

Abstract
The finding that the systemic renin-angiotensin system (RAS) is not activated in most types of chronic renal disease has led to the suggestion that a local, intrarenal RAS may be an important determinant in the relentless progression of renal disease. Therefore, cell specific changes in various components of the RAS in response to renal mass reduction and angiotensin converting enzyme (ACE) inhibition were examined. Thirty Sprague-Dawley rats were randomly assigned to sham surgery, subtotal nephrectomy (STNx) alone or STNx treated with the ACE inhibitor, perindopril, and sacrificed after 12 weeks. In sham rats, renin mRNA and protein were only present in the juxtaglomerular apparatus. In contrast, in STNx kidneys, renin and angiotensin II expression were noted predominantly in renal tubular epithelial cells in association with overexpression of the prosclerotic cytokine, transforming growth factor-β1 (TGF-β1). In perindopril-treated STNx rats expression of renin and TGF-β1 were similar to control animals. These finding indicate that following renal mass reduction there is pathological tubular expression of various components of the RAS. Furthermore, in contrast to the juxtaglomerular apparatus, tubular renin expression was reduced with ACE inhibition. These changes within the intrarenal RAS may be pathogenetically linked to the development of tubulointerstitial injury.
The relentless progression of renal injury following initial insult remains incompletely understood, as do the mechanisms that lead to the tubulointerstitial injury which accompanies glomerular disease. Despite the beneficial effects of angiotensin converting enzyme (ACE) inhibition, 1,2 the finding that the systemic renin-angiotensin system (RAS) is not activated in most types of chronic renal disease has led to the suggestion that a local, intrarenal RAS may be an important determinant in the progression of renal disease.
The existence of an intrarenal RAS is suggested by the high concentration of angiotensin II in the glomerular filtrate and proximal tubular lumen relative to plasma. 3 Angiotensinogen and angiotensin converting enzyme are both present in the kidney and while the juxtaglomerular apparatus is the main source of systemic renin it may also contribute in a paracrine fashion to the intrarenal RAS . 4 While the glomerulus is often the primary site of injury in renal disease, it is the extent of tubulointerstitial rather than glomerular injury which correlates most closely with and predicts future loss of renal function in patients with primary glomerular disease. 5,6 Indeed, recent studies indicate that production of various cytokines by the proximal tubular epithelium may be a critical factor in the development of tubulointerstitial fibrosis 7 as indicated by the formation of the prosclerotic cytokine, transforming growth factor-β (TGF-β), predominantly in this region. 8 However, under physiological conditions renin can only be detected in the proximal tubule with the use of ultrasensitive, reverse transcriptase polymerase chain reaction (RT-PCR) methods. 9
Apart from its actions on glomerular hemodynamics and electrolyte transport, angiotensin II (AII), the effector molecule of the RAS, may also function as a growth factor leading to the stimulation of extracellular matrix synthesis. 10 While the precise mechanisms underlying this effect have not been fully delineated, several studies suggest that the ability of AII to stimulate extracellular matrix production may be mediated by increased expression of TGF-β. 11
Previous studies examining the intrarenal RAS in models of renal disease such as the remnant kidney have yielded conflicting results that may reflect differences in methodology. 12,13 The present study sought to examine in detail the cell specific changes in the renin-angiotensin system following renal mass reduction, and furthermore to investigate the relationship between the RAS and TGF-β expression and to determine the effects of ACE inhibition on the intrarenal RAS in this model of kidney disease.
Materials and Methods
Animals
Thirty male Sprague-Dawley rats weighing 200–250 g were randomized to three groups of 10 animals each. Anesthesia was achieved by the intraperitoneal administration of pentobarbital (6 mg/100 g body weight, Boehringer Ingelheim, Artarmon, NSW, Australia). The control group underwent sham surgery consisting of laparotomy and manipulation of both kidneys before wound closure. The other 20 rats all underwent subtotal nephrectomy (STNx) performed by right subcapsular nephrectomy and infarction of approximately two-thirds of the left kidney by selective ligation of two of three to four extrarenal branches of the left renal artery. 14 Animals were then randomly assigned to two groups: STNx alone or STNx with the ACE inhibitor perindopril 8 mg/l drinking water (Servier, Neuilly, France). Rats were housed in a temperature (22°C) controlled room with ad libitum access to commercial standard rat chow (Norco Co-Operative Ltd., Lismore N.S.W., Australia) and water during the entire study. Rats from each group were sacrificed at 12 weeks after surgery. At sacrifice the remnant (left) kidney of STNx and the left kidney of sham animals were excised and the renal capsule removed. The kidney was then immersion fixed in 10% neutral buffered formalin and embedded in paraffin for later in situ hybridization and immunohistochemical studies. Experiments were approved by the Animal Welfare and Ethics Committee of the Austin and Repatriation Medical Center.
Renal Function
Body weight was measured weekly. Plasma urea and creatinine were measured by autoanalyzer (Beckman Instruments, Palo Alto, CA) at the beginning and end of the study. Glomerular filtration rate was measured at the end of the study by single shot Tc99m-DTPA clearance. 15 Systolic blood pressure was measured in conscious rats using an occlusive tail-cuff plethysmograph attached to a pneumatic pulse transducer (Narco Bio-System Inc., Houston, Texas). 16 Before sacrifice, rats were housed in metabolic cages for 24 hours for subsequent measurement of urinary protein excretion using the Coomassie Brilliant Blue method. 17 Plasma renin activity was measured by radioimmunoassay. 15
Tubulointerstitial Morphology
Paraffin-embedded sections were stained with hematoxylin and eosin, periodic acid-Schiff (PAS) and Masson’s Trichrome. Tubulointerstitial injury was defined as tubular dilatation and/or atrophy, interstitial fibrosis, and inflammatory cell infiltrates. Morphological analyses were performed as previously described 18 with the extent of injury graded on a scale of 0 to 4 by an observer blinded to the animal treatment group. With this method injury is graded as 0 for normal; 1 for involvement of <10% of the cortex; 2 for involvement of 10 to 25% of the cortex; 3 for involvement of 25 to 75% of the cortex, 4 for extensive damage involving >75% of the cortex.
Glomerular Morphology
Glomerular injury was assessed in PAS-stained sections by examining 50 glomeruli in PAS-stained sections. Each glomerulus was graded as normal (0); mildly sclerotic (1+, lesion occupying <25% of glomerular tuft); moderately sclerotic (2+, lesion occupying 25 to 50% of glomerular tuft); severely sclerotic (3+, lesion occupying more than 50% of glomerular tuft); or globally sclerotic (4+, lesion occupying 100% of glomerular tuft). A semiquantitative score was thus derived for each animal using the formula:
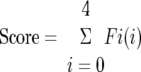 |
where Fi is the percentage of glomeruli in the rat with a given score i. 19
In Situ Hybridization
The 1.4-kb cDNA probe for rat renin (gift of Dr. D.J. Campbell, Melbourne, Australia) was cloned into pGEM 4 (Stratagene, La Jolla, CA) and linearized with BamHI to produce an antisense riboprobe with T7 RNA polymerase. The 945-bp cDNA probe for TGF-β1 (gift of Dr. Qian, Bethesda, MD) and the 600-bp cDNA probe for α1 (IV) collagen (gift of Dr. R. Timpl, Martinsried, Germany) were also cloned into pBluescript KS+ (Stratagene) and linearized with XbaI and HindIII to produce an antisense riboprobes with T7 RNA polymerase. In situ hybridization was performed as previously described. 20 In brief, sections 4 μm thick cut from formalin-fixed paraffin-embedded kidney tissue were placed onto slides precoated with 3-aminopropyltriethoxysilane and baked overnight at 37°C. Tissue sections were dewaxed and rehydrated in graded ethanol and milliQ water, equilibrated in P buffer (50 mmol/L Tris-HCl, pH 7.5, 5 mmol/L EDTA) and incubated in 125 μg/ml Pronase E in P buffer for 10 minutes at 37°C. Sections were then washed in 0.1 mol/L sodium phosphate buffer (pH 7.2), briefly refixed in 4% paraformaldehyde for 10 minutes, rinsed in milliQ water, dehydrated in 70% ethanol, and air dried. Hybridization buffer containing 2 × 10 4 cpm/μl riboprobe in 300 mmol/L NaCl, 10 mmol/L Tris-HCl (pH 7.5), 10 mmol/L Na2HPO4, 5 mmol/L EDTA (pH 8.0), 1× Denhardt’s solution, 50% formamide, 17 mg/ml yeast RNA, 10% w/v dextran sulfate was heated to 85°C for 5 minutes. Twenty-five μl of this solution was then added to each section. Hybridization was performed overnight at 60°C in 50% formamide humidified chambers. Sections hybridized with sense probe for renin, TGF-β1 and α1 (IV) collagen were used as controls for nonspecific binding. After hybridization, slides were washed in 2× SSC containing 50% formamide prewarmed to 50°C to remove coverslips. Sections were then washed in the above solution for 1 hour at 55°C, rinsed three more times in RNase buffer (10 mmol/L Tris-HCl, pH 7.5, 1 mmol/L EDTA, pH 8.0, 0.5 mol/L NaCl) and then incubated with RNase A (150 μg/ml) for 1 hour at 37°C. Sections were later washed in 2× SSC for 45 minutes at 55°C, dehydrated in graded ethanol, air dried, and exposed to Kodak X-Omat autoradiographic film for 1–3 days. Slides were then dipped in Ilford K5 nuclear emulsion (Ilford, Mobberley, Cheshire, UK), stored in a light-free box with desiccant at room temperature for 2 to 3 weeks, immersed in Kodak D19 developer, fixed in Ilford Hypam, and stained with hematoxylin and eosin or periodic acid-Schiff (PAS).
Quantitative Autoradiography
Densitometry of autoradiographic images obtained by in situ hybridization was performed by computer-assisted image analysis as previously described 14,21,22 in accordance with guidelines for computer assisted densitometric quantitative autoradiography for in situ hybridization with 33P. 23 In brief, using the MCID system (Image Research, Ontario, Canada), in situ autoradiographic images were placed on a uniformly illuminating fluorescent light box (Northern Light Precision Luminator Model C60, Image Research, Ontario, Canada) and captured using a video camera (Sony Video Camera Module CCD, Japan) connected to an IBM AT computer with a 512 × 512 pixel array imaging board with 256 gray levels. Following appropriate calibration by constructing a curve of optical density versus radioactivity density, 24,25 quantitation of digitized autoradiographic images was performed with the MCID software and expressed as relative optical density (ROD). Control kidneys were arbitrarily assigned a value of 1. TGF-β1 and α1 (IV) collagen mRNA were quantitated only in the superficial cortex, the most distant area from the infarct-related scar in STNx animals. In addition to quantitation of gene expression, localization and relative abundance of transcript were visualized using pseudocolorized computer images.
Immunohistochemistry
Immunohistochemistry for renin and angiotensin II was performed as previously described. 26 In brief, sections 4 μm thick were placed onto slides, deparaffinized, and rehydrated. To block endogenous peroxidase sections were pretreated with 1% H2O2/methanol. Sections were next incubated in protein blocking agent (Lipshaw-Immunon, Pittsburgh, PA) for 20 minutes at room temperature followed by incubation with either renin or angiotensin II antiserum. Polyclonal renin anti-serum 27 was raised by immunizing rabbits with renin purified from mouse submandibular gland. Angiotensin II antibody (gift of Dr. D. Caranzaro, Cornell University Medical Center, New York) 28 was raised by immunizing rabbits with a conjugate of synthetic angiotensin II combined with rabbit serum albumin. 29 Sections were incubated with specific anti-serum for 18 hours at 4°C, washed in PBS and then incubated with universal biotinylated immunoglobulin (DAKO, Carpinteria, CA) and avidin-biotin complex (Vector, Burlingame, CA). Peroxidase conjugates were subsequently localized using diaminobenzidine tetrahydrochloride (DAB) as a chromogen and counterstained with Mayer’s hematoxylin. Negative controls included omitting the primary antibody or replacing it with normal rabbit IgG at an equivalent protein concentration.
To quantify the presence of renin in the juxtaglomerular apparatus, 50 JGAs were counted in kidney sections from each rat and assessed by a blinded observer. The number of JGAs with positive immunostaining for renin was then expressed as a percentage of the total. 13 The extent of tubular staining was assessed according to the percentage of the gridfield showed positive staining using a scoring system where 0 = absent staining, I = 1 to 5%, II = 5 to 25%, III = 25 to 50%, IV = 50 to 75%, and V = >75% as previously described. 18
Statistics
Results are expressed as mean ± SEM unless stated otherwise. Data were analyzed by ANOVA with comparisons between groups using Fisher’s least significant difference method. 30 Semiquantitative data obtained from immunohistochemical studies were expressed as median (range) and between group comparisons were analyzed using the Kruskal-Wallis test. Analyses were performed using the Statview SE+ Graphics package (Abacus Concepts, Berkeley, CA) on an Apple Macintosh Performa 6360 computer. A p value less than 0.05 was considered statistically significant.
Results
Renal Function
Functional data are shown in Table 1 ▶ . Rats that had undergone STNx became hypertensive. This was ameliorated by perindopril treatment. Similarly STNx rats developed chronic renal impairment characterized by elevated serum creatinine and heavy proteinuria, both of which were reduced by treatment with perindopril. Receipt of ACE inhibitor was confirmed by elevation of plasma renin activity in comparison with untreated STNx rats.
Table 1.
Renal Function at 12 Weeks after Subtotal Nephrectomy in Treated and Untreated Animals
| Sham | STNx | STNx + perindopril | |
|---|---|---|---|
| Body weight (g) | 510 ± 20 | 435 ± 12† | 467 ± 11‡ |
| Kidney weight (g) | 1.6 ± 0.1 | 2.5 ± 0.1† | 2.0 ± 0.1§‡ |
| Systolic BP (mm Hg) | 138 ± 4 | 190 ± 4† | 141 ± 5‡ |
| Plasma creatinine (μmol/L) | 50 ± 2 | 140 ± 15† | 92 ± 5§‡ |
| Urinary protein (mg/dl) | 21 ± 2 | 328 ± 66† | 171 ± 49*‡ |
| Plasma renin (ng AI/ml/h) | 11.9×/÷ 1.7 | 2.3×/÷ 0.4† | 11.3×/÷ 1.6‡ |
Data are expressed as mean ± SEM except for plasma renin which is shown as geometric mean ×/÷ tolerance factor.
*p = 0.07 versus sham; §p < 0.05 versus sham; †p < 0.01 versus sham. ‡p < 0.01 versus STNx.
Histopathology
Kidneys from STNx rats demonstrated glomerulosclerosis. The proportion of normal tubules was also substantially reduced in STNx animals as a result of tubular dilatation, tubular atrophy, and interstitial pathology (Table 2 ▶ , Figure 1 ▶ ). Treatment with perindopril significantly reduced the extent of both glomerular and tubulointerstitial injury in STNx rats.
Table 2.
Renin Immunostaining, TGF-β1 and α1(IV) Collagen Gene Expression and Glomerular and Tubulointerstitial Injury after Subtotal Nephrectomy in Treated and Untreated Animals
| Sham | STNx | STNx perindopril | |
|---|---|---|---|
| JGA renin (% positive staining) | 42 (32–50) | 12 (8–20)* | 30 (12–56)† |
| Tubular renin (mean score) | 0.2 (0.0–0.6) | 4.8 (2.8–7.4)* | 1.3 (0.6–2.0)† |
| TGF-β1 mRNA OD (AU) | 1.0± 0.01 | 3.1± 0.4* | 1.4± 0.2† |
| α1(IV) col mRNA OD (AU) | 1.0± 0.01 | 5.4± 2.1* | 1.8± 0.3† |
| Glomerulosclerosis index | 0.08± 0.02 | 3.39± 0.13* | 0.98± 0.16†* |
| Tubulointerstitial injury score | 0 | 58.7± 9.3* | 15.5± 2.9†* |
Data for JGA and tubular renin and angiotensin II immunostaining are expressed as median (range). TGF-β1 and α1(IV) col mRNA are expressed as mean ± SEM of optical density (OD) in arbitrary units (AU). mRNA was quantitated only in the superficial cortex, the most distant area from the infarct-related scar in STNx animals.
*p < 0.01 versus sham.
†p < 0.01 versus STNx.
Figure 1.

Assessment of renal injury in PAS-stained sections in sham (A), STNx (B) and perindopril-treated STNx kidney (C). G denotes glomerulus. Magnification, ×300.
Renin mRNA
Subtotal nephrectomy and its treatment with ACE inhibition was associated with altered distribution of renin gene expression in the kidney (Figure 2) ▶ . In sham kidneys abundant expression of renin mRNA was noted in the juxtaglomerular apparatus and not in the tubular epithelium (Figure 3A) ▶ . In contrast, in STNx kidneys, in areas distant from the site of infarction, de novo renin expression was noted in renal tubular epithelial cells with minimal or absent expression in the JGA (Figure 3, B and C) ▶ . This aberrant expression of renin by the tubular epithelium was particularly apparent in areas of marked structural injury in comparison with areas in which only minor damage was noted (Figure 3, B and D) ▶ . In perindopril-treated STNx rats, areas distant from the infarct scar demonstrated a pattern of renin gene transcription similar to that of control animals (Figures 2 and 3E) ▶ ▶ . Sections incubated with renin sense probe showed no hybridization (Figure 3F) ▶ .
Figure 2.

In situ hybridization of renin, TGF-β, and type IV collagen mRNA in autoradiographs showing distribution and intensity of transcript in sham operated controls, STNx, and perindopril-treated STNx rat kidneys. Magnitude of gene expression is indicated semiquantitatively in pseudocolorized computer images (blue, none; green, low; yellow, moderate; red, high). Magnification, ×4. Punctate cortical renin mRNA was noted in sham animals consistent with expression in the juxtaglomerular apparatus. In STNx kidneys, renin mRNA was noted only in the peri-infarct scar region (left margin). In perindopril-treated STNx kidneys renin mRNA was present both in the infarct scar region (lower margin) and also within the cortex in a punctate distribution. Low levels of TGF-β1 and α1 (IV) collagen mRNA were present in sham animals. Diffuse overexpression of TGF-β1 in both scar and non-scar adjacent areas in STNx kidneys was noted. α1 (IV) collagen mRNA was also diffusely overexpressed in STNx kidneys, particularly in areas adjacent to the infarct scar (lower right). Residual overexpression of TGF-β1 (left margin) and α1 (IV) collagen (left margin and upper pole) mRNA in the region of the infarct scar was seen in perindopril-treated STNx kidneys.
Figure 3.

Assessment of renin mRNA by in situ hybridization in control (A), STNx (B–D), and perindopril-treated STNx rats (E). Abundant expression of renin mRNA was noted in the juxtaglomerular apparatus (arrow) of sham operated controls (A) and in perindopril-treated STNx (E) but not in untreated STNx rats (C). In STNx rats, renin mRNA was observed in tubules (B) with only minimal expression in the juxtaglomerular apparatus (C). Tubular renin (arrow) was particularly apparent in areas of marked injury (B) compared with regions in which only mild structural injury was sustained (D). Sections incubated with renin sense probe showed no hybridization (F). G denotes glomerulus. Magnification, ×640.
Immunohistochemistry
The pattern of renin immunostaining corresponded to that of its mRNA in sham, STNx and perindopril-treated STNx rats in areas distant from the site of infarction (Figure 4) ▶ and in the peri-infarct region (Figure 5) ▶ . In the non infarct-adjacent regions, the proportion of JGAs containing immunostainable renin was significantly reduced in STNx rats compared with sham and perindopril-treated STNx rats (Table 2) ▶ . However, in the peri-infarct area JGA and vascular renin was present and similar in both untreated and perindopril-treated STNx rat kidneys (Figure 5) ▶ .
Figure 4.

Assessment of renin protein by immunohistochemistry in kidneys of sham (A) and STNx (B) and perindopril-treated STNx rats (C). Renin protein was detected in the juxtaglomerular apparatus in sham operated controls (A) and in perindopril-treated STNx (C), but not in untreated STNx rats (B). Immunostainable renin (arrow) was observed in tubules in STNx (B) but not in sham (A) or perindopril-treated STNx rat kidneys (C). Sections incubated with normal rabbit IgG showed no specific labeling (D). G denotes glomerulus. Magnification, ×300.
Figure 5.

Assessment of renin protein (A) and mRNA (B) by in situ hybridization in the scar-adjacent region of STNx kidney. Expression of renin protein and mRNA was noted in the juxtaglomerular apparatus and in the afferent arteriole (arrow) G denotes glomerulus. Magnification, ×300.
Angiotensin II immunostaining was present in the JGA in sham animals but not elsewhere in the nephron (Figure 6A) ▶ . In STNx kidneys, distant from the site of infarction, AII was noted in renal tubular epithelial cells and not in the JGA (Figure 6B) ▶ . In perindopril-treated STNx rats AII staining was not detected in the kidney being absent from both scar-adjacent region and in areas distant from the site of infarction (Figure 6C) ▶ . Sections incubated with normal serum or normal rabbit IgG showed no specific labeling (Figure 6D) ▶ .
Figure 6.

Angiotensin II immunostaining in sham (A), STNx (B), and perindopril-treated STNx rat kidneys (C). Angiotensin II was present in the juxtaglomerular apparatus in sham operated controls (A, arrow) and in tubules in STNx rat kidneys (B, arrow). No immunostainable angiotensin II was detected in perindopril-treated STNx rats (C) including the JGA (arrow). Sections incubated with normal rabbit IgG showed no specific labeling (D). G denotes glomerulus. Magnification, ×340.
Transforming Growth Factor-β1 and α1 (IV) Collagen
Transforming growth factor-β1 mRNA was expressed at low levels in glomeruli and tubulointerstitium of sham animals (Figure 7, A and B) ▶ . In contrast, diffuse overexpression of TGF-β was noted in subtotal nephrectomy rat kidneys (Figures 2, 7C, and 7D) ▶ ▶ in comparison with sham and perindopril-treated STNx rats. Abundant TGF-β transcript was noted in both in the epithelium of dilated and atrophic tubules (Figure 7C) ▶ and in glomeruli (Figure 7D) ▶ . In areas adjacent to the infarct diffuse overexpression of TGF-β1 was noted in all glomerular, tubular and vascular structures. In perindopril-treated STNx rats TGF-β1 expression was reduced compared with untreated STNx animals in non-infarct adjacent tissue (Figure 7, E and F) ▶ , paralleling the amelioration of histological injury (Table 2) ▶ . However, TGF-β1 transcript was still overexpressed in the region adjacent to the infarct scar (Figure 2) ▶ . Sections incubated with TGF-β1 sense probe showed no specific hybridization (Figure 7G) ▶ . The pattern of gene expression for α1 (IV) collagen was similar that of TGF-β1 in sham, STNx, and perindopril-treated STNx rats (Figure 2) ▶ .
Figure 7.

In situ hybridization for transforming growth factor-β in sham (A, B), STNx (C, D), and perindopril-treated STNx rats (E, F). Abundant TGF-β1 mRNA was noted in kidneys of STNx rats in comparison with sham and perindopril-treated STNx animals. Kidney section incubated with TGF-β1 sense probe showed no specific hybridization (G). G denotes glomerulus. Magnification, ×640.
Discussion
It is now more than a decade since the existence of a local as distinct from a systemic RAS was confirmed using molecular biological techniques. 31,32 By combining molecular biological, immunohistochemical, and morphological techniques the present study has extended this concept by demonstrating that even within a single organ, the kidney, different regions have their own locally active, renin-angiotensin systems. Indeed, not only do tubules have all of the components of the RAS, including the ability to synthesize renin, but, at this site the RAS and in particular renin synthesis appears to be regulated in a different manner from the major site of its production in the kidney, the juxtaglomerular apparatus. Following STNx, renin synthesis is suppressed at the juxtaglomerular apparatus but appears de novo in the tubular epithelium. Furthermore, in the present study, the juxtaglomerular apparatus and tubule responded in a divergent manner to ACE inhibition with regard to renin synthesis. As in the intact kidney, 33 ACE inhibition led to increased JGA renin with proximal extension into the afferent arteriole while in the tubule this intervention led to suppression of renin production. These findings provide evidence that within the kidney the regulation of the RAS differs between the juxtaglomerular apparatus and the tubules. The relevance of this altered pattern of renin synthesis is confirmed by concomitant de novo appearance of the effector molecule of the RAS, angiotensin II in the renal tubule in response to subtotal nephrectomy. These findings represent the first in vivo demonstration that the tissue-based local renin-angiotensin system is activated within renal tubules in the setting of renal mass reduction and suggest that this tubular RAS may be pathogenetically involved in the progressive tubulointerstitial injury that accompanies glomerular disease.
The intrarenal renin-angiotensin system has been the subject of intense investigation following the appreciation of the paracrine effects of angiotensin II and the dissociation between its plasma and intrarenal concentrations. 34,35 The substrate, angiotensinogen is primarily formed in the liver leading to circulating levels that are in excess of 1000-fold greater than angiotensins I and II. 36 In addition, angiotensinogen is also synthesized by epithelial cells of the proximal tubule 37,38 as is ACE 39 so that under most circumstances renin activity is the main determinant of angiotensin II formation from its abundant substrate. 40
Studies using in situ hybridization have not found renin mRNA outside the JGA and afferent arteriole in the normal kidney. 31,32 This was interpreted as indicating that the immunohistochemical localization of renin in subapical vessels of the proximal tubule 41 reflects the endocytotic activity of these cells rather than endogenous synthesis of renin. More recently, with the use of ultra-sensitive techniques such as RT-PCR (reverse transcription polymerase chain reaction) low levels of renin mRNA have been detected in the microdissected tubules of normal rats. 9 In addition, using quantitative competitive RT-PCR, modulation of renin expression in response to uninephrectomy, 9 salt depletion 42 and ACE inhibition 43 have also been noted. However, in other studies using RT-PCR, proximal tubule renin mRNA has not been found either under basal conditions, or after ACE inhibition and given the ultrasensitivity of RT-PCR and the abundance of renin in the JGA it is possible that contamination with JGA transcripts may account for these divergent findings. 44
In the present study, renal mass reduction was associated with dramatic changes in the microtopography of renin expression with a substantial reduction in JGA renin and its de novo appearance in the tubular epithelium. The presence of renin mRNA in the tubular epithelium indicates that the renin protein detected immunohistochemically within these cells was synthesized intracellularly rather than derived by pinocytosis. Indeed, the paucity of juxtaglomerular renin synthesis in this model suggests that little, if any, of the renin detected immunohistochemically would have been derived from the glomerular filtrate. Furthermore, the co-localization of angiotensin II to the tubular epithelial cells suggests that all of the necessary components of the renin-angiotensin system are present within these cells in the setting of renal injury following subtotal nephrectomy. However, despite its presence in the tubular epithelium and throughout the peri-infarct region, JGA renin expression was substantially reduced thus accounting for the relative reduction in total kidney and non-scar adjacent renin mRNA noted previously. 45
Angiotensin II has several non-hemodynamic actions which may contribute to the progression of renal disease. These include induction of oxidant stress by stimulating sodium transport, 46 increased macromolecular trafficking 47 and the increased secretion of various peptide growth factors, most notably TGF-β. 48 In vitro, angiotensin II induces TGF-β expression in a variety of cell types that may contribute to the pathogenesis of progressive renal injury including proximal tubular cells 48 and renal interstitial fibroblasts 49 as well as mesangial 50 and vascular smooth muscle cells. 51 Furthermore, the stimulation of matrix protein synthesis which accompanied AII-induced induction of TGF-β transcription can be blocked by either neutralizing TGF-β antibody or by AII-receptor antagonism. 50 Similarly, in vivo, infusion of angiotensin II leads to increased TGF-β and matrix protein expression 52 further suggesting a link between activation of the renin-angiotensin system, TGF-β expression and extracellular matrix (ECM) synthesis.
There is substantial evidence to support a pathogenetic role for TGF-β in orchestrating the accumulation of ECM, leading to fibrosis and ultimately, renal dysfunction in several models of experimental renal injury and in human kidney disease (reviewed in Ref. 52 ). Its prosclerotic effects include stimulation of ECM production, inhibition of its degradation, up-regulation of cell-matrix adhesion molecules and chemoattraction of macrophages and fibroblasts. 53 In the present study, renal mass reduction was associated with greatly increased expression of TGF-β1 mRNA throughout the nephron.
In many primary glomerular diseases the extent of interstitial damage correlates better with the degree of renal dysfunction than do indices of glomerular injury 54 and the mechanisms by which such glomerular changes lead to tubulointerstitial injury have been the focus of considerable recent attention. 55-57 The model of renal mass reduction, as used in the present study, is believed to lead to progressive renal injury as a consequence of altered intraglomerular hemodynamics. 58 The finding that in this form of primarily glomerular injury, there is activation of the renin-angiotensin system and TGF-β1 expression within the tubular epithelium, as demonstrated in the present study, provides a pathophysiological link between glomerular injury and the development of tubulointerstitial fibrosis.
The findings of the present study do not suggest that intrarenal-derived AII is the only stimulus for TGF-β expression following renal mass reduction. Indeed, in this study neither renin nor AII were detected in glomeruli of subtotal nephrectomized rats despite abundant TGF-β1 mRNA at this site suggesting that other factors such as physical hemodynamic forces 59 and other vasoactive hormones such as the endothelins 60 may also be involved in up-regulating TGF-β expression. Furthermore, although tubular expression of renin and AII were noted following subtotal nephrectomy this does not indicate that AII is the only stimulus for TGF-β expression and fibrosis at this site. For instance, as in the glomerulus other factors such as protein trafficking may also be involved in tubulointerstitial TGF-β expression and fibrosis. 7
The expression of renin by tubular epithelial cells, described in the present study may reflect a phenotypic change that occurs as a nonspecific response to injury. Indeed, this phenomenon of cell transdifferentiation in which there is de novo expression of proteins such as osteopontin 61 and SPARC 21 has been well described in the setting of renal disease.
Interruption of the renin-angiotensin system by either ACE inhibition or angiotensin II-receptor antagonism reduces renal injury in several models of experimental and human kidney disease. 52 In the absence of activation of the systemic RAS, these pharmacological intervention studies provide further evidence for the role of the local, intrarenal RAS in the progression of kidney disease. In the present study, administration of the ACE inhibitor, perindopril was associated with disappearance of aberrant tubular expression of renin and angiotensin II along with the restoration of high level expression to the JGA. In addition, ACE inhibition was associated with reduced expression of the AII-inducible mediator of renal fibrosis, TGF-β along with the amelioration of the structural and functional manifestations of renal injury.
Acknowledgments
We thank Servier Laboratories for providing perindopril and for their assistance with the cost of color reproduction. Dr Gilbert is the recipient of a career development award from the Juvenile Diabetes Foundation International.
Footnotes
Address reprint requests to Dr. Richard Gilbert, Department of Medicine, Repatriation Campus, University of Melbourne, Heidelberg West 3081, Victoria, Australia. E-mail: gilbert@austin.unimelb.edu.au.
Supported in part by a grant from the Baxter Health Corporation Extramural Grant Program.
References
- 1.The GISEN Group (Gruppo Italiano di Studi Epidemiologici in Nefrologia): Randomised placebo-controlled trial of effect of ramipril on decline in glomerular filtration rate and risk of terminal renal failure in proteinuric, non-diabetic nephropathy. Lancet 1997, 349:1857–1863 [PubMed]
- 2.Lewis EJ, Hunsicker LG, Bain RP, Rhode RD, : the Collaborative Study Group: The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. N Engl J Med 1993, 329:1456-1462 [DOI] [PubMed] [Google Scholar]
- 3.Seikaly MG, Arant BS, Seney FD: Endogenous angiotensin concentrations in specific intrarenal fluid compartments of the rat. J Clin Invest 1990, 86:1352-1357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murlow PJ: The intrarenal renin-angiotensin system. Curr Opin Nephrol Hypertens 1993, 2:41-44 [DOI] [PubMed] [Google Scholar]
- 5.Risdon RA, Sloper JC, de Wardener HE: Relationship between renal function and histological changes found in renal biopsy specimens from patients with persistent glomerulonephritis. Lancet 1968, ii:363-366 [DOI] [PubMed] [Google Scholar]
- 6.Schainuck LI, Striker GE, Cutler RE, Benditt EP: Structural-functional correlations in renal disease. Part II. The correlations. Hum Pathol 1970, 1:631-641 [DOI] [PubMed] [Google Scholar]
- 7.Remuzzi G, Ruggenenti P, Benigni A: Understanding the nature of renal disease progression. Kidney Int 1997, 51:2-15 [DOI] [PubMed] [Google Scholar]
- 8.Gilbert RE, Cox A, Wu LL, et al: Expression of transforming growth factor-β1 and type IV collagen in the renal tubulointerstitium in experimental diabetes: effects of angiotensin converting enzyme inhibition. Diabetes 1998, 47:414-422 [DOI] [PubMed] [Google Scholar]
- 9.Tank JE, Moe OW, Star RA, Henrich WL: Differential regulation of rat glomerular and proximal tubular renin mRNA following uninephrectomy. Am J Physiol 1996, 270:F776-F783 [DOI] [PubMed] [Google Scholar]
- 10.Ruiz-Ortega M, Gomez-Garre D, Alcazar R, et al: Involvement of angiotensin II and endothelin in matrix protein production and renal sclerosis. J Hypertens 1994, Suppl 12:S51-S58 [PubMed] [Google Scholar]
- 11.Wolf G: Link between angiotensin II and TGF-β in the kidney. Miner Electrolyte Metab 1998, 24:174-180 [DOI] [PubMed] [Google Scholar]
- 12.Rosenberg M, Correa-Rotter R, Inagami T, Kren S, Hostetter T: Glomerular renin synthesis and storage in the remnant kidney in the rat. Kidney Int 1991, 40:677-683 [DOI] [PubMed] [Google Scholar]
- 13.Pupilli C, Chevalier R, Carey R, Gomez A: Distribution and content of renin and renin mRNA in remnant kidney of adult rat. Am J Physiol 1992, F731–F738 [DOI] [PubMed]
- 14.Wu L, Cox A, Roe C, Dziadek M, Cooper ME, Gilbert RE: Transforming growth factor β1 and renal injury following subtotal nephrectomy in the rat: Role of the renin-angiotensin system. Kidney Int 1997, 51:1553-1567 [DOI] [PubMed] [Google Scholar]
- 15.Cooper ME, Rumble JR, Allen T, O’Brien RC, Jerums G, Doyle AE: Antihypertensive therapy in a model combining spontaneous hypertension with diabetes. Kidney Int 1992, 41:898-903 [DOI] [PubMed] [Google Scholar]
- 16.Pfeffer JM, Pfeffer MA, Frohlich ED: Validity of an indirect tail-cuff method for determining systolic arterial pressure in unanesthetized normotensive and spontaneously hypertensive rats. J Lab Clin Med 1971, 78:957-962 [PubMed] [Google Scholar]
- 17.Lott JA, Stephan VA, Pritchard KJ: Evaluation of the Coomassie Brilliant Blue G-250 method for urinary protein. Clin Chem 1983, 29:1946-1950 [PubMed] [Google Scholar]
- 18.Kliem V, Johnson RJ, Alpers CE, et al: Mechanisms involved in the pathogenesis of tubulointerstitial fibrosis in 5/6 nephrectomized rats. Kidney Int 1996, 49:666-678 [DOI] [PubMed] [Google Scholar]
- 19.Scholey JW, Miller PL, Rennke HG, Meyer TW: Effect of converting enzyme inhibition on the course of adriamycin-induced nephropathy. Kidney Int 1989, 36:816-822 [DOI] [PubMed] [Google Scholar]
- 20.Gilbert RE, McNally PG, Cox A, et al: SPARC gene expression is reduced in early diabetes related kidney growth. Kidney Int 1995, 48:1216-1225 [DOI] [PubMed] [Google Scholar]
- 21.Wu L, Cox A, Roe C, Dziadek M, Cooper ME, Gilbert RE: Secreted protein acidic and rich in cysteine expression after subtotal nephrectomy and blockade of the renin-angiotensin system. J Am Soc Nephrol 1997, 8:1374-1382 [DOI] [PubMed] [Google Scholar]
- 22.Gilbert RE, Cox A, McNally PG, et al: Increased epidermal growth factor expression in diabetes related kidney growth. Diabetologia 1997, 40:778-785 [DOI] [PubMed] [Google Scholar]
- 23.Baskin DG, Stahl W: Fundamentals of quantitative autoradiography by computer densitometry for in situ hybridization with emphasis on 33P. J Histochem Cytochem 1993, 41:1767-1773 [DOI] [PubMed] [Google Scholar]
- 24.Wookey PJ, Tikellis C, Du H-C, Qin H-F, Sexton PM, Cooper ME: Amylin binding in rat renal cortex, stimulation of adenyl cyclase and activation of plasma renin. Am J Physiol 1996, 270:F289-F294 [DOI] [PubMed] [Google Scholar]
- 25.Mendelsohn F, Dunbar M, Allen A, et al: Localization of angiotensin II receptors in rat and monkey kidney by in vitro autoradiography. Kidney Int Suppl 1987, 31:S40-S44 [PubMed] [Google Scholar]
- 26.Rumble JR, Cooper ME, Soulis T, et al: Vascular hypertrophy in experimental diabetes: role of advanced glycation end products. J Clin Invest 1997, 99:1016-1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berka JL, Alcorn D, Ryan GB, Skinner SL, Weaver DA: Renin processing in cultured juxtaglomerular cells of the hydronephrotic mouse kidney. J Histochem Cytochem 1993, 41:365-373 [DOI] [PubMed] [Google Scholar]
- 28.Bragat AC, Blumenfeld J, Sealey JE: Effect of high-performance liquid chromatography on plasma angiotensin II measurements in treated and untreated normotensive and hypertensive patients. J Hypertens 1997, 15:459-465 [PubMed] [Google Scholar]
- 29.Gocke DJ, Gerten J, Sherwood LM, Laragh JH: Physiological and pathological variations in plasma angiotensin II in man. Circ Res 1969, 24 and 25:Supplement I131–I148 [PubMed]
- 30.Snedecor GW, Cochran WG: Statistical Methods. 1980:228-236 Iowa State University Press Iowa
- 31.Deschepper CF, Mellon SH, Cumin F, Baxter JD, Ganong WF: Analysis by immunocytochemistry and in situ hybridization of the distribution of renin and its mRNA in kidney, testis, adrenal, and pituitary of the rat. Proc Natl Acad Sci USA 1986, 83:7552-7556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dzau VJ, Ellison KE, Brody T, Ingelfinger J, Pratt RE: A comparative study of the distribution of renin and angiotensinogen messenger ribonucleic acids in rat and mouse tissues. Endocrinology 1987, 120:2334-2338 [DOI] [PubMed] [Google Scholar]
- 33.Berka JLA, Alcorn D, Coghlan JP, et al: Granular juxtaglomerular cells and prorenin synthesis in mice treated with enalapril. J Hypertens 1990, 8:229-238 [PubMed] [Google Scholar]
- 34.Mendelsohn FAO: Angiotensin II: evidence for its role as an intrarenal hormone. Kidney Int 1992, 22:S78-S81 [PubMed] [Google Scholar]
- 35.Navar LG, Inscho EW, Majid SA, Imig J, Harrison-Bernard LM, Mitchell KD: Paracrine regulation of the renal microcirculation. Physiol Rev 1996, 76:425-536 [DOI] [PubMed] [Google Scholar]
- 36.Brasier AR, Li J: Mechanisms for inducible control of angiotensinogen gene transcription. Hypertension 1996, 27:465-475 [DOI] [PubMed] [Google Scholar]
- 37.Richoux JP, Cordonnier JL, Bouhnik J, et al: Immunocytochemical localization of angiotensinogen in rat liver and kidney. Cell Tissue Res 1983, 233:439-451 [DOI] [PubMed] [Google Scholar]
- 38.Ingelfinger JR, Zuo WM, Fon EA, Ellison KE, Dzau VJ: In situ hybridization evidence for angiotensinogen messenger RNA in rat proximal tubule. J Clin Invest 1990, 85:417-423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sibony M, Gasc JM, Soubrier F, Alhenc-Gelas F, Corvol P: Gene expression and tissue localization of the two isoforms of angiotensin I converting enzyme. Hypertension 1993, 21:827-835 [DOI] [PubMed] [Google Scholar]
- 40.Navar LG, Imig JD, Zou L, Wang CT: Intrarenal production of angiotensin II. Semin Nephrol 1997, 17:412-422 [PubMed] [Google Scholar]
- 41.Taugner R, Hackenthal E, Rix E, Nobiling R, Poulsen K: Immunocytochemistry of the renin-angiotensin system: renin, angiotensinogen, angiotensin I, angiotensin II, and converting enzyme in the kidneys of mice, rats and tree shrews. Kidney Int 1982, 22:S33-S43 [PubMed] [Google Scholar]
- 42.Tank JE, Henrich WL, Moe OW: Regulation of glomerular and proximal tubule renin mRNA by chronic changes in dietary NaCl. Am J Physiol 1997, 273:F892-F898 [DOI] [PubMed] [Google Scholar]
- 43.Moe OW, Ujiie K, Star RA, et al: Renin expression in proximal tubule. J Clin Invest 1993, 91:774-779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen M, Harris M, Smart A, et al: Renin and renin mRNA in proximal tubules of the rat kidney. J Clin Invest 1994, 94:237-243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Correa-Rotter R, Hostetter TH, Manivel JC, Rosenberg ME: Renin expression in renal ablation. Hypertension 1992, 20:483-490 [DOI] [PubMed] [Google Scholar]
- 46.Nath KA, Croatt AJ, Hostetter TH: Oxygen consumption and oxidant stress in surving nephrons. Am J Physiol 1990, 258:F1354-F1362 [DOI] [PubMed] [Google Scholar]
- 47.Keane WF, Raij L: Relationship among altered glomerular barrier permselectivity, angiotensin II, and mesangial uptake of macromolecules. Lab Invest 1985, 52:599-604 [PubMed] [Google Scholar]
- 48.Wolf G, Mueller E, Stahl RAK, Ziyadeh FN: Angiotensin II-induced hypertrophy of cultured murine proximal tubular cells is mediated by endogenous transforming growth factor-β. J Clin Invest 1993, 92:1366-1372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ruiz-Ortega M, Egido J: Angiotensin II modulates cell growth-related events, and synthesis of matrix proteins in renal interstitial fibroblasts. Kidney Int 1997, 52:1497-1510 [DOI] [PubMed] [Google Scholar]
- 50.Kagami S, Border WA, Miller DE, Noble NA: Angiotensin II stimulates extracellular matrix protein synthesis through induction of transforming growth factor-β expression in rat glomerular mesangial cells. J Clin Invest 1994, 93:2431-2437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gibbons GH, Pratt RE, Dzau VJ: Vascular smooth muscle hypertrophy versus hyperplasia. J Clin Invest 1992, 90:456-461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Noble NA, Border WA: Angiotensin II in renal fibrosis: should TGF-β rather than blood pressure be the therapeutic target. Semin Nephrol 1997, 17:455-466 [PubMed] [Google Scholar]
- 53.Sharma K, Ziyadeh FN: The emerging role of transforming growth factor-β in kidney diseases. Am J Physiol 1994, 266:F829-F842 [DOI] [PubMed] [Google Scholar]
- 54.Nath KA: Tubulointerstitial changes as a major determinant in the progression of renal damage. Am J Kidney Dis 1992, 20:1-17 [DOI] [PubMed] [Google Scholar]
- 55.Ong ACM, Fine LG: Loss of glomerular function and tubulointerstitial fibrosis. Kidney Int 1994, 45:245-351 [DOI] [PubMed] [Google Scholar]
- 56.Eddy AA: Experimental insights into the tubulointerstitial disease accompanying primary glomerular lesions. J Am Soc Nephrol 1994, 5:1273-1287 [DOI] [PubMed] [Google Scholar]
- 57.Couser WG, Johnson RJ: Mechanisms of progressive renal disease in glomerulonephritis. Am J Kidney Dis 1994, 23:193-198 [DOI] [PubMed] [Google Scholar]
- 58.Brenner BM: Hemodynamically mediated glomerular injury and the progressive nature of kidney disease. Kidney Int 1983, 23:647-655 [DOI] [PubMed] [Google Scholar]
- 59.Yasuda T, Kondo S, Homma T, Harris RC: Regulation of extracellular matrix by mechanical stress in rat glomerular mesangial cells. J Clin Invest 1996, 98:1991-2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Egido J: Vasoactive hormones and renal sclerosis. Kidney Int 1996, 49:578-597 [DOI] [PubMed] [Google Scholar]
- 61.Lan HY, Yu XQ, Yang NS, et al: De novo glomerular osteopontin expression in rat crescentic glomerulonephritis. Kidney Int 1998, 53:136-145 [DOI] [PubMed] [Google Scholar]