung section provided its own internal control for MSA (bronchial smooth muscle cells) and FVIII-r.ag and KDR (alveolar septal capillaries).
Computer-Aided Three-Dimensional Reconstruction
Each slide was again examined to confirm the presence of plexiform and/or concentric-obliterative lesions. Plexiform lesions were defined, in part, by the intense FVIII-r.ag and/or KDR staining of the endothelial cells (Figure 1) ▶ . Most lesions consisted of slit-like multiple channels within small muscular pulmonary arteries; others showed partial destruction of the media, with the proliferating endothelial cells appearing to break through the wall of the artery. Concentric, intraluminal, onionskin-like layers that stained intensely with FVIII-r.ag but lacked an MSA signal (Figure 2) ▶ identified concentric-obliterative lesions.
Figure 1.
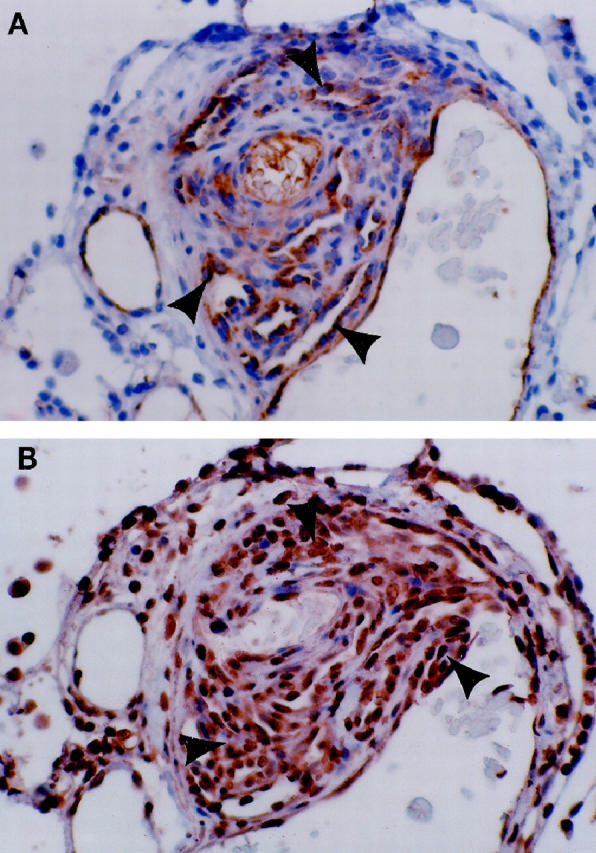
Plexiform lesion from a patient with PPH. A: The multichanneled, cellular lesions stain positive for the FVIII-r.ag endothelial cell marker (arrowheads). B: Same lesion as A, immunostained for the VEGF receptor KDR. All of the endothelial cells stain positive for this endothelial cell-specific marker (arrowheads).
Figure 2.
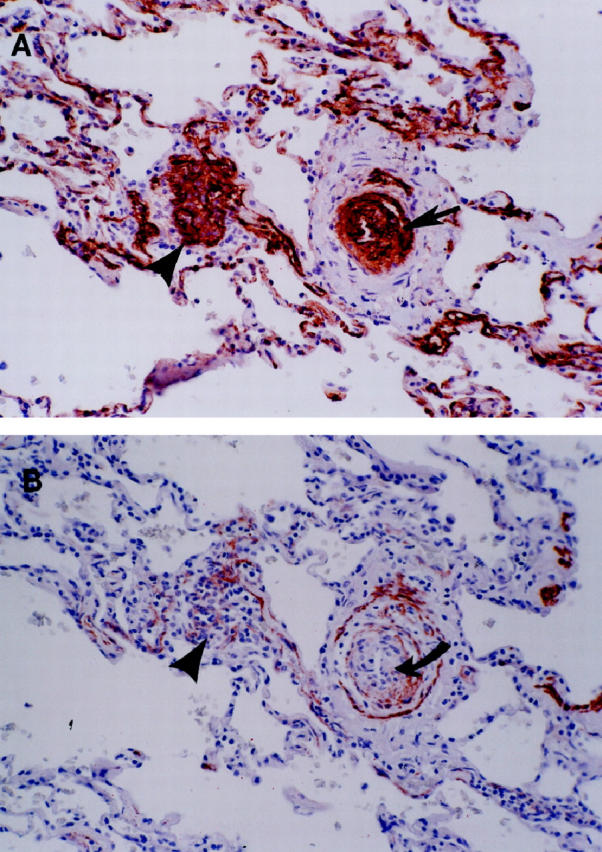
A: An illustration of the multiple concentric onion-skin pattern of the FVIII-r.ag-positive cells in a concentric-obliterative lesion (arrow). On the left is a conglomerate of positively stained endothelial cells, consistent with a partial slice through a plexiform lesion (arrowhead). B: For contrast, an adjacent section is stained for MSA. Note that the positive endothelial cells of the concentric-obliterative lesion do not stain with MSA (curved arrow). The surrounding smooth muscle coat is positive for MSA and negative for FVIII-r.ag. Also note that the plexiform lesion on the left is now more apparent in this deeper cut (arrowhead).
The lesions were magnified by ×100 to ×200 and then sequentially photographed using color print film. The lumina, endothelial cells, and smooth muscle cell layers of the serially sectioned vessels were then traced with color-coded inks onto corresponding serial transparent overlays. The locations of VEGF receptor (KDR) and p27/kip1 immunostained cells were similarly copied onto overlays. Adding a fourth and fifth marker greatly increased the complexity and memory requirements for the three-dimensional reconstructions; therefore only one set of serial sections from case 2 was chosen for computerized reconstruction using all five markers.
Adjacent bronchioles, alveoli, and large arteries were used as guides to maintain the correct x-y coordinates of the lesions. The resultant maps were then scanned into the computer with a HP ScanJet IIcx/T scanner.
The two-dimensional map images were automatically and manually edited to eliminate scanning errors and verified by eye with the original maps. The edited images were then combined into a three-dimensional model in which the orientation and spacing of the sections were preserved. The resulting model was encoded in a format suitable for use with Vis5D, a three-dimensional visualization package.
Vis5D is public domain software made available by the University of Wisconsin. Its primary use is the visualization of the results of numerical weather simulations. The graphics capabilities can be extended to general three-dimensional visualization, which was applied in the present study. The rendering algorithm allows the user to interactively choose any viewing angle and encode different regions of the maps in any color with any degree of opacity. In addition, two-dimensional slices may be taken from any angle. This greatly facilitates our three-dimensional conception of these lesions and the functional components’ spatial relationships within.
Results
Table 2 ▶ summarizes the sequence and spacing of the vascular changes.
Table 2.
Summary of Reconstructed Vascular Lesions
| Case no. and block | Diagnosis | Total length of reconstruction (μm) | Branching pattern (axial* or dichotomous†) | Sequence and spacing of vascular changes after branch point (N, P, C, or D × distance in μm) |
|---|---|---|---|---|
| 1a | PPH | 445 | Dichotomous | Branch 1: C× 230, P× 200 |
| Branch 2: P× ? | ||||
| 1b | PPH | 245 | Dichotomous | Branch 1: N× 35, C× 105, P× 100 |
| Branch 2: medial hypertrophy× ? | ||||
| 1c | PPH | 170 | Dichotomous | Branch 1: N× 35, C× 65, P× 70 |
| Branch 2: medial hypertrophy× ? | ||||
| 1d | PPH | 250 | Dichotomous | Branch 1: N× 40, C× 55, P× 80 |
| Branch 2: N× 30, P× 145 | ||||
| 2 | PPH | 260 | Dichotomous | Branch 1: N× 25, P× 120, D× 80 |
| Branch 2: N× 35, P× 110, D× 80 | ||||
| 3 | Cirrhosis | 410 | Axial | P× 400, D× ? |
| 4 | AIDS | 540 | Dichotomous | Branch 1: N× 200, P× 70 |
| Branch 2: N× 55, P× 200 | ||||
| 5a | Scleroderma | 340 | Axial | N× 10, C× 50, P× 300 |
| 5b | Scleroderma | 340 | Dichotomous | Branch 1: P× 200 |
| Branch 2: N× 35, C× 70, P× 100 | ||||
| 6 | Normal | 145 | Dichotomous | Branch 1: N |
| Branch 2: N |

*Axial = A medium-sized axial pulmonary artery gives rise to a branching, smaller-sized artery.
†Dichotomous = A normal-appearing vessel bifurcates into two equally sized branches.
N = Normal pulmonary artery.
C = Concentric-obliterative lesion.
P = Plexiform lesion.
D = Dilatation lesion.
? = Vessel disappears in distal sections.
Both cases of PPH (yielding a total of five reconstructed lesions) demonstrated a dichotomous branching pattern. Concentric-obliterative and/or plexiform lesions became visible just distal to branch points of small- to medium-sized pulmonary arteries (Figure 3) ▶ . In the reconstructed vessels, a concentric-obliterative lesion was always associated with a distal plexiform lesion. However, plexiform lesions were either isolated lesions or were associated with a proximal concentric-obliterative lesion. Plexiform lesions terminated in aneurysmal-like dilatation lesions, or simply disappeared in the subsequent sections. The length of the longest fully reconstructed plexiform lesion was 200 μm. The lengths of the concentric-obliterative lesions were similar to those of the downstream plexiform lesions within the same vessel. Medial hypertrophy was more prominent in PPH case 1 than in case 2. Figure 4 ▶ illustrates the three-dimensional view of case 1. The initial concentric-obliterative lesion of the left branch is followed distally by a plexiform lesion. The right branch consists only of a plexiform lesion. The extensive intravascular, FVIII-r.ag-positive, endothelial cell layers obliterate normal blood flow, leaving only small channels and slit-like openings. Figure 4B ▶ shows a longitudinal view of the same lesion. For comparison, Figure 5 ▶ shows a similar-sized, branching pulmonary blood vessel from a normal lung (case 6). In the normal vessels, the lumina are unobstructed and the cross-sectional area available for blood flow is much greater than that in the abnormal vessel shown in Figure 4 ▶ .
Figure 3.

Longitudinal views of plexiform lesions in PPH. (A) Low-power magnification of a pulmonary artery stained for MSA. Distal to the bifurcation of the muscular artery, the two branching vessels are occluded by plexiform lesions (arrows). The thin smooth muscle layers of the bronchiolar and pulmonary arteries are highlighted by the MSA stain (short arrows). (B) Higher-power magnification of the same lesion stained with FVIII-r.ag. In this example, the endothelial cells lining the multiple lumina of the plexiform lesions stain positive for FVIII-r.ag. Dilatation lesions can be seen within and adjacent to the plexiform lesions (arrows).
Figure 4.
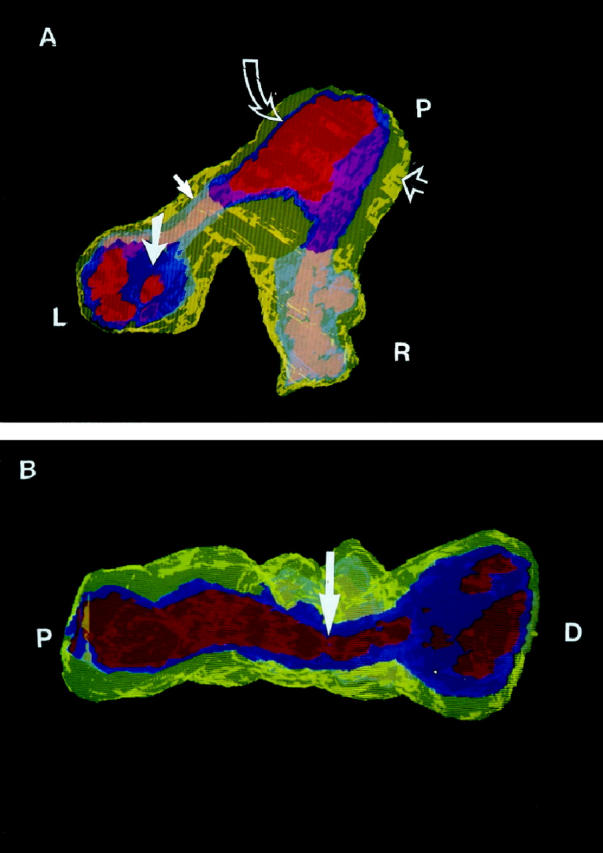
(A) A three-dimensional cutaway view of plexiform and concentric-obliterative lesions in a bifurcating vessel from case 1 (PPH). Yellow, smooth muscle cells; ultramarine or gray-blue, endothelial cells; red or flesh-toned, lumen. (The apparent color changes are a result of the transparent overlying yellow smooth muscle and blue endothelial cell layers.) The proximal end (P) shows slight medial hypertrophy (yellow, open arrow), a normal endothelial cell monolayer (blue, curved open arrow), and an unobstructed lumen (red). At the left branch (L), the patent vessel turns into a concentric proliferation of endothelial cells (gray-blue, closed arrow), opening further distally into the multichanneled plexiform lesion (ultramarine, closed large arrow) . At the right branch (R), the patent vessel turns into a plexiform lesion (gray-blue). The blue endothelial cells of the plexiform lesions severely disrupt the lumina, best seen in (B). (B) A longitudinal view of the same vessel shows the proximal unobstructed lumen (P), a constriction distal to the branch point (arrow), and the proliferating and obstructing endothelial cells (blue) at the distal end (D) of one of the branches. This view highlights the transition of the patent pulmonary artery to concentric and plexiform changes.
Figure 5.
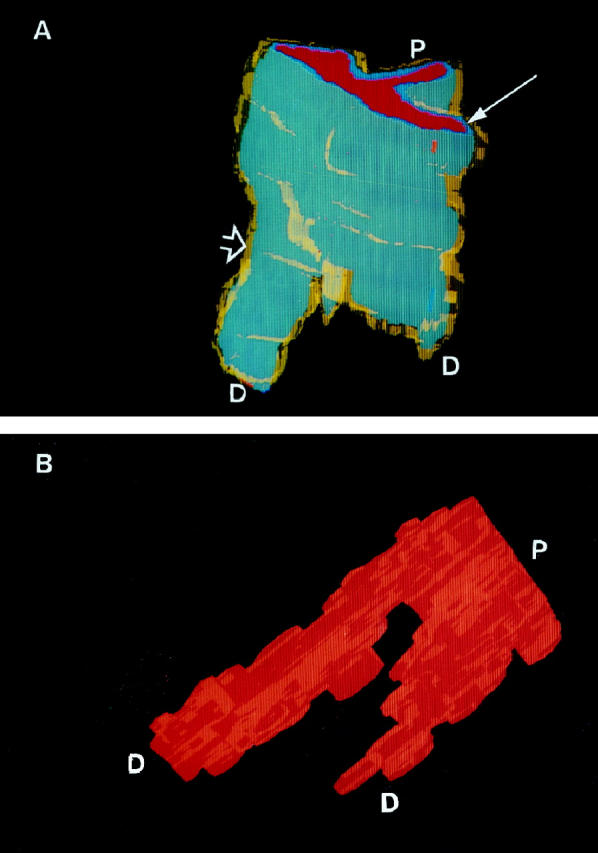
(A) Three-dimensional view of a normal small pulmonary artery at a branch point (case 6). Note the thin yellow smooth muscle cell coat (open arrow) and the gray-blue endothelial cell monolayer (thin arrow). P, Proximal end; D, distal end. (B) Luminal cast of a normal vessel. Rotated view of A. Note the large, unobstructed channels, even distal to the branch point. P, Proximal end; D, distal end.
A reconstructed pulmonary vessel from the patient with cirrhosis (case 3) demonstrated an axial pulmonary artery giving rise to a single, smaller side branch (see illustration in Table 2 ▶ ). In this case, the plexiform lesion appeared to arise directly from the wall of the larger vessel and then terminated as a dilatation lesion. The length of this plexiform lesion was 400 μm, the longest lesion identified.
Case 4 (AIDS-related PH) was unique in that one of the branched vessels remained normal for 200 μm before evolving into a 70-μm-long plexiform lesion. The other branch appeared normal for 55 μm, and then became plexiform for 200 μm. Concentric-obliterative lesions were not present in this case of AIDS-associated plexiform arteriopathy.
Both axial and dichotomous branching patterns were reconstructed in the lung tissue from the scleroderma patient (case 5). Similar to the PPH cases, concentric-obliterative or plexiform lesions occurred at or just distal to the branch points. Again, solitary plexiform lesions were identified, but the concentric-obliterative lesions occurred only proximal to the plexiform lesions.
All non-PPH cases (cases 3–5) demonstrated a prominent endothelial cell proliferative component similar to that seen in the two PPH cases. Medial hypertrophy was present to a variable degree. Although not shown in these reconstructions, mononuclear inflammatory cells tended to cluster around affected vessels. The lumina of the reconstructed vessels were severely compromised, ranging from total obliteration to a few remaining slit-like openings. The dilatation lesions also showed extensive structural disorder. No thrombotic lesions were identified in the vessels of these patients, perhaps because of the treatment of these patients with anticoagulants.
We examined two blocks from case 2, each serially sectioned (50 sections per block), and sequentially stained with H&E, FVIII-r.ag, MSA, KDR, and p27/kip1 (Figures 6 and 7) ▶ ▶ . On average, each histological section contained 10 plexiform lesions. Because KDR is endothelial cell specific, 29-31 immunostaining of all endothelial cells (including the proliferating ones) should be positive. Indeed, the KDR immunomarker stained all of the cells comprising the plexiform lesion. Interestingly, the KDR marker appears to be more sensitive for the endothelial cell population in the plexiform lesion than the FVIII-r.ag immunostain. Figures 1 and 6 ▶ ▶ illustrate the endothelial cells stained by KDR versus FVIII-r.ag. The FVIII-r.ag-positive cells tend to be more prevalent adjacent to the multiple lumina, whereas KDR-positive cells are present throughout.
Figure 6.
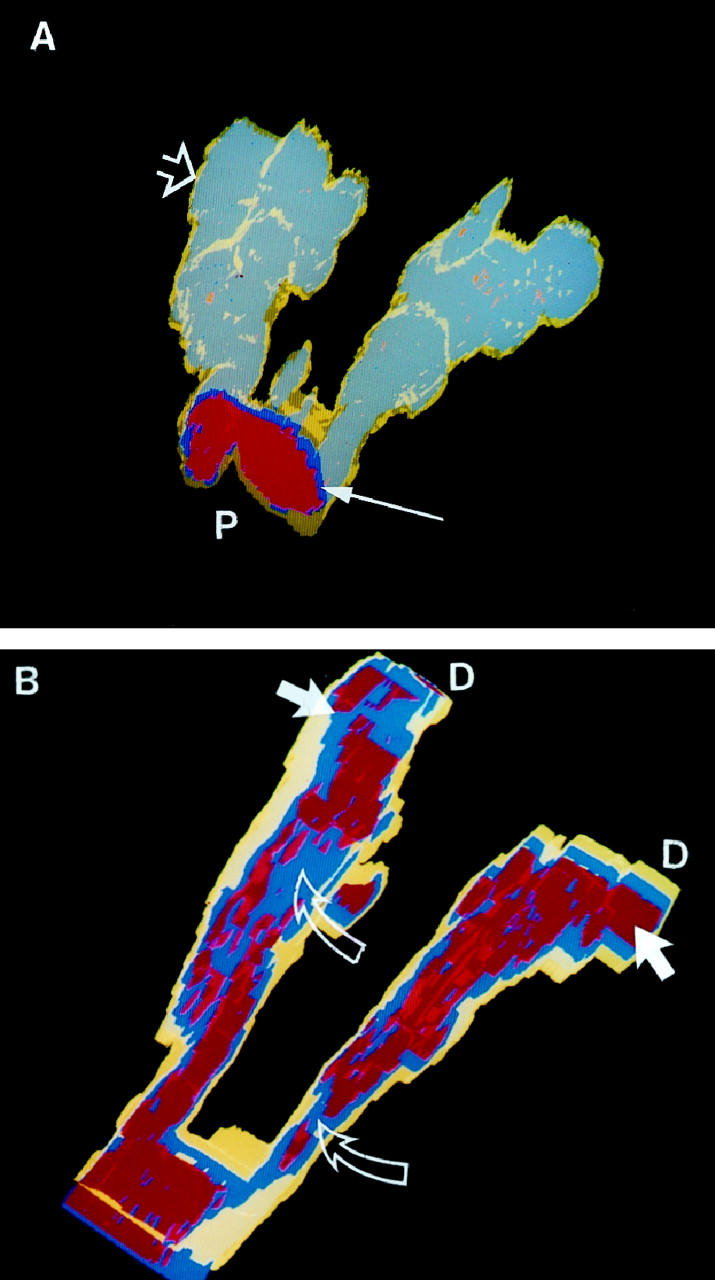
Lesion reconstructed from PPH case 2. (A) The gray-blue represents the endothelial cells stained with antibody directed against KDR. The yellow smooth muscle layer (open arrow) is thin, in marked contrast to PPH case 1 (Figure 4) ▶ . The distorted endothelial cell layers begin distal to the bifurcation of the vessel and can be seen through the translucent yellow smooth muscle coat. The proximal lumen (P) is lined by an endothelial cell monolayer (thin arrow). (B) Rotated and cutaway view of the same lesion. Note the severely disrupted red lumina distal to the bifurcation and the distal (D) dilatation lesions (arrows). Blue endothelial cells (open arrows) obstruct the lumina. Yellow, Smooth muscle; blue, KDR positive cells; red, lumen.
Figure 7.
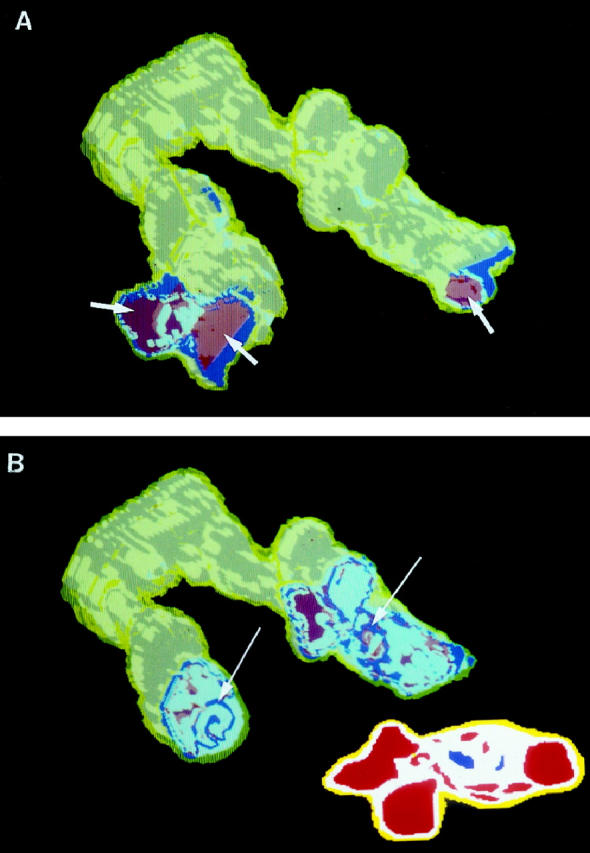
KDR and p27/kip1 staining pattern of the two plexiform lesions that are part of the complex vascular lesion depicted in Figure 6 ▶ . In these views, the distal portions of the arteries have been cut away. Yellow, Smooth muscle cells; red, lumen; white, KDR-positive/p27kip1-positive endothelial cells; blue, KDR-positive/p27/kip1-negative endothelial cells. (A) A view of the dilatation lesions (arrows) at the distal ends of the two pulmonary artery branches. (B) A more proximal cutaway view showing small foci of proliferating (p27/kip1-negative) endothelial cells (arrows). The inset shows a cross-sectional view of a two-dimensional map image used in the computerized reconstruction of case 2.
The KDR-positive endothelial cells that stain positive for the cell quiescence marker p27/kip1 are shown in Figure 7 ▶ . The p27/kip1 staining pattern mimicked that of the FVIII-r.ag; that is, the elongated, more mature-appearing endothelial cells nearest the red cell containing lumina stained positive, whereas the core of plump endothelial cells stained negative (see Figure 7B ▶ , inset).
The terminal dilatation lesions of this PPH case stained positive for the endothelial cell markers KDR and FVIII-r.ag, as well as the p27/kip1 cell cycle inhibitory protein marker. In marked contrast to PPH case 1 (Figure 4) ▶ , the smooth muscle layers around the plexiform lesions in case 2 were extremely thin.
Discussion
Our three-dimensional analysis of plexiform arteriopathy allowed us to examine the cellular and structural topographic relationships of diseased vessels, the extent and location of endothelial cell growth in plexiform and concentric-obliterative lesions, the heterogeneity of the smooth muscle cell coat in plexiform arteriopathy, and, by inference, the impact of vessel remodeling on blood flow. Because of the complexity associated with the three-dimensional computer reconstruction of microscopic structures, we limited our reconstruction efforts to five patients and nine vascular lesions.
Our study indicates that the plexiform lesion is functionally important because the vascular lumen (and consequently blood flow) is severely occluded along the entire length of a vessel affected by a single plexiform lesion. A bidimensional view only shows a limited representation of the pulmonary vessels. Three-dimensional reconstructions illustrate that a vessel with an apparently normal cross section may become severely occluded by downstream plexiform arteriopathy. Therefore, numerous histological sections may be needed to adequately evaluate plexiform arteriopathy.
We found a correlation between arterial branch points and intraluminal vascular remodeling, which was similar to findings of previous morphometric studies of plexiform arteriopathy. 10,11,32 The specific distribution of the plexiform and concentric-obliterative luminal lesions at branch points and only in pulmonary (not bronchial) arteries suggests that shear stress and/or turbulent flow may influence the pathogenesis of the lesions.
Although all of the lung sections we studied exhibited plexiform arteriopathy, not all showed concentric-obliterative changes or smooth muscle hypertrophy. In the nine vessels reconstructed, concentric-obliterative lesions (n = 6) were never isolated lesions. If present, they were proximal to plexiform changes. However, plexiform lesions (n = 14) did appear as solitary lesions. From this, one could hypothesize that the plexiform lesion forms first, independent of a component of medial smooth muscle cell hypertrophy. Over time, the plexiform lesion may transform into a tight network of intraluminal concentric obstruction composed of endothelial cells and recruited myofibroblast cells, beginning at the proximal end of the arterial branch and proceeding distally. Thus it is possible that concentric lesions represent a scar that temporally follows the plexiform lesion. Interestingly, two of our cases that lacked concentric-obliterative changes (case 2, PPH; case 4, AIDS-associated PH) also lacked a hypertrophic smooth muscle component, suggesting perhaps that concentric intimal fibrosis and medial thickening are related.
Some authors suggest either no association between dilatation and plexiform lesions or that the dilatation lesions are simply dilated capillaries or shunts. 8,32,33 Our three-dimensional reconstructions illustrate that the dilatation lesion is found within a short distance downstream of the plexiform lesion, adjacent to or within the same vessel. A dilatation lesion may represent another manifestation of the overall angiogenic-like process occurring at the site of the plexiform lesion.
If a vascular lesion in PPH can evolve over time and progressively mature, then the individual cells that make up the lesion might also mature and differentiate. To examine phenotypic differences in the individual endothelial cells of a plexiform lesion, we reconstructed a vessel using two additional, more functional, immunohistochemical markers. One marker, the VEGF receptor KDR, identifies vascular endothelial cells. KDR, unlike its ligand VEGF, is present in all endothelial cells and is an early marker of angiogenesis and vasculogenesis. 25 We also used p27/kip1 expression to identify quiescent, nonproliferative cells. Cells that are stimulated by growth factors demonstrate a decreased level of p27/kip1. 26-28 FVIII-r.ag expression was used as a marker for a mature endothelial cell phenotype—a cell that contains Weibel-Palade bodies. In our reconstructed plexiform vessel, positive staining for p27/kip1 occurred primarily in the endothelial cells lining the peripheral multiple lumina of the plexiform lesion. Decreased or absent staining occurred in the central core of the endothelial cell layers, suggesting that in these central cores, endothelial cells proliferate. Notably, the expression of FVIII-r.ag paralleled the p27/kip1 findings, that is, whereas all endothelial cells stained positive for the endothelial cell-specific receptor KDR, only the apparently quiescent (p27/kip1-positive) and mature (FVIII-r.ag-positive) endothelial cells lined the multiple lumina. Thus there was a central core of cells positive for KDR but negative for FVIII-r.ag and p27/kip1, indicating that these cells 1) were endothelial cells (KDR-positive), 2) did not express a marker of maturation (FVIII-r.ag-negative), and 3) were likely under stimulation by growth factors (p27/kip1-negative).
In summary, we present evidence that plexiform lesions are composed of phenotypically distinct endothelial cells and are not static end-stage lesions. This phenotypic diversity is expressed in the evolution of the plexiform lesion. We propose that plexiform lesions begin and evolve within the blood vessel lumen (ie, a plexiform lesion evolves into a concentric-obliterative lesion), and that there is evolution on a cellular level. At a minimum, there are two distinct KDR-positive endothelial cells: 1) a quiescent FVIII-r.ag- and p27/kip1-positive phenotype that lines structures resembling early blood vessels and 2) a proliferating FVIII-r.ag- and p27/kip1-negative endothelial cell phenotype in the solid central core of cells of the plexiform lesion. The developmental relationships between the two endothelial cell phenotypes and the surrounding smooth muscle cells of the plexiform lesions, along with the identification of the specific roles of the endothelial cell phenotypes in the process of “misguided angiogenesis,” may shed further light on the pathogenesis of the vascular remodeling in severe pulmonary hypertension and may provide tools to identify earlier stages of endothelial cell abnormalities in this disease.
Note added in proof
We with to thank Dr. Alfred P. Fishman, University of Pennsylvania, for bringing to our attention the 1961 publication by Moschcowitz, Rubin, and Strauss in The American Journal of Pathology entitled “Hypertension of the pulmonary circulation due to congenital glomoid obstruction of the pulmonary arteries,” specifically its authors’ statement: “Four cases of pulmonary hypertension are reported in which we believe the hypertension to be caused by obstruction of innumerable small branches of the pulmonary artery by glomoid lesions arising within the vessel lumens.” (Am J Pathol 1961, 39:75–87)
Acknowledgments
We thank Vickie McHenry, HT (ASCP) IHC, for invaluable assistance with the immunohistochemistry.
Footnotes
Address reprint requests to Dr. Carlyne D. Cool, University of Colorado Health Sciences Center, Department of Pathology, Box B216, 4200 East Ninth Avenue, Denver, CO 80262. E-mail: carlyne.cool@uchsc.edu.
Supported in part by a grant from the PPH Cure Foundation and the RO1HL60913-01 grant from the Heart, Lung and Blood Institute, National Institutes of Health, to NFV and RMT, and in part by a grant from the Witham Family Foundation.
References
- 1.Wagenvoort CA, Wagenvoort N: Primary pulmonary hypertension. A histopathologic study of the lung vessels in 156 clinically diagnosed cases. Circulation 1970, 42:1163-1184 [Google Scholar]
- 2.Edwards, WD: Pathology of pulmonary hypertension. Cardiovasc Clin 1987, 18:321–354 [PubMed]
- 3.Saunders JB, Constable TJ, Heath D, Smith P, Paton A: Pulmonary hypertension complicating portal vein thrombosis. Thorax 1978, 34:281-283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robalino BD, Moodie DS: Association between primary pulmonary hypertension and portal hypertension: analysis of its pathophysiology and clinical, laboratory and hemodynamic manifestations. J Am Coll Cardiol 1991, 17:492-498 [DOI] [PubMed] [Google Scholar]
- 5.Thomas SH, Butt AY, Corris PA, Egan JJ, Higenbottam TW, Madden BP, Waller PC: Appetite suppressants and primary pulmonary hypertension in the United Kingdom. Br Heart J 1995, 74:660-663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brenot F, Herve P, Petitpretz P, Parent F, Duroux P, Simonneau G: Primary pulmonary hypertension and fenfluramine use. Br Heart J 1993, 70:537-541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petitpretz P, Brenot F, Azarian R, Parent F, Rain B, Herve P, Simonneau G: Pulmonary hypertension in patients with human immunodeficiency virus infection. Comparison with primary pulmonary hypertension. Circulation 1994, 89:2722-2727 [DOI] [PubMed] [Google Scholar]
- 8.Pietra GG: Histopathology of primary pulmonary hypertension. Chest 1994, 105:2S-6S [DOI] [PubMed] [Google Scholar]
- 9.Pietra GG, Ruttner JR: Specificity of pulmonary vascular lesions in primary pulmonary hypertension. A reappraisal. Respiration 1987, 52:81-85 [DOI] [PubMed] [Google Scholar]
- 10.Ogata T, Iijima T: Structure and pathogenesis of plexiform lesion in pulmonary hypertension. Chin Med J 1993, 106:45-48 [PubMed] [Google Scholar]
- 11.Yaginuma G, Mohr H, Takahashi T: Distribution of arterial lesions and collateral pathways in the pulmonary hypertension of congenital heart disease: a computer-aided reconstruction study. Thorax 1990, 45:586-590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heath D, Edwards JE: The pathology of hypertensive pulmonary vascular disease. A description of six grades of structural changes in the pulmonary arteries with special reference to congenital cardiac defects. Circulation 1958, 18:533-547 [DOI] [PubMed] [Google Scholar]
- 13.Fuster V, Steele PM, Edwards WD, Gersh BJ, McGoon MD: Primary pulmonary hypertension: natural history and the importance of thrombosis. Circulation 1984, 70:580-587 [DOI] [PubMed] [Google Scholar]
- 14.Tuder RM, Groves B, Badesch DB, Voelkel NF: Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am J Pathol 1994, 144:275-285 [PMC free article] [PubMed] [Google Scholar]
- 15.Cool CD, Kennedy D, Voelkel NF, Tuder RM: Pathogenesis and evolution of plexiform lesions in pulmonary hypertension associated with scleroderma and human immunodeficiency virus infection. Hum Pathol 1997, 28:434-442 [DOI] [PubMed] [Google Scholar]
- 16.Haworth SG, Rabinovitch M, Meyrick B, Michel R, Pietra GG, Polak JM, Reid LM, Tuder R: The pathology of pulmonary hypertension. Primary Pulmonary Hypertension: Executive Summary from the World Symposium—Primary Pulmonary Hypertension 1998. Edited by S Rich. (Available from the World Health Organization via the Internet (http://www.who.int/ncd/cvd/pph.html).)
- 17.Loscalzo J: Endothelial dysfunction in pulmonary hypertension. N Engl J Med 1992, 327:70-75 [DOI] [PubMed] [Google Scholar]
- 18.Tuder RM, Cool CD, Geraci MW, Wang J, Abman SH, Wright L, Badesch DB, Voelkel NF: Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med 1999, 159:1925-1932 [DOI] [PubMed] [Google Scholar]
- 19.Giaid A, Saleh D: Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med 1995, 333:214-221 [DOI] [PubMed] [Google Scholar]
- 20.Giaid A, Yanagisawa M, Langleben D, Michel RP, Levy R, Shennib H, Kimura S, Masaki T, Duguid WP, Steward DJ: Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Engl J Med 1993, 328:1732-1739 [DOI] [PubMed] [Google Scholar]
- 21.Wright L, Tuder RM, Wang J, Cool CD, Lepley RA, Voelkel NF: 5-Lipoxygenase and 5-lipoxygenase activating protein (FLAP) immunoreactivity in lungs from patients with primary pulmonary hypertension. Am J Respir Crit Care Med 1998, 157:219-229 [DOI] [PubMed] [Google Scholar]
- 22.Lee S-D, Shroyer KR, Markham NE, Cool CD, Voelkel NF, Tuder RM: Monoclonal endothelial cell proliferation is present in primary but not secondary pulmonary hypertension. J Clin Invest 1998, 101:927-934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tuder RM, Radisavljevic Z, Shroyer KR, Polak JM, Voelkel NF: Monoclonal endothelial cells in appetite suppressant-associated pulmonary hypertension. Am J Respir Crit Care Med 1998, 158:1999-2001 [DOI] [PubMed] [Google Scholar]
- 24.Folkman J, D’Amore PA: Blood vessel formation: what is its molecular basis? Cell 1996, 87:1153-1156 [DOI] [PubMed] [Google Scholar]
- 25.Risau W: Differentiation of endothelium. FASEB J 1995, 9:926-933 [PubMed] [Google Scholar]
- 26.Sanchez-Beato M, Saez AI, Martinez-Montero JC, Sol Mateo M, Sanchez-Verde L, Villuendas R, Troncone G, Piris MA: Cyclin-dependent kinase inhibitor p27kip1 in lymphoid tissue: P27kip1 expression is inversely proportional to the proliferative index. Am J Pathol 1997, 151:151-160 [PMC free article] [PubMed] [Google Scholar]
- 27.Kato J, Matsuoka M, Polyak K, Massague J, Sherr CJ: Cyclic AMP-induced G1 phase arrest mediated by an inhibitor (p27KIP1) of cyclin-dependent kinase 4 activation. Cell 1994, 79:487-496 [DOI] [PubMed] [Google Scholar]
- 28.Nourse J, Firpo E, Flanagan WM, Coats S, Polyak K, Lee M-H, Massague J, Crabtree GR, Roberts JM: Interleukin-2-mediated elimination of the p27kip1 cyclin-dependent kinase inhibitor prevented by rapamycin. Nature 1994, 372:570-573 [DOI] [PubMed] [Google Scholar]
- 29.Patterson C, Perrella MA, Hsieh C-M, Yoshizumi M, Lee M-E, Haber E: Cloning and functional analysis of the promoter for KDR/flk-1, a receptor for vascular endothelial growth factor. J Biol Chem 1995, 39:23111-23118 [DOI] [PubMed] [Google Scholar]
- 30.Waltenberger J, Claesson-Welsh L, Siegbahn A, Masabumi S, Heldin C-H: Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J Biol Chem 1994, 269:26988-26995 [PubMed] [Google Scholar]
- 31.Ferrara N, Davis-Smyth T: The biology of vascular endothelial growth factor. Endocr Rev 1997, 18:4-25 [DOI] [PubMed] [Google Scholar]
- 32.Jamison BM, Michel RP: Different distribution of plexiform lesions in primary and secondary pulmonary hypertension. Hum Pathol 1995, 26:987-993 [DOI] [PubMed] [Google Scholar]
- 33.Pietra GG: The pathology of primary pulmonary hypertension. Primary Pulmonary Hypertension. Lung Biology in Health and Disease, vol 99. Edited by LJ Rubin and S Rich. New York, Marcel Dekker, 1997, pp 19–61