

Abstract
The inflammatory pathogenesis in airways of patients with cystic fibrosis (CF) is still unresolved. We demonstrate here that in in situ human ΔF508 homozygous CF bronchial tissues, submucosal gland cells exhibit an absence of inhibitor factor κBα (IκBα) and high levels of chemokine interleukin-8 (IL-8) expression. These results were confirmed by cultured human CF bronchial gland cells in which a lack of cytosolic IκBα and high levels of constitutively activated nuclear factor κB (NFκB) associated with an up-regulation of IL-8 production (13-fold increase) were found when compared to non-CF (control) disease bronchial gland cells. We also demonstrated that the isoflavone genistein, a well known CFTR mutant Cl− channel stimulator, significantly reduces the endogenous and Pseudomonas aeruginosa lipopolysaccharide-induced IL-8 production in cultured CF bronchial gland cells by increasing cytosolic IκBα protein levels. Overall, results show that genistein is a potent inhibitor of the activated NFκB identified in CF gland cells. This strong inhibition of constitutively activated NFκB and the resulting down-regulation of IL-8 production by genistein in the CF gland cells highlights the key role played by cytosolic IκBα in the regulation of inflammatory processes in CF human airway cells.
Cystic fibrosis (CF) is a genetic disease caused by mutations in a single gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR), which has been shown to be a cAMP-activated Cl− channel 1 and to regulate the activity of other channels in airway epithelial cells. 2 Despite considerable progress in our understanding of the structure and functions of CFTR, the mechanism by which the absence or dysfunction of the CFTR protein causes numerous pathological manifestations, including early chronic airway inflammation, remains unexplained in CF disease. Recently, we demonstrated that CF mice raised in pathogen-free conditions exhibited more lymphocytes in the airway submucosa compared with wild-type littermates. 3 Recent studies 4,5 have shown evidence that CF human bronchial submucosal tissues in situ and primary cultures of CF bronchial gland cells as well as established CF cell lines 6 constitutively expressed significantly high levels of proinflammatory cytokines, particularly the chemokine interleukin (IL)-8, which is one of the most potent neutrophilic chemoattractants in CF human airways. Thus, it is possible that chronic endobronchial inflammation in CF patients may be related to constitutive abnormalities in the regulation of proinflammatory cytokine expression by CF airway epithelial cells, even in the absence of bacterial infection. The abnormal regulation of some components of one or more inflammatory cascades used in local immune defenses may be a direct consequence of mutant CFTR in CF respiratory epithelial cells. Nuclear factor κB (NFκB) is a central mediator that can rapidly activate transcription of various inflammatory cytokines, chemokines and adhesion molecules in lung epithelial cells. 7 To date, there is no evidence that a constitutive NFκB activation associated with IL-8 up-regulation is present in airway epithelial cells from CF patients.
In a recent study, 4 we demonstrated that eight different ΔF508 homozygous CF bronchial tissues and CF human bronchial submucosal gland (HBG) cells subcultured in a resting (unstimulated) state exhibited consistently high mRNA and protein IL-8 expression. This abnormally high IL-8 production at the bronchial submucosal level in CF secretory glands was selective and was not observed for other cytokines such as IL-1β, IL-6, or the anti-inflammatory cytokine IL-10. Consequently, it was suggested that the exaggerated production of IL-8 identified in CF-HBG cells might be occurring primarily because of the abnormal regulation of an endogenous pathway, rather than as a general response to airway inflammation. High levels of endogenous IL-8 detected in CF bronchial gland cells suggests that this up-regulated expression of IL-8 might result from constitutively activated NFκB in CF gland cells. In most cell types, NFκB exists as an inactive complex in the cytoplasm bound to its natural cytoplasmic inhibitor, IκBα. On activation, IκBα rapidly degrades and allows translocation of free, active NFκB dimers into the nucleus to activate target genes. 8,9
Genistein is an isoflavonoid abundant in legumes, particularly soybeans. 10 It was introduced as a specific inhibitor of protein tyrosine kinase 11 and other ATP-binding enzymes 12 and has been shown to suppress Pseudomonas aeruginosa lipopolysaccharide (LPS)-induced MUC2 mucin gene transcription in CF respiratory epithelial cells via the activation of NFκB. 13 Whether genistein exhibits anti-inflammatory properties in native CF human airway epithelial cells, in particular CF bronchial gland cells in which high constitutive IL-8 expression is selectively up-regulated compared to non-CF bronchial glands, is not known. To answer this question, we examined the ability of genistein to inhibit the constitutive and P. aeruginosa LPS-induced NFκB activation and subsequent IL-8 production in cultures of ΔF508 homozygous CF and non-CF human bronchial gland cells.
In the present study, we demonstrated that in situ CF bronchial submucosal gland cells express both a high level of endogenous chemokine IL-8 and a total absence of inhibitor factor IκBα in contrast to non-CF disease bronchial tissues, in which a strong immunoreactivity for IκBα, but not the endogenous IL-8, was identified. We have also shown that treatment of cultured CF bronchial gland cells with genistein reverses the constitutive and P. aeruginosa LPS-induced nuclear translocation of NFκB by increasing cytosolic IκBα protein levels and decreasing IL-8 production.
Experimental Procedures
Human Bronchial Tissues
Human CF bronchial tissue was obtained from eight recipients undergoing lung transplant operations (all of the CF patients were ΔF508 homozygous; four females and four males; mean age 17.3 years; range, 9–27 years). Tissues for control experiments were obtained from four non-CF disease patients (two males with primary pulmonary hypertension, aged 28 and 29 years, and two males with pulmonary idiopathic fibrosis, aged 40 and 61 years). To evaluate the level of airway inflammation for each CF and non-CF patient in the study, we first analyzed the number of inflammatory cells and polymorphonuclear neutrophils surrounding the bronchial submucosal glands by extensive histological examinations. Data of histological examinations did not demonstrate a significant increase in the mean number of inflammatory cells in the CF patient group compared with the non-CF disease control group. 4
Immunohistochemistry
For the immunohistochemical analysis of CF and non-CF bronchial tissues, frozen tissue samples were embedded in OCT (Miles Tissue Tek, Elkhart, IN), immersed in liquid nitrogen, and stored at −80°C. Bronchial cryosections (5 μm thick) deposited onto gelatin-coated glass slides were stored at −20°C after air-drying and rehydrated in 0.1 mol/L phosphate buffered saline (PBS) at pH 7.2. Sets of serial cryofixed sections were then blocked with PBS-1% bovine serum albumin for 10 minutes and stained for IL-8 and IκBα. Areas of the submucosal connective tissue showing glands in bronchial cryosections of four non-CF disease patients were selected and analyzed. A minimum of 24 microscopy fields (>450 mm 2 of submucosal tissues) were examined. The percentage of IL-8-positive glands was calculated. Monoclonal antibodies against IL-8 (dilution 1:50) were purchased from Biosource International (Camarillo, CA). Rabbit antiserum to human IκBα (dilution 1:60) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). In all immunofluorescence experiments, bound antibodies were detected using the streptavidin-fluorescein isothiocyanate (FITC) system (Amersham International, Amersham, UK). Secondary antibodies of goat biotinylated anti-mouse and anti-rabbit IgG fractions (Boehringer Mannheim, Mannheim, Germany) and streptavidin-FITC were used at a dilution of 1:50. Negative controls were performed using either non-immune mouse or rabbit IgG fractions (Sigma Chemical Co., St. Louis, MO). Specimens were counterstained with Harris hematoxylin solution, mounted in citifluor antifading solution (Agar Scientific, Stanted, UK), and observed by using a Zeiss Axiophot microscope (Zeiss, Le Pecq, France) employing epifluorescence and Nomarski differential interference illumination.
Cell Culture
Cell isolation and subculture procedures of HBG cells were performed on bronchial tissues collected from eight ΔF508 homozygous CF patients and four non-CF patients, as described previously. 4 Briefly, HBG cells were isolated by enzymatic digestion from bronchial submucosa and grown onto type I collagen-coated 25-cm 2 tissue culture flasks in a DMEM/Ham’s F12-mixture (50/50%, v/v) supplemented with 1% Ultroser G (a serum substitute from Sepracor, Villeneuve-la-Garenne, France), glucose (10 g/l), and sodium pyruvate (0.33 g/l). Penicillin G (100 U/ml) and streptomycin (100 μg/ml) were also added. After 4 weeks in culture, second and third-passage CF-HBG and non-CF HBG cells had proliferated and exhibited characteristics of homogenous submucosal epithelial and secretory gland cells. Using the halide-sensitive fluorescent dye 6-methoxy-N-(3-sulfopropyl)-quinolinium, we have previously shown a significant increase in Cl− channel activity via the CFTR protein in non-CF HBG cells in response to forskolin treatment (demonstrating a cAMP-dependent activation of a Cl− efflux), which is not preserved in cultured CF HBG cells. 4
Enzyme-Linked Immunosorbent Assay (ELISA) for IL-8 Determination
Primary cultures of confluent ΔF508 homozygous CF and non-CF HBG cells grown on type I collagen-coated coverslips at the same passage were incubated at the for 16 hours in a Ultroser G-free control medium (DMEM/Ham’s F12, alone). Following this incubation period, CF and non-CF HBG cells were cultured for 6 hours in either DMEM/Ham’s F12 alone, or DMEM/Ham’s F12 with 1.0 μg/ml P. aeruginosa lipopolysaccharide (P. aeruginosa LPS, serotype 10, Sigma). CF and non-CF cells were also cultured under each of these two conditions in the presence or absence of various concentrations (20–100 μmol/L) of genistein (Sigma), a specific tyrosine kinase inhibitor with broad-spectrum activity. 9 Genistein was added 2 hours before the addition of P. aeruginosa LPS. Immediately after each period of cell exposure, supernatants were collected and stored at −80°C until tested for the presence of cytokine IL-8. The ELISAs for IL-8 detection, which were sensitive down to a level of 5 pg/ml, were performed by following the manufacturer’s instructions in commercially available ELISA kits (Biosource International). To assess cell viability, lactate dehydrogenase (LDH) released into the cell supernatant was measured immediately after incubation using a Sigma LDH kit. LDH release never exceeded 5% of the total LDH content of cells under the experimental conditions used here. All results are expressed as pg/ml/10 6 cells.
Immunofluorescence
After each period of cell treatment as described above, CF and non-CF HBG cell monolayers were fixed in situ in cold methanol for 10 minutes at −20°C, air-dried, and rehydrated in 0.1mol/L PBS at pH 7.4 before immunodetection studies. Cells were stained for IκBα expression using rabbit antiserum to human IκBα (Santa Cruz Biotechnology) for 1 hour at room temperature and a donkey anti-rabbit FITC-conjugated antibody for 45 minutes at room temperature. Negative controls were obtained using either nonspecific IgG as the primary antibody (ref. M7769, Sigma) or with FITC-conjugated antibody alone. After rinsing in three changes of PBS-1% bovine serum albumin for 10 minutes each time, all specimens were counterstained with Harris hematoxylin solution for 10 seconds, mounted in citifluor antifading solution (Agar Scientific), and observed using a Zeiss Axiophot microscope (Zeiss, Le Pecq, France) employing epifluorescence and Nomarski differential interference illumination. Representative fields of resting and stimulated CF and non-CF HBG cells were digitized with 256 gray levels and printed using color photo paper (Hewlett-Packard, Palo Alto, CA).
Cell Extracts and Western Blot Analysis
Non-CF and CF HBG cell monolayers treated or not (controls) by genistein as previously described were washed in PBS (pH 7.2), harvested by scraping, centrifuged (300 × g, 5 minutes, 4°C), and total protein extracted (30 minutes, 4°C) in RIPA buffer (50 mmol/L Tris, pH 8.0, 150 mmol/L NaCl2, and 1% Nonidet P-40 (Sigma), 0.5% deoxycolate (Sigma), 0.1% sodium dodecyl sulfate supplemented with 0.1 mmol/L phenylmethylsulfonyl fluoride (Sigma), 5 μg/ml aprotinine, 1 μg/ml chymostatin, 4 μg/ml pepstatin, 5 μg/ml leupeptin, and 0.1 mg/ml α-1 antitrypsin (Boehringer Mannheim). Protein extracts were centrifuged (12,000 × g, 30 minutes, 4°C) and protein concentrations were measured using the Bradford assay (Bio-Rad, Hercules, CA). Equal amounts of protein were boiled for 4 minutes in Laemmli buffer, and electrophoresis was carried out under denaturing using 4 to 15% polyacrylamide gels (Pharmacia Biotech, Orsay, France). Proteins were transferred onto a nitrocellulose membrane (Millipore, Bedford, MA) by electroblotting and detected using rabbit antiserum to human IκBα (Santa Cruz Biotechnology). Proteins were visualized using horseradish peroxidase-conjugated donkey anti-rabbit IgG (Boehringer Mannheim) and the enhanced chemiluminescence detection kit (Amersham Life Science, Arlington Heights, IL) according to the manufacturer’s instructions. Prestained molecular weight markers (Bio-Rad) were loaded on each gel to verify effective transfer of proteins to membranes. Densitometric analysis of Western blots was performed on a Bio-Rad model GS-690 imaging densitometer using Molecular Analyst software, version 1.4.1. The gels were scanned in the transmittance mode at a resolution setting of 800 dpi. The intensities of bands were compared on the basis of adjusted volume (mean optical density × area in square millimeter).
Preparation of Nuclear Extracts and Electrophoretic Mobility Shift Assay (EMSA)
Nuclear extracts were prepared using a procedure slightly modified from that of Israel et al. 14 Briefly, 5–6 × 10 6 cells were washed with cold PBS and cells were resuspended in 1.5 ml of hypotonic buffer (10 mmol/L HEPES, pH 7.9, 1.5 mmol/L, MgCl2, 10 mmol/L KCl, 0.5 mmol/L DTT (Sigma), and 0.1% Nonidet P-40 (Boehringer Mannheim). After incubating for 10 minutes on ice, the homogenate was centrifuged at 10,000 rpm and the resulting pellet was resuspended by gentle pipetting in 30 μl of lysis buffer (20 mmol/L HEPES, 420 mmol/L NaCl, 1.5 mmol/L MgCl2, 0.2 mmol/L EDTA, 25% [v/v] glycerol, and 0.5 mmol/L DTT). This suspension was incubated for 20 minutes at 4°C followed by centrifugation at 14,000 rpm for 10 minutes. The nuclear extract was divided into aliquots and stored at −80°C for subsequent use. To minimize proteolysis, all buffers contained 0.5 mmol/L phenylmethylsulfonyl fluoride, 5 μg/ml aprotinin, 1 μg/ml chymostatin, 4 μg/ml pepstatin, 5 μg/ml leupeptin, and 0.1 mg/ml α-1 antitrypsin (Boehringer Mannheim). The consensus κB DNA sequence was used for the EMSA (5′AGT TGA GGG GAC TTT CCC AGG C3′, Promega Corp., Madison, WI). Probe DNA (with 5′ overhangs) was end-labeled by the T4 polynucleotide kinase (Pharmacia Biotech, Paris, France) enzyme with [α32P] ATP. Nuclear extracts (4 μg) were incubated with 50 kcpm of 32P-labeled NFκB oligonucleotide in binding reaction mixture (20% Ficoll, 175 mmol/L NaCl, 300 mmol/L KCl, 0.05% Nonidet P-40, pH 7.0) in a final volume of 15 μl. After 15 minutes on ice and 15 minutes at room temperature, the protein-DNA complexes were resolved on a nondenaturing 5% polyacrylamide gel in a 1× TBE buffer (89 mmol/L Tris-HCl, 89 mmol/L boric acid, and 2 mmol/L EDTA) and electrophoresed for 1.5 hours at room temperature. Gels were then dried and subjected to autoradiography for analysis. Identity of the different NFκB heterodimeric proteins was carried out by incubations of the nuclear extracts with polyclonal antibodies against the NFκB proteins NFκB1 (p50) and the Rel proteins (p65) RelA (Santa Cruz Biotechnology), before addition of the labeled κB probe. These antibodies were added to the above reaction mixture at a concentration of 10 μg/100 μl. In competition studies, a 100-fold molar excess of unlabeled oligonucleotide was added to the binding reaction mixture. All samples were then incubated at room temperature for 1 hour before gel loading.
Statistical Analysis
Results were expressed as means ± SD. Each data point was confirmed in triplicate at least, and each cell culture experiment performed at least three times. Differences in IL-8 levels were analyzed using the Student’s t-test for paired and unpaired samples.
Results
Lack of IκBα Protein in Bronchial Gland Cells from Patients with CF
To demonstrate that both the high levels of IL-8 and activated NFκB were expressed in CF human submucosal secretory gland cell type, we investigated the immunoreactivity of endogenous IL-8 and IκBα protein in serial bronchial cryofixed sections obtained from eight CF patients and compared them to four non-CF disease controls in which IL-8 immunoreactivity was undetectable in most of the bronchial submucosal glands. When sections of bronchial submucosal tissues from CF patients were analyzed (Figure 1) ▶ , submucosal gland cells in all of the CF samples showing a high immunoreactivity for IL-8 ( Ref. 4 and Figure 1A ▶ ) were negative for IκBα (Figure 1C) ▶ . This is in contrast to bronchial submucosal glands from non-CF disease patients, in which no detectable IL-8 is observed in 90% of submucosal glands ( Ref. 4 and Figure 1E ▶ ) but strong immunoreactivities for IκBα were found (Figure 1G) ▶ . Similarly, a weak immunostaining for IκBα protein was detected in cultured CF HBG cells (Figure 1D) ▶ in comparison to non-CF HBG cells in which dense IκBα staining was identified in the cytoplasm of cultured cells (Figure 1H) ▶ .
Figure 1.
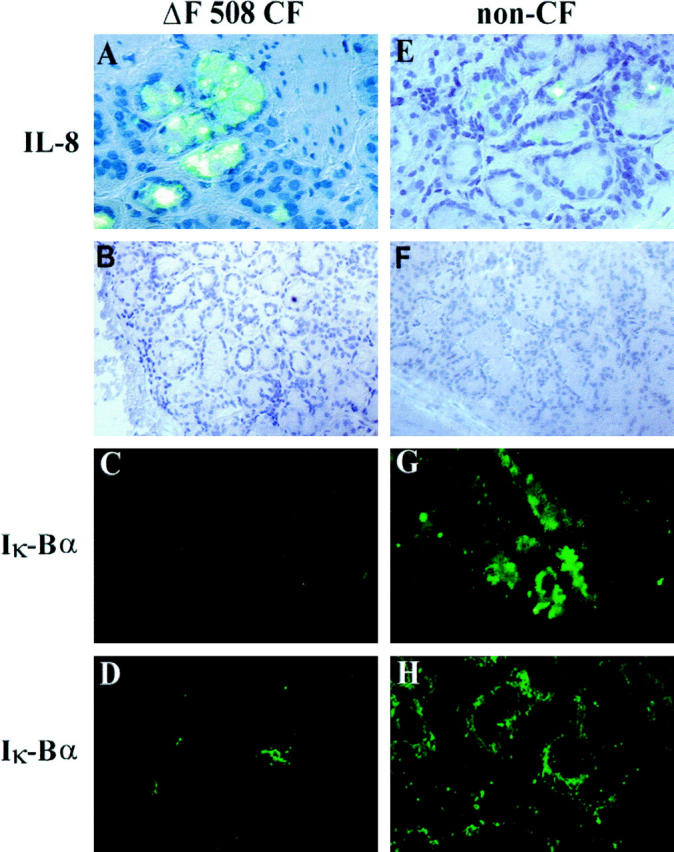
Lack of IκBα protein in in situ and in vitro bronchial gland cells from patients with CF. Analysis of serial frozen tissue sections (5 μm thick) in bronchial submucosal gland structures from ΔF508 homozygous CF patients (A-C) and non-CF (E-G) patients. Shown are Nomarski photomicrographs (B and F, magnification, ×200) and respective immunofluoresence micrographs for detection of IκBα (C and G). In CF submucosal glands exhibiting dense IL-8 staining (A, magnification, ×400), an absence of immunostaining for IκBα was seen for most CF submucosal bronchial gland cells in adjacent serial bronchial sections (C, magnification, ×200). By contrast, more of 90% of submucosal gland cells in non-CF bronchial sections showing no endogenous IL-8 immunoreactivity (E, magnification, ×400) revealed a positive and strong immunoreactivity for IκB α (G, magnification, ×200). Note that subcultures of resting CF HBG cells showed no detectable immunoreactivity for IκBα protein (D) in comparison to resting non-CF HBG cells in which a strong immunoreactivity for IκBα was detected in all cells (H, magnification, ×400). No immunostaining was detected in non-CF HBG cells when the primary antibody was replaced by nonimmune serum (data not shown).
Genistein Is a Potent Inhibitor of High Constitutive IL-8 Production by CF Bronchial Gland Cells
It has been reported that the phosphorylation and proteolysis of IκBα is a prerequisite for the process of NFκB activation. 8,9 To rule out the possibility that high constitutive IL-8 production by CF HBG cells may be secondary to the constitutive activation of NFκB factor through IκBα degradation, cultured CF HBG cells and non-CF HBG cells were incubated with increasing doses of genistein (20–100 μmol/L). Under similar control culture conditions (ie, an unstimulated resting state), the spontaneous secretion of IL-8 by CF HBG cells was 13-fold higher compared to non-CF HBG cells (Figure 2A) ▶ . Interestingly, exposure of both CF and non-CF HBG cells to genistein treatment significantly (P < 0.001) blocked the IL-8 production in a dose- and time-dependent manner. Treatment of CF HBG cells with the lowest concentration of genistein (20 μmol/L, 16 hours) resulted in a significant decrease (P < 0.001) in basal IL-8 production down to those levels of IL-8 production observed in untreated non-CF HBG cells (360 ± 40 pg/ml/10 6 cells and 242 ± 20 pg/ml/10 6 cells, respectively). In parallel, it could be demonstrated by immunofluoresence analysis (Figure 2, B and C) ▶ that a marked accumulation of IκBα protein takes place in the cytoplasm of CF HBG cells after treatment with genistein (Figure 2C) ▶ compared with that seen in untreated CF HBG cells (Figure 2B) ▶ . These results were confirmed by Western blot analysis of cytoplasmic extracts obtained from non-CF HBG cells when compared to CF HBG cells in the presence and absence of genistein. Using the lowest concentration of genistein (20 μmol/L, 16 hours), we demonstrated that cytoplasmic IκBα levels in treated CF HBG cells, evaluated by densitometric analyses, were nearly 80% of IκBα levels measured in cytoplasmic extracts from (control) non-CF HBG cells (Figure 2D ▶ , lane 3 compared with lane 1).
Figure 2.
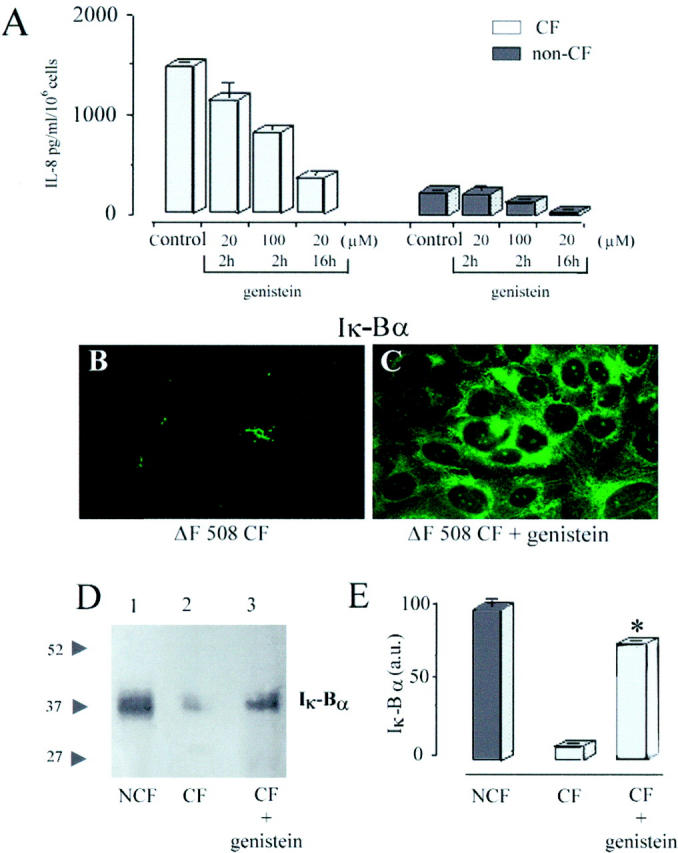
Levels of IL-8 production and expression of immunoreactive IκBα protein in cultured CF and non-CF HBG cells after their exposure to genistein. ELISA assays (A) of the spontaneous (control) production of IL-8 in supernatants after 6 hours shows that the level of IL-8 release is significantly higher (a 13-fold increase) in CF HBG cells compared with non-CF HBG cells. IL-8 release was reduced as indicated in a dose- and time-dependent manner with increasing doses of genistein. Values in ELISA assays represent means ± SD of 8 ΔF508 homozygous CF HBG and 4 non-CF HBG cell cultures, respectively, with each assayed in triplicate. In CF HBG cells (B and C), treatment of cells with genistein (20 μmol/L, 16 hours) was accompanied by the de novo presence of cytosolic IκBα, as demonstrated by immunofluoresence (C), compared to untreated CF HBG cells (B). D: Equal amounts of cytoplasmic protein from non-CF gland cells (lane 1) , CF gland cells (lane 2), and genistein-treated CF gland cells (lane 3) were analyzed for levels of IκBα by Western blotting with anti-IκBα antibodies. Relative positions of size markers are shown at the left in kilodaltons. E shows densitometric analyses of the data represented in D, combined with three similar studies, expressed in arbitrary units (a.u.). Note the presence of high endogenous IκBα protein levels in (control) non-CF gland cells compared to CF gland cells. Treatment of CF gland cells with genistein (20 μmol/L, 16 hours) resulted in a significant increase of cytosolic IκBα protein, up to 80% of IκBα protein levels when compared to (control) non-CF gland cells. *, significant difference from nontreated CF gland cells (P < 0.05).
Genistein Is a Potent Inhibitor of Constitutive NFκB Activation in CF Bronchial Gland Cells
It was, therefore, of interest to determine whether or not genistein affects constitutive and inducible NFκB activation in CF and non-CF HBG cells, respectively. EMSAs were performed on both CF and non-CF HBG cells in unstimulated conditions and after stimulation with P. aeruginosa LPS in the presence or absence of genistein. Nuclear extracts obtained from CF and non-CF HBG cells were prepared and incubated with an end 32P-labeled DNA oligonucleotide containing the recognition site for NFκB. As shown in Figure 3 ▶ , EMSA of nuclear extracts harvested from resting (unstimulated) CF HBG cells (Figure 3A ▶ , lane 3) demonstrated high constitutive amounts of activated NFκB compared with non-CF HBG cells, in which no evidence of constitutive NFκB activation was found for the same culture conditions (Figure 3B ▶ , lane 7). The specificity of NFκB DNA binding was confirmed in competition experiments. Incubation of the same nuclear extracts of resting CF HBG cells with a 100-fold excess of unlabeled (cold κB) NFκB oligonucleotide led to complete inhibition of binding activity (Figure 3A ▶ , lane 1). We determined the components of the NFκB DNA binding-protein complex by performing supershift assays with antibodies to the p50 and p65 subunits of human NFκB. As shown in Figure 3A ▶ , the addition of antibody to p65 caused a supershift (Figure 3A ▶ , lane 2). A similar result was observed with the addition of antibody to p50 (data not shown). Exposure of CF HBG cells to genistein led to a significant down-regulation of NFκB activation in cells that were identified in unstimulated conditions (Figure 3A ▶ , lane 4) or after stimulation with P. aeruginosa LPS (Figure 3A ▶ , lane 6). Although no specific NFκB binding activity was identified in unstimulated (control) non-CF HBG cells (Figure 3B ▶ , lane 7), P. aeruginosa LPS induced a strong NFκB binding activity (Figure 3B ▶ , lane 7). The pretreatment of non-CF HBG cells with genistein abolished P. aeruginosa LPS-induced NFκB activation (Figure 3B ▶ , lanes 9 and 10).
Figure 3.

Comparison of NFκB activation in CF and non-CF HBG cells. EMSAs were performed on nuclear extracts from CF HBG (A, lanes 1–6) and non-CF HBG (B, lanes 7–10) cells in the basal state and after exposure to 1 μg/ml P. aeruginosa LPS for 4 hours in the presence or absence of genistein (100 μmol/L). To demonstrate the specificity of binding of the NFκB oligonucleotide, a 100-fold molar excess of unlabeled NFκB (lane 1, cold κB) was used to compete with the labeled NFκB probe. The addition of antibody to Re1a (p65) component (lane 2, p65) caused a supershift with a reciprocal decrease in the intensity of the NFκB band. Note the presence of high endogenous NFκB activity in resting (unstimulated) CF HBG cells (lane 3) compared to resting non-CF HBG cells in which no evidence of endogenous NFκB activity was observed (lane 7). Interestingly, exposure of resting CF HBG cells to genistein treatment completely blocked constitutive NFκB activity (lane 4). The addition of P. aeruginosa LPS maintained a significantly high NFκB activity in CF HBG cells (lane 5) which could be mostly inhibited by the addition of genistein (100 μmol/L, 2 hours, lane 6). In non-CF HBG cells, the P. aeruginosa LPS-induced NFκB activity (lane 8) was completely blocked by genistein treatment (lane 9). Note that genistein alone did not induce NFκB activation in non-CF HBG cells (lane 10).
Restoration of IκBα Protein Is Associated with IL-8 Reduction in CF HBG Cells
Having demonstrated that genistein can abolish P. aeruginosa LPS-induced NFκB activation, we next analyzed the IL-8 production and the expression of cytosolic IκBα protein after P. aeruginosa LPS stimulation in the presence or absence of genistein. As shown in Figure 4A ▶ , P. aeruginosa LPS (1 μg/ml) alone induced approximately a sixfold increase of IL-8 production in CF and non-CF HBG cell types. This increase in IL-8 production induced by P. aeruginosa LPS was significantly diminished (P < 0.001) by genistein in a dose-dependent fashion in both cell types (Figure 4A) ▶ .
Figure 4.

Levels of IL-8 production and expression of immunoreactive I-kBα protein in P. aeruginosa LPS-stimulated CF and non-CF HBG cells in the presence or absence of genistein. ELISA assays (A) of the stimulated production of IL-8 in supernatants after 6 hours shows that the level of IL-8 release is sixfold higher in P. aeruginosa LPS-stimulated CF and non-CF HBG cells compared to control (unstimulated) CF and non-CF HBG cells, respectively. Genistein significantly (P < 0.001) reduces the production of IL-8 by P. aeruginosa LPS-stimulated CF and non-CF HBG cells in a dose and time-dependent manner, as indicated. Values in ELISA assays represent means ± SD of eight ΔF508 homozygous CF HBG and four non-CF HBG cell cultures, respectively, with each assayed in triplicate. Compared to resting (B1) and LPS-stimulated (B2) ΔF508 CF HBG cells and resting (B4) and LPS-stimulated (B5) non-CF HBG cells, exposure of P. aeruginosa LPS stimulated ΔF508 CF and non-CF HBG cells to genistein (100 μmol/L, 2 hours) led to the de novo presence of cytosolic IκBα protein (B3 and B6, respectively).
Because CF bronchial submucosal glands in situ and subcultures of resting CF HBG cells exhibit an absence of cytosolic IκBα protein and high constitutive IL-8 protein expression, we postulated that the treatment of CF HBG cells with genistein should increase the IκBα protein level in the cytoplasm of CF HBG cells. For this reason, the immunoreactivity of IκBα protein was monitored in unstimulated and LPS-stimulated CF HBG cells, in the presence or absence of genistein and compared with similarly treated non-CF HBG cells.
Compared with resting (Figure 4B, 1) ▶ ▶ and LPS-stimulated CF HBG cells (Figure 4B, 2) ▶ ▶ , the treatment of CF HBG cells with genistein (100 μmol/L, 2 hours) permitted the induction and maintenance of IκBα protein expression in the LPS-stimulated CF HBG cells (Figure 4B, 3) ▶ ▶ . Exposure of non-CF HBG cells to P. aeruginosa LPS alone (1 μg/ml, 4 hours) caused a marked depletion of cytoplasmic IκBα (Figure 4B ▶ , 5) compared with unstimulated non-CF HBG (Figure 4B, 4) ▶ . Interestingly, the exposure of non-CF HBG cells to P. aeruginosa LPS plus genistein (Figure 4B ▶ , 6) prevented the P. aeruginosa LPS-induced degradation of IκBα in cells. These results, which agree with our EMSA data (Figure 3A and B) ▶ , indicate that the high constitutive NFκB activation exhibited by resting CF HBG cells is prevented by genistein treatment via the induction and maintenance of cytosolic IκBα protein.
Discussion
The pathogenesis of early chronic airway inflammation in CF patients is an important topic that remains the subject of much debate. In the present study, we have demonstrated for the first time a lack of IκBα protein expression associated with constitutive NFκB activation in bronchial gland cells from patients with CF compared with non-CF disease patients. The in situ immunolabeling of native CF human bronchial sections highlights an absence of cytoplasmic IκBα protein in bronchial gland secretory cells compared with that seen in similar tissue from non-CF disease patients. Consistent with these in vivo findings, we show that no significant cytoplasmic IκBα protein is expressed in subcultures of resting CF HBG cells compared with non-CF HBG cells under identical growth conditions. This lack of cytoplasmic IκBα was associated with high levels of constitutively activated NFκB and an aberrant secretion of IL-8 by resting CF HBG cells. These findings are consistent with our previous report showing a selective in vivo and in vitro up-regulation of IL-8 expression in CF bronchial gland cells. 4
To date, constitutive nuclear NFκB-RelA activation in the absence of obvious exogenous stimuli or viral infection has been detected only in in vivo situations associated with abnormal tumor cell proliferation in Hodgkin’s disease 15 and breast cancers. 16 Although it is tempting to speculate that the lack of cytoplasmic IκBα protein in CF HBG cells could be related to a mutant CFTR-dependent mechanism in CF airway disease, the relationships between the mislocalization of CFTR protein, the lack of cytoplasmic IκBα protein, elevated levels of constitutively activated NFκB, and the up-regulated IL-8 production demonstrated in CF HBG cells remain open to interpretation.
Using different criteria, we have shown that genistein is a potent inhibitor of NFκB activation in CF bronchial gland cells. Firstly, genistein suppressed activated NFκB in resting CF gland cells. Secondly, genistein inhibited the P. aeruginosa LPS-induced activation of NFκB in CF and non-CF gland cells. Thirdly, inhibition of genistein was the result of reversing and preventing nuclear translocation of NFκB due to an increased accumulation of cytosolic IκBα. Finally, the significant accumulation of cytosolic IκBα protein was paralleled by a strong reduction in IL-8 release by genistein-treated CF gland cells.
The balance between inactive and active NFκB relies mostly on IκB factors. 17 It has recently been demonstrated 18 that in type II pulmonary epithelial cells, IκBα degradation and resynthesis is required for endogenous IL-8 transcriptional activation and termination. High nuclear Re1A levels are expected to cause a strong transcriptional up-regulation of the IκBα gene, leading to a sequestering of excess free RelA. This has been demonstrated in transgenic animals with overexpressed RelA, which is masked efficiently by up-regulated IκBα and maintained in the cytoplasm. 19 Consequently, the continuous elevated degradation of IκBα in CF bronchial gland cells should account for the constitutive RelA activity in this cell phenotype.
NFκB can be activated by a variety of signals relevant to respiratory epithelial cell physiology. Although there are different early events involved in the activation of NFκB, all of them may converge to phosphorylate IκBα, which is essential for its rapid degradation by a proteasome-dependent pathway and the subsequent translocation of NFκB into the nucleus. 8,9 To date, the effects of tyrosine kinase inhibition on the activation of NFκB and cytokine release are controversial and vary according to the cell types analyzed. In human monocytes and alveolar macrophages exposed to LPS, NFκB activation and cytokine release have been shown to be inhibited by genistein. 20,21 By contrast, it has been reported that LPS-induced NFκB translocation is not blocked by tyrosine kinase inhibitors in THP-1 cells, 22 Chinese hamster ovary (CHO) cells, and RAW 264.7 cells. 23
We propose that the novel anti-inflammatory properties of genistein shown in the present study may relate to an early common signal in the different signal transduction cascades in CF and non-CF bronchial gland cells. How does genistein work? Genistein can mediate its effects by several mechanisms, including either inhibition of IκBα phosphorylation or increased dephosphorylation of IκBα, by increased synthesis of IκBα, or by decreased degradation of IκBα. 9 Further investigations are required to determine whether or not the suppression of constitutive and inducible nuclear translocation of NFκB through the inhibition or reversal of IκBα degradation by genistein treatment of ΔF508 homozygous CF HBG cells is due to variability in the inhibition of protein tyrosine kinases or to direct protein-protein interactions with mutated CFTR protein involved in regulating the activation of NFκB and subsequent IL-8 production.
In conclusion, we have demonstrated that submucosal gland cells from CF bronchial tissues in vivo and in vitro constitutively produce high levels of IL-8 chemokine and activated NFκB through a lack of cytoplasmic IκBα protein. This may represent the first signal that initiates the early and sustained mucosal inflammation by releasing elevated levels of IL-8 chemokine in human CF airways. 4-6 Keeping in mind that genistein is a well known mutated CFTR Cl− channel stimulator in CF respiratory epithelial cells 24,25 and a potent inhibitor of constitutive and inducible NFκB activation in human ΔF508 CF bronchial gland cells as shown in the present study, the future development of genistein and/or other isoflavonoid derivatives may provide future alternatives for the treatment of chronic airway inflammation in CF patients.
Acknowledgments
We thank the team of Service du Departement de Chirurgie Cardio-Vasculaire (Pr. A. Carpentier), Hôpital Broussais, Paris, France, for their cooperation in providing lung transplant tissues. We also thank Ms. M. Adib-Conquy (Laboratoire d’Immuno-Allergie, Dr. J. M. Cavaillon, Institut Pasteur, Paris, France) for help with the EMSA technique and Mr. S. Jarry (Laboratoire de Biochimie, Pr. B. Haye, UFR Sciences, Reims, France) for help with the Western blot technique.
Footnotes
Address reprint requests to Dr. Jacky Jacquot, INSERM Unité 514, IFR 53, Maison Blanche 45, rue Cognacq-Jay, 51092 Reims Cédex, France. E-mail: jacky.jacquot@univ-reims.fr.
Supported by a grant from the Association Française de Lutte contre la Mucoviscidose (AFLM). S. Escotte is a predoctoral fellow of AFLM. The GOEMAR Laboratories (Saint Malo, France) fund O. Tabary.
O. Tabary and S. Escotte contributed equally to this work.
References
- 1.Riordan JR, Rommens JM, Kertem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL, Drumm ML, Iannuzzi MC, Collins FS, Tsui LC: Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 1989, 245:1066-1073 [DOI] [PubMed] [Google Scholar]
- 2.Stutts MJ, Canessa CM, Olsen JC, Hamrick M, Cohn JA, Rossier BC, Boucher RC: CFTR as a cAMP-dependent regulator of sodium channels. Science 1995, 269:847-850 [DOI] [PubMed] [Google Scholar]
- 3.Zahm JM, Gaillard D, Dupuit F, Hinnrasky J, Porteous D, Dorin JR, Puchelle E: Early alterations in airway mucociliary clearance and inflammation of the lamina propria in CF mice. Am J Physiol 1997, 272:C853-C859 [DOI] [PubMed] [Google Scholar]
- 4.Tabary O, Zahm JM, Hinnrasky J, Couetil JP, Cornillet P, Guenounou M, Gaillard D, Puchelle E, Jacquot J: Selective upregulation of chemokine IL-8 expression in cystic fibrosis bronchial gland cells in vivo and in vitro. Am J Pathol 1998, 153:921-930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kammouni W, Figarella C, Marchand S, Merten M: Altered cytokine production by cystic fibrosis tracheal gland serous cells. Infect Immun 1997, 65:5176-5183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DiMango E, Zar HJ, Bryan R, Prince A: Diverse Pseudomonas aeruginosa gene products stimulate respiratory epithelial cells to produce interleukin-8. J Clin Invest 1995, 96:2204-2210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blackwell TS, Christman JW: The role of nuclear factor-kB in cytokine gene regulation. Am J Respir Cell Mol Biol 1997, 17:3-9 [DOI] [PubMed] [Google Scholar]
- 8.Baeuerle PA, Baltimore D: NF-kB: ten years after. Cell 1996, 87:13-20 [DOI] [PubMed] [Google Scholar]
- 9.May MJ, Ghost S: Signal transduction through NF-kB. Immunol Today 1998, 19:80-88 [DOI] [PubMed] [Google Scholar]
- 10.Hutchins AM, Lampe JW, Martini MC, Cambell DR, Slavin JL: Vegetables, fruits, and legumes: effects on urinary isoflavonoid phytoestrogen and lignan excretion. J Am Diet Assoc 1995, 95:769-774 [DOI] [PubMed] [Google Scholar]
- 11.Akiyama T, Ishida J, Nakagawa S, Ogawara H, Watanabe S, Itoh N, Shibuya M, Fukami Y: Genistein, a specific inhibitor of tyrosine-specific protein kinase. J Biol Chem 1987, 262:5592-5595 [PubMed] [Google Scholar]
- 12.Huang J, Naasr M, Kim Y, Matthews HR: Genistein inhibits protein histidine kinase. J Biol Chem 1992, 267:15551-15515 [PubMed] [Google Scholar]
- 13.Li JD, Feng W, Gallup M, Him JH, Gum J, Kim Y, Basbaum CB: Activation of NF-κB via rc-dependent Ras-MAPKC-pp90rsk pathway is required for Pseudomonas aeruginosa-induced mucin overproduction epithelial cells. Proc Natl Acad Sci USA 1998, 95:5718-5723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Israel N, Gougerot-Pocidalo A, Virelizier JL: Redox status of cells influences constitutive and induced NF-kappa B translocation and HIV long terminal repeat activity in human T and monocytes cell lines. J Immunol 1992, 149:3386-3393 [PubMed] [Google Scholar]
- 15.Bargou RC, Emmerich F, Krappmann D, Bommert K, Mapara MY, Arnold W, Royer HD, Grinstein E, Greiner A, Scheidereit C, Dörken B: Constitutive nuclear factor-kappaB-RelA activation is required for proliferation and survival of Hodgkin’s disease tumor cells. J Clin Invest 1997, 100:2961-2969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sovak MA, Bellas RE, Kim DW, Zanieski GJ, Rogers AE, Triash AM, Sonensheim GE: Aberrant nuclear factor-kappaB/Rel expression and the pathogenesis of breast cancer. J Clin Invest 1997, 100:2952-2960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Henkel TT, Machleidt I, Kroncke Y, Ben-Neriah Y, Bauerle P: Rapid proteolysis of I kappaB-α is necessary for activation of transcription factor NF-kappa B. Nature 1993, 365:182-185 [DOI] [PubMed] [Google Scholar]
- 18.Brasier AR, Jamaluddin M, Casola A, Duan W, Shen Q, Garofalo RP: A promoter recruitment mechanism for tumor necrosis factor-a-induced interleukin-8 transcription in type II pulmonary epithelial cells. J Biol Chem 1998, 273:3551-3561 [DOI] [PubMed] [Google Scholar]
- 19.Perez P, Lira SA, Bravo R: Overexpression of Rela in transgenic mouse thymocytes: specific increase in levels of the inhibitor protein I-kB α. Mol Cell Biol 1995, 15:3523-3530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gentz Y, Zhang B, Lotz M: Protein tyrosine kinase activation is required for lipopolysaccharide induction of cytokines in human blood monocytes. J Immunol 1993, 151:6692-6700 [PubMed] [Google Scholar]
- 21.Carter AB, Monick MM, Hunninghake GW: Lipopolysaccharide-induced NF-kB activation, and cytokine release in human alveolar macrophages is PKC-independent, and TK-, and PC-PLC dependent. Am J Respir Cell Mol Biol 1998, 18:384-391 [DOI] [PubMed] [Google Scholar]
- 22.Yoza BK, Hu JYQ, McCall CE: Protein-tyrosine kinase activation is required for lipoplolysaccharide induction of interleukin 1 and NF-kB activation, but not NF-kB nuclear translocation. J Biol Chem 1996, 271:18306-18309 [DOI] [PubMed] [Google Scholar]
- 23.Delage RL, Fenton MJ, Savedra R, Perera P, Vogel SN, Thieringer P, Golenbock DT: CD14-mediated translocation of nuclear factor-kB induced by lipopolysaccharide does not require tyrosine kinase activity. J Biol Chem 1994, 269:22253-22260 [PubMed] [Google Scholar]
- 24.Illek B, Fischer H, Santos G, Widdicombe JH, Machen TE, Reenstra WW: Cyclic AMP-independent activation of CFTR Cl channels by the tyrosine kinase inhibitor genistein. Am J Physiol 1995, 268:C886-C893 [DOI] [PubMed] [Google Scholar]
- 25.Wang F, Zeltwanger S, Yang ICH, Nairn AC, Hwang TC: Actions of genistein on cystic fibrosis transmembrane conductance regulator channel gating. J Gen Physiol 1998, 111:477-490 [DOI] [PMC free article] [PubMed] [Google Scholar]