

Abstract
Apoptosis was induced in human monocytic THP.1 cells by the use of chemicals with disparate mechanisms of action. Apoptotic cells were characterized by a reduced inner mitochondrial membrane potential, increased cytosolic cytochrome c, ultracondensed mitochondria, condensed chromatin, cytoplasmic inclusions of β-actin, and fragmentation of the Golgi apparatus. All of these changes, except the release of cytochrome c, were prevented by caspase inhibition. Cells were separated into two populations, with either normal or low inner mitochondrial membrane potential, using fluorescence-activated cell sorting. Ultracondensed mitochondria were observed only in the cells with low inner mitochondrial membrane potential, whereas noncondensed mitochondria were found in the cells with a normal inner mitochondrial membrane potential. We have demonstrated a sequence of related biochemical and ultrastructural changes, commencing with the release of mitochondrial cytochrome c, followed by activation of caspases and a reduction of inner mitochondrial membrane potential. These changes involved the formation of ultracondensed but not swollen mitochondria. Thus the release of mitochondrial cytochrome c was not the result of the mitochondrial permeability transition, reduction of inner mitochondrial membrane potential, or rupture of the outer mitochondrial membrane. Discontinuities in the outer membrane of ultracondensed mitochondria may, however, facilitate the further release of caspase-activating proteins, thereby amplifying the apoptotic process.
The process of apoptosis, which was originally recognized and described according to morphological criteria, 1 is now the subject of intense biochemical investigation. 2,3 Apoptotic cells are characterized by the presence of a condensed nucleus and a shrunken cytoplasm, closely packed with organelles that may themselves be condensed. 1 This cytoplasmic condensation has been used as a key criterion for distinguishing apoptotic cells from those undergoing oncosis, the contrasting form of cell death, and from those undergoing the secondary changes associated with necrosis. 4 Nevertheless, the general absence of swollen or disrupted mitochondria in apoptotic cells resulted in this organelle’s role in apoptosis being largely ignored. The striking nuclear changes have thus tended to dominate most morphological studies, particularly since their correlation with internucleosomal fragmentation. The formation of the resulting DNA “ladders” provided the first biochemical correlate for the morphological changes associated with apoptosis 5 and proved to be a useful marker, although various examples of apoptosis that do not involve this internucleosomal cleavage have subsequently been described. 6 Recent biochemical studies have now focused attention on the mitochondria during the initiation of apoptosis. One popular hypothesis for this initiation involves the possible release from these organelles of cytochrome c, which may combine with ATP/dATP and Apaf-1 to activate a cascade of caspase activity. 7 Several mitochondrial changes have been correlated with the development of apoptosis, and two of them, the reduction in inner mitochondrial membrane potential (ΔΨm) and the closely related mitochondrial permeability transition 8,9 have recently been implicated in the redistribution of cytochrome c. The mitochondrial permeability transition, which is usually associated with a loss of ΔΨm, involves the opening of a channel in the inner mitochondrial membrane. This channel allows the ingress of water into the mitochondrial matrix, resulting in osmotic swelling and the subsequent rupture of the outer membrane. 10 Ultrastructural evidence for mitochondrial swelling and discontinuities in the outer membrane have been reported during the redistribution of cytochrome c, but, surprisingly, these changes occurred in the absence of any loss of ΔΨm. 11 The presence of a specific channel in the outer mitochondrial membrane has also been suggested as a route for the egress of cytochrome c10, and such a mechanism may not necessarily involve mitochondrial swelling.
The involvement of the cytoskeleton in the development of apoptosis has also been reported by numerous groups, 12-14 and particular emphasis has been placed on changes in actin cleavage/distribution. 15,16 Caspase-dependent cleavage of the actin-regulatory protein gelsolin has even been implicated in the release of cytochrome c from mitochondria. 17
We have recently reported the presence of discontinuities in the outer membrane of ultracondensed mitochondria in apoptotic THP.1 cells. 18 In the present study we clearly demonstrate that these ultracondensed mitochondria occurred only in cells exhibiting a reduced ΔΨm. Furthermore, we show that both of these changes, together with all other morphological indicators of apoptosis, were prevented by the inhibition of caspase activity. The redistribution of mitochondrial cytochrome c was unaffected by this inhibition and thus preceded all of the other changes.
Materials and Methods
Cell Culture and Treatment
Media and serum were purchased from Gibco (Paisley, UK). The broad-spectrum caspase inhibitor benzyloxycarbonyl-Val-Ala-Asp (OMe) fluoromethyl ketone (Z-VAD.fmk) was purchased from Enzyme Systems (Dublin, CA), and the protease inhibitors N-tosyl-l-phenylalanyl chloromethyl ketone (TPCK) and N-tosyl-l-lysine chloromethyl ketone (TLCK) were from Boehringer-Mannheim (Lewes, UK). The fluorescent probe 3,3′-dihexyloxacarbocyanine iodide (DiOC6(3)) and the mouse monoclonal antibody recognizing mitochondrial membrane-bound cytochrome c oxidase (subunit II) were purchased from Molecular Probes (Eugene, OR). The mouse monoclonal antibody recognizing human cytochrome c was from PharMingen (San Diego, CA). All other chemicals and primary antibodies were obtained from Sigma Chemical Company (Poole, UK).
THP.1 cells were maintained in RPMI 1640 supplemented with 10% heat-inactivated fetal bovine serum and 2 mmol/L glutamine in an atmosphere of 5% CO2 in air at 37°C. 19 Logarithmically growing cells were used for all experiments. To induce apoptosis, 0.5 × 10 6 cells/ml were incubated in the presence of cycloheximide (25 μmol/L), etoposide (25 μmol/L), or TPCK (75 μmol/L) as previously described. 19,20 The proportion of cells undergoing apoptosis was determined by flow cytometry after staining with Hoechst 33342/propidium iodide 20 or labeling with Annexin V, as previously described. 18 To assess the effects of caspase inhibition on apoptosis, THP.1 cells were treated with Z-VAD.fmk (50 μmol/L) 5 minutes before exposure to the apoptotic stimulus.
Flow Cytometric Analysis of ΔΨm
Suspensions of 0.5 × 10 6 cells were incubated for 20 minutes at 37°C with DiOC6,(3) (50 nmol/L). Control experiments were performed by incubating cells for a further 10 minutes at 37°C with m-carbonyl cyanide m-chlorophenyl hydrazone (50 μmol/L), an uncoupling agent that abolishes the ΔΨm. Experimental samples were incubated, for 4 hours, with etoposide or TPCK, in the presence or absence of Z-VAD.fmk, before being analyzed and sorted, into subpopulations of cells with normal and decreased ΔΨm, in a Becton-Dickinson “Vantage” flow cytometer. To differentiate between these subpopulations each experiment was calibrated, using cells treated with the uncoupling agent, to select the subpopulation of cells (about 80%) with a decreased ΔΨm.
Electron Microscopy and Immunocytochemistry
Sorted cells from the flow cytometer were collected in 4% glutaraldehyde in 0.1 mol/L sodium cacodylate buffer (pH 7.4), until the volume of the fixative had been doubled by the carrier solution. Samples were collected for 2 hours, combined, and spun down in a swing-out rotor at 3000 × g. Cells in culture (3 × 106) were spun down before the supernatant was gently replaced with 2% glutaraldehyde in 0.1 mol/L sodium cacodylate buffer (pH 7.4). All samples were fixed overnight, at 4°C, and postfixed with 1% osmium tetroxide/1% potassium ferrocyanide overnight at 4°C. Fixed pellets were stained en bloc with 5% aqueous uranyl acetate overnight at room temperature, dehydrated, and embedded in Agar 100 epoxy resin. Sections up to 1 μm were examined unstained by electron spectroscopic imaging with a Zeiss 902A electron microscope. Ultrathin sections were stained with lead citrate and examined in a Jeol 100-CXII electron microscope equipped with a rotating stage/eucentric goniometer. All quantitative assessments were based on counts of at least 500 cells at each treatment/time point.
Duplicate pellets were fixed with 4% formaldehyde (pH 7.4), freshly made up from paraformaldehyde in Dulbecco’s phosphate-buffered saline (PBS), for 1 hour at room temperature. They were rinsed in PBS, dehydrated in ethanol, and infiltrated with Unicryl resin from British Biocell International (Cardiff, Wales). The resin was polymerized with UV radiation (λ360 nm) at 4°C, according to the manufacturer’s instructions.
Ultrathin sections were blocked with normal goat serum and diluted 1:50 in PBS containing 1% bovine serum albumin and 1% Tween 20, for 4 hours at room temperature. They were incubated in primary antibody, a mouse IgG1 monoclonal antibody (clone AC-15) raised against a slightly modified β-cytoplasmic actin N-terminal peptide from Sigma Chemical Company, diluted 1:10 in PBS containing 1% normal goat serum, 1% bovine serum albumin, and 1% Tween 20 (PBSGAT), for 18 hours at 4°C. In control incubations the primary antibody was replaced with mouse IgG1 from Dako (Ely, UK). Thorough washing in PBSGAT was followed by incubation in a goat-derived anti-mouse IgG from British Biocell International (Cardiff, Wales), which had been absorbed against human serum proteins and conjugated with 20 nm colloidal gold, diluted 1:50 in PBSGAT, for 18 hours at 4°C. Serial sections were incubated with a range of dilutions of antibodies to α-actin, tubulin, and vimentin before treatment with the appropriate colloidal gold-conjugated IgG. Ultrathin sections were examined unstained or after staining with uranyl acetate and lead citrate.
Preparation of Cytosolic Extracts
Cytosolic extracts were isolated as described previously. 18,21 Briefly, 50 × 10 6 THP.1 cells were washed twice in ice-cold PBS and resuspended in 200 μl of extraction buffer (220 mmol/L mannitol, 68 mmol/L sucrose, 50 mmol/L piperazine-N,N′-bis(2-ethanesulfonic acid/KOH, pH 7.5, 50 mmol/L KCl, 5 mmol/L EGTA, 2 mmol/L MgCl2, 1 mmol/L dithiothreitol, 1 mmol/L phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 10 μg/ml pepstatin A, and 10 μg/ml aprotinin). They were incubated, on ice, for 30 minutes and then homogenized, using a glass Dounce homogenizer, with 40 strokes of the “B” pestle. Homogenates were centrifuged at 14,000 × g, for 15 minutes at 4°C, and the resulting supernatants were used as the cytosolic extracts.
Western Blot Analysis
Cellular proteins were resolved on 10% sodium dodecyl sulfate polyacrylamide gels and blotted onto “Hybond-C” nitrocellulose membranes from Amersham International (Little Chalfont, UK). Cytochrome c was detected using a mouse monoclonal antibody (7H8.2C12) to human cytochrome c, and parallel samples were checked for mitochondrial contamination, using the antibody (12C4-F12) to cytochrome c oxidase. 18 Actin was detected in samples of 0.2 × 10 6 cells, prepared as previously described, 21 with both the monoclonal (AC-15) antibody that was used for immunocytochemistry and with a rabbit-derived polyclonal antibody to the C-terminal actin fragment, attached to Multiple Antigen Peptide. The bound primary antibodies were visualized with an enhanced chemiluminescence kit from Amersham International, using an appropriate secondary antibody conjugated with horseradish peroxidase from Sigma Chemical Company.
Results
Diverse Apoptotic Stimuli Induce Similar Morphological Changes
Untreated THP.1 cells were large (12–16 μm in diameter) and irregular in outline, with many short, irregular microvilli and a multilobed nucleus (Figure 1a) ▶ . The diffuse perinuclear and perinucleolar heterochromatin was weakly differentiated from the euchromatin, but the nucleolus was clearly visible in most sections. Several mitochondria, with irregular transverse or obliquely arranged cristae, were evident in cell profiles. Treatment of cells for 4 hours with cycloheximide (25 μmol/L), an inhibitor of protein synthesis, resulted in several of the cells showing the characteristic features of apoptosis, including shrinkage, loss of microvilli, increased cytosolic density, vacuolation, and perinuclear clumping of condensed chromatin (Figure 1b) ▶ . Small ultracondensed mitochondria were present within most of these cells, but this change was also observed in a few cells that showed no other morphological signs of apoptosis (Figure 1b) ▶ . The cytoplasm of apoptotic cells also included spheroidal inclusions (1.0–2.5 μm in diameter) of a fine amorphous material (Figure 1b) ▶ and clusters of small vesicles (Figure 1c) ▶ . These clusters were found in 5–10% of treated cells; usually only one cluster was visible in a cell profile, but six were found in one cell. A few of these clusters were adjacent to the cell membrane (Figure 1c) ▶ , but most were restricted to the perinuclear region. The majority of clusters consisted of a large proportion (>90%) of remarkably uniform vesicles 35–45 nm in diameter; the remaining vesicles were larger and oblate. A few clusters were closely associated with distorted membranous cisternae, some of which resembled the trans elements of the Golgi apparatus. Combinations of two or more of these morphological changes were usually evident, even in the absence of an overall increase in cytoplasmic staining density. Cytoplasmic and nuclear changes similar to those observed after cycloheximide were also detected after the treatment of cells, for 4 hours, with etoposide, a DNA topoisomerase II inhibitor (Figure 1d) ▶ . The spheroidal cytoplasmic inclusions of fine amorphous material were often closely associated with the cell membrane, and some were observed within cellular protrusions or apoptotic buds. A few of these membrane-limited structures, containing spheroidal inclusions, were found free in the extracellular medium (Figure 2a) ▶ . In agreement with previous results, 19 the morphological changes (Figure 2b) ▶ and the incidence of apoptosis (Figure 3) ▶ were particularly pronounced in cells after treatment, for 4 hours, with a combination of the trypsin-like protease inhibitor TLCK (100 μmol/L) with cycloheximide (25 μmol/L). The chymotrypsin-like protease inhibitor TPCK (75 μmol/L) was also a potent inducer of apoptosis (Figure 3) ▶ and resulted in cytoplasmic changes indistinguishable from those observed after etoposide, cycloheximide, or cycloheximide and TLCK (Figure 2c) ▶ . Despite these similarities, however, treatment with TPCK for 4 hours resulted in different nuclear changes, with dispersal of the nucleolar dense fibrillar component and small clumps of partially condensed chromatin throughout the nucleoplasm. This morphology has been correlated with the formation of large kilobase pair fragments of DNA, in the absence of internucleosomal cleavage. 20
Figure 1.
Ultrastructural changes in apoptotic THP.1 cells. a: Untreated cell with numerous mitochondria and a well-developed Golgi apparatus (curved black arrow ). b: The cytoplasm of cells treated with cycloheximide (25 μmol/L), for 4 hours, contains many small ultracondensed mitochondria (black arrowheads). These are evident not only in cells showing perinuclear clumping of condensed chromatin (*) and intracytoplasmic spheroidal inclusions (black arrows), but also in a cell devoid of other morphological signs of apoptosis. c: A cycloheximide-treated cell contains spheroidal inclusions (black arrows) and a cluster of small vesicles (curved black arrow). d: A cell treated with etoposide (25 μmol/L), for 4 hours, shows chromatin condensation, partial nucleolar disintegration, ultracondensed mitochondria (black arrowheads), and spheroidal inclusions (black arrow). All bars = 1 μm.
Figure 2.

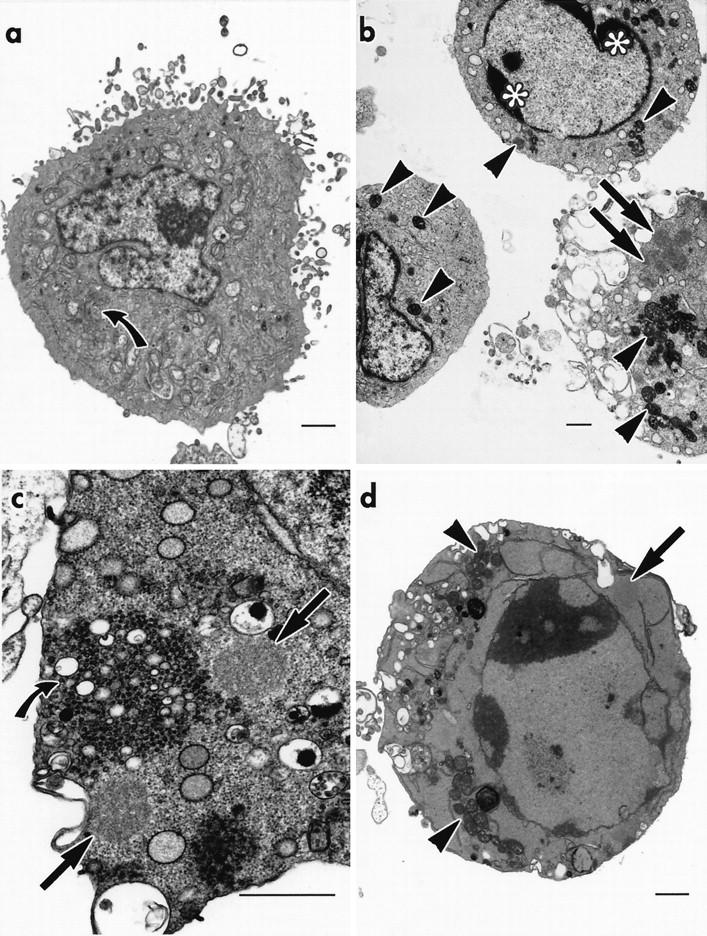
Cytoskeletal changes in apoptotic THP.1 cells. a: A cell treated with etoposide (25 μmol/L), for 4 hours, shows a spheroidal inclusion (black arrow) within an apoptotic bud that is probably free in the extracellular medium. b: A cell treated with cycloheximide (25 μmol/L), for 4 hours, in the presence of TLCK (100 μmol/L) shows chromatin condensation and a spheroidal inclusion (black arrow). Mitochondria exhibiting various degrees of condensation display bizarre deformities and cytoplasmic invaginations (white arrowheads); one ultracondensed mitochondrion shows a discontinuity in its outer membrane (black arrowhead ). c: Cells treated with TPCK (75 μmol/L) show dispersal of the dense fibrillar component of the nucleolus and indistinct clumps of partially condensed chromatin throughout the nucleoplasm. Cytoplasmic changes include ultracondensed mitochondria (black arrowheads) and spheroidal inclusions (black arrows ). d: A spheroidal inclusion (black arrow) and numerous deformed, ultracondensed mitochondria (black arrowheads), many with an incomplete outer membrane, are present in the partially condensed cytoplasm of a cell treated with TPCK (75 μmol/L) for 4 h. All bars = 1 μm.
Figure 3.

Incidence (% ± SEM) of apoptotic THP.1 cells, after incubation with a range of chemical stimuli, for 4 hours, in either the presence (□) or the absence (▪) of the caspase inhibitor Z-VAD.fmk (50 μmol/L). Apoptotic cells were stained with Hoechst 33342 and detected by flow cytometry.
Z-VAD.fmk Inhibits the Morphological Changes of Apoptosis but Not the Release of Mitochondrial Cytochrome c
The development of all of the detectable morphological changes induced by the three agents (Figures 1 and 2) ▶ ▶ was prevented by Z-VAD.fmk (50 μmol/L). Cells treated with this broad-spectrum caspase inhibitor before incubation with etoposide (Figure 4a) ▶ or TPCK (Figure 4b) ▶ were morphologically indistinguishable from controls, thus demonstrating the key role of caspase activation in these structural changes. Both etoposide and TPCK induced a time-dependent increase in the cytosolic level of cytochrome c, which was first clearly detected after 2 hours of treatment (Figure 5 ▶ , lane 4). Cytochrome oxidase subunit II, a marker for mitochondrial contamination, was not detected in any of these samples (data not shown). Treatment with Z-VAD.fmk did not block the increase in cytosolic cytochrome c induced by either stimulus (Figure 5 ▶ , lane 7). Thus, as Z-VAD.fmk blocked the morphological changes without inhibiting this increase, the release of mitochondrial cytochrome c is upstream, or independent of caspase activation.
Figure 4.

THP.1 cells incubated for 4 hours with either (a) etoposide (25 μmol/L) or (b) TPCK (75 μmol/L) in the presence of Z-VAD.fmk (50 μmol/L) show none of the morphological features associated with apoptosis (bars = 1 μm). Cells incubated for 3 hours with cycloheximide (25 μmol/L) in the presence of TLCK (100 μmol/L) (c and d) show rare examples of the coexistence of ultracondensed (black arrowheads) and noncondensed mitochondria. The cristae are evident in some of these ultracondensed mitochondria, as electron-lucent spaces between regions of the dense inner compartment. They are, however, indistinct in the small but noncondensed mitochondrial profile (curved white arrow), which may be undergoing condensation (bars = 500 nm).
Figure 5.

Z-VAD.fmk does not inhibit the etoposide- or TPCK-induced release of mitochondrial cytochrome c. Western blots of 30-μg samples of cytosolic proteins from THP.1 cells incubated for up to 4 hours with either etoposide (25 μmol/L) or TPCK (75 μmol/L), alone or in the presence of Z-VAD.fmk (50 μmol/L). Immunolabeling for cytochrome c shows low levels of this protein in control samples (lane 1) and in cells incubated for up to 1 hour (lanes 2 and 3), but much higher levels are evident after incubations of 2, 3, and 4 hours (lanes 4–7). Coincubation with Z-VAD.fmk had no detectable effect on this increase (lane 7).
The Formation of Ultracondensed Mitochondria
Ultracondensed mitochondria were the most consistent feature of cells treated, for 4 hours, with cycloheximide, etoposide, or TPCK. Most of these ultracondensed mitochondria appeared in sections as uniform short rods or circles, 200–600 nm in diameter (Figures 1b, 1d, 2a, and 4c) ▶ ▶ ▶ , with an increased electron density that was entirely restricted to the matrix of the inner compartment (Figure 4d) ▶ . The organelles were rarely more than 1.5 μm long, but in a few instances bizarre elongated forms, over 4 μm long, were observed (Figure 2b) ▶ . In a few cells, reorganization of the inner membrane resulted in the formation of discrete globular profiles within the poorly defined outline of the outer membrane (Figures 2d and 4d) ▶ ▶ . This membrane often disappeared into obliquity, but discontinuities were identified where it could not be traced, even by tilting the section or by electron spectroscopic imaging of thick (1-μm) sections. Time-course studies showed that less than 5% of cells fixed after 1 hour of treatment with cycloheximide, etoposide, or TPCK contained ultracondensed mitochondria. No significant increase in the incidence of ultracondensed mitochondria was detected after 2 hours of treatment, but most (55–60%) cells examined after 3 hours contained these organelles. Only at this time did a few cells contain both noncondensed and ultracondensed mitochondria (Figure 4c) ▶ together with some rare, possibly transitional forms, in which a slight increase in density was evident despite the irregular disposition of many of the cristae (Figure 4d) ▶ . Thus most cells were devoid of any recognizable transitional form and contained homogeneous populations of either noncondensed or ultracondensed mitochondria. The presence of only small numbers of untreated THP.1 cells with ultracondensed mitochondria is consistent with the low background incidence of apoptosis in these cells. The virtual absence of mixed populations of noncondensed and ultracondensed mitochondria, or of intermediate forms, indicates that a rapid transition probably occurs throughout all of the mitochondria within each affected cell. To establish the significance of these ultrastructural changes, cells were examined by flow cytometry, so that the incidence of apoptosis, as indicated by staining with Hoechst 33342/propidium iodide (Figure 3) ▶ , Annexin V, and DiOC6(3), could be quantified. Treatment with either etoposide or TPCK resulted in marked increases in the percentage of apoptotic cells, as assessed by these three independent methods. This etoposide- and TPCK-induced apoptosis was abolished by Z-VAD.fmk (Table 1) ▶ . Staining with DiOC6(3) was also used to monitor changes in ΔΨm. Many of the cells with low ΔΨm were smaller than the control cells, as indicated by a decrease in forward light scatter (Figure 6) ▶ , another characteristic of apoptotic cells. As these cells constituted only a small proportion of the total cell population, any structural changes could not be correlated directly with these particular cells. The cells were therefore sorted and then examined by electron microscopy to establish whether ultracondensed mitochondria were present in either or both of the subpopulations.
Table 1.
Comparative Rates of Apoptosis
| Treatment | % Apoptosis | ||
|---|---|---|---|
| Low ΔΨm | High Hoechst | High PS | |
| Control | 5.4 ± 0.9 | 2.4 ± 0.9 | 4.3 ± 0.2 |
| Etoposide | 42.0 ± 2.2 | 29.7 ± 5.7 | 39.8 ± 0.8 |
| Etoposide+ Z-VAD.fmk | 3.7 ± 1.1 | 3.8 ± 1.2 | 1.9 ± 0.3 |
| TPCK | 40.8 ± 4.4 | 55.2 ± 3.2 | 56.2 ± 5.2 |
| TPCK+ Z-VAD.fmk | 4.2 ± 1.5 | 4.1 ± 1.1 | 2.3 ± 0.3 |
THP.1 cells were incubated for 4 h alone (Control), with etoposide (25 μmol/L) or with TPCK (75 μmol/L), in the presence or absence of Z-VAD.fmk (50 μmol/L). The percentage of apoptotic cells was determined by a decreased mitochondrial membrane potential (ΔΨm), an increased uptake of Hoechst 33342 (Hoechst), or an increased externalization of phosphatidylserine (PS). Results are expressed as the mean (± SEM) of at least three separate experiments.
Figure 6.

Etoposide induces an increase in the proportion of cells with reduced ΔΨm. THP.1 cells were incubated for 4 hours either alone (control) or with etoposide (25 μmol/L) and then incubated with DiOC6(3) to measure ΔΨm. The intensity of DiOC6(3) fluorescence is plotted, on a logarithmic scale, against forward light scatter (cell size). A subpopulation of cells exhibiting a normal (a) and reduced (b) ΔΨm were separated using a fluorescence-activated cell sorter. The subpopulation with a reduced ΔΨm is increased by etoposide treatment and contains many small cells
Cells Sorted According to ΔΨm
Almost all of the cells (>95%) isolated because of their reduced ΔΨm were apoptotic and contained ultracondensed mitochondria. The incidence of other signs of apoptosis was also greatly enhanced in these cells, particularly the cytoplasmic inclusions of amorphous material. Many of the sorted cells had chromatin condensed against the inner membrane of the nuclear envelope and only a few had normal, intact Golgi stacks. Although sorted for their low ΔΨm, they exhibited a wide range of morphologies (Figure 7a) ▶ , ranging from cells with a dense, shrunken cytoplasm to others showing signs of lysis. In contrast, the cells sorted for normal ΔΨm did not exhibit any morphological signs of apoptosis. The mitochondria of most of these cells (>80%) had a normal appearance, and those present in the remaining cells were swollen rather than ultracondensed (Figure 7b) ▶ . Thus these results demonstrate that a reduction in ΔΨm is definitely not dependent on the development of swollen mitochondria and, in this system, involves their ultracondensation.
Figure 7.

Cells treated with etoposide and sorted for reduced DiOC6(3) fluorescence yield two different subpopulations of cells. Cells exhibiting reduced fluorescence (a) all have ultracondensed mitochondria (black arrowheads), despite differences in cytoplasmic density. Chromatin condensation, cytoplasmic vacuoles (curved white arrow), vesicle clusters (curved black arrow), and spheroidal inclusions (black arrow) were all present in these cells. A cell sorted for normal DiOC6 fluorescence (b) has a well-developed Golgi apparatus (curved black arrow). The mitochondrial swelling (white arrowheads), observed in a subset of this population, is also evident. Cells treated with TPCK (75 μmol/L), for 4 hours (c and d), show immunogold labeling for β-actin over several spheroidal inclusions (black arrows). All bars = 1 μm.
Immunolocalization of Actin
The cytoplasmic inclusions of amorphous material were readily apparent in nonosmicated samples that had been embedded in acrylic resin (Figure 7c) ▶ . These inclusions were not labeled by antibodies to α-actin, tubulin, or vimentin (data not shown), but they were heavily labeled by the antibody to the β-cytoplasmic actin N-terminal peptide (Figure 7, c and d) ▶ . In untreated THP.1 cells, diffuse labeling with this antibody was observed throughout the cytoplasm, particularly against the inner leaflet of the cell membrane (data not shown).
Actin Cleavage
To determine whether the ultrastructural changes were the result of actin cleavage, cellular proteins from both controls and treated cells were examined by Western blotting. The presence of intact actin was indicated by the presence of a ∼42-kd band that reacted with both the monoclonal antibody to the β-cytoplasmic actin N-terminal peptide and confirmed with the polyclonal antibody to the C-terminal residues of actin. This polyclonal antibody also detected several subsidiary bands, but no differences could be detected between samples from control and treated cells (Figure 8) ▶ and thus no sign of actin cleavage was detected.
Figure 8.

Western blots of cellular proteins from THP.1 cells. Control samples (A and D) and samples from cells treated, for 4 hours, with etoposide (B and E) or TPCK (C and F) show the presence of a ∼42-kd band. Lanes A, B, and C were incubated with the monoclonal antibody to the β-cytoplasmic actin N-terminal peptide, whereas lanes D, E, and F were incubated with a polyclonal antibody to the C-terminal residues of actin. This antibody also resulted in weak labeling of various other bands, but no differences could be detected between any samples from control and treated cells.
Discussion
Ultracondensed Mitochondria and the Reduction in ΔΨm
Ultracondensed mitochondria, the most striking ultrastructural feature of the apoptotic THP.1 cells studied in our experiments, have been reported previously, but their significance was not fully established. 18 Fluorescence-activated cell sorting has now enabled us to demonstrate, unequivocally, that these organelles are only present in cells exhibiting a low ΔΨm. The uniformity of the cytoplasmic response to the three disparate agents used in our study is consistent with these changes being part of a common apoptotic pathway. 2
The ultracondensed mitochondria described in the present study are distinct from those of the condensed conformation. 22 Highly condensed mitochondria have been reported as a short-term response to cell injury. 23,24 In these early studies, however, condensation was followed by a rapid return to the orthodox configuration, followed by the development of the high-amplitude swelling and flocculent densities that are normally associated with oncosis. Unlike this transient response to injury, the changes observed in the present study resemble the “pleomorphic micromitochondriosis” described in apoptotic nodal myocytes 25 and the “mitochondrial pyknosis” or “hypercondensation” observed during apoptosis in a colon carcinoma cell line. 26 Interestingly, both of these studies also reported mitochondrial proliferation, and the latter detected a reduction in ΔΨm. Other studies have described “ultracondensed” mitochondria in apoptotic lymphoblastic leukemic cells and implicated them in the signaling of the apoptotic process. 27,28 The ultracondensed mitochondria observed in THP.1 cells were not transient, as they were present at all stages of apoptosis and in cells undergoing secondary necrosis. They were not an artifact of the sorting procedure, as they were absent from cells sorted for normal ΔΨm (Figure 7b) ▶ . Strikingly, all of the mitochondria in cells sorted for low ΔΨm were ultracondensed (Figure 7a) ▶ , whereas those in cells with normal ΔΨm were not condensed. Thus, in THP.1 cells, a reduction in ΔΨm accompanied the development of ultracondensed mitochondria. Reduction in ΔΨm has been proposed as a critical early step in the development of lymphocyte apoptosis, 8 but in the present study, as in most models of apoptosis, this change is preceded by the redistribution of cytochrome c. 10
Redistribution of Mitochondrial Cytochrome c
A failure of electrical and osmotic homeostasis, resulting in swelling and subsequently in rupture of the outer mitochondrial membrane, has been proposed as a mechanism for the release of cytochrome c into the cytosol. 11 The mitochondrial permeability transition is usually associated with swollen mitochondria 9,10 and with loss of ΔΨm. 10 The predominance of ultracondensed mitochondria, in apoptotic THP.1 cells, indicated that overdistension of mitochondria may be discounted as the mechanism for cytochrome c release in the present study.
The presence of Bcl-xL, an anti-apoptotic pore-forming member of the Bcl-2 family, may prevent osmotic disruption of the outer mitochondrial membrane, 11 but other studies have suggested that a pore formed by Bax, a pro-apoptotic member of this family, may make possible the release of cytochrome c. 10 The presence of such a pore may account for the absence of any detectable morphological changes in the mitochondria of apoptotic THP.1 cells, before the redistribution of cytochrome c.
The prevention of all of the ultrastructural changes by Z-VAD.fmk (Figure 4, a and b) ▶ , without blocking the release of mitochondrial cytochrome c (Figure 5) ▶ , clearly shows that this release precedes ultracondensation. Although Z-VAD.fmk does not inhibit the release of mitochondrial cytochrome c during chemical-induced apoptosis, it does prevent this redistribution during receptor-mediated apoptosis. 29 Thus activation of caspases, which is critical for the initiation of receptor-mediated apoptosis, may occur after commitment to cell death during chemical-induced apoptosis. In chemical-induced apoptosis, the increase in cytosolic cytochrome c (Figure 5) ▶ , in the presence of Apaf-1, activates caspase-9, which, in turn, activates the “effector” caspases-3 and -7. These enzymes are responsible for most of the biochemical and morphological changes associated with the apoptotic phenotype. 30,31
The caspase-dependent ultracondensation of mitochondria and, particularly, the attendant discontinuities in the outer mitochondrial membrane may result in the liberation of more cytochrome c and other caspase activators, including apoptosis-inducing factor, thus amplifying the initial apoptotic stimulus.
This interpretation is consistent with the divergent morphologies that occurred in cells sorted for reduced ΔΨm. Despite the consistent presence of ultracondensed mitochondria, these cells represented many stages of the apoptotic process, thus reflecting the delay between the activation of the caspase cascade and the instant of fixation for each cell.
Golgi Fragmentation in Apoptotic Cells
The appearance of the clusters of cytoplasmic vesicles in apoptotic THP.1 cells is very similar to that reported in mitotic cells and confirmed as the transitional form of the Golgi apparatus. 32,33 The suggested origin of the clusters is supported by their absence from THP.1 cells with an intact Golgi apparatus. The presence of intact Golgi cisternae in some cells with ultracondensed mitochondria indicates that the clusters probably develop after the mitochondrial changes. Despite the retention of the nuclear membrane, there is a profound reorganization of the cell architecture during apoptosis, and so modification of the Golgi apparatus in these cells is unsurprising if, to our knowledge, unreported.
Actin Redistribution in Apoptotic Cells
The presence of numerous cytoplasmic inclusions of actin was particularly surprising, as little attention has been given to the redistribution of this protein during apoptosis, despite the large literature concerning its cleavage. Inhibitors of calpain may prevent actin proteolysis during apoptosis, but the extent of this actin fragmentation is very low. 16,34 Actin cleavage, by caspase-1 and -3, has been demonstrated in cell-free systems, 15,35,36 but its degradation in intact human cells is still controversial. 37 Cleavage of actin by caspase-3 results in a “fractin” fragment that can be identified in the cell processes of apoptotic neurones. 37 We cannot exclude the possibility that a similar fragment was present in our samples, as an antibody similar to that used in the current study did not detect “fractin.” 38 Similarly, Guenal et al 39 reported caspase-dependent actin cleavage and, using immunofluorescence microscopy, suggested that the resulting fragments accumulated in apoptotic bodies. The present study provides conclusive evidence that actin-rich bodies are extruded from the cytoplasm of THP.1 cells and that these bodies consist of intact β-actin. The absence of significant fragmentation was confirmed by our immunoblotting results, with the nonspecific antibody used previously to detect actin cleavage in cell extracts. 15 Caspase-dependent disorganization of the actin cytoskeleton has been attributed to the cleavage of β-catenin rather than actin itself, 13 and similar effects have been attributed to the depletion of ezrin/radixin/moesin proteins after activation of the caspase cascade. 40 Surprisingly, disruption of the cytoskeleton has also been associated with changes in the permeability of the mitochondrial outer membrane. 41 Actin redistribution, including the formation of cytoplasmic aggregations, has also been observed, by light microscopy, during the apoptosis of several cell types. 12,42-45 Recently, Jaunin et al 14 have shown aggregates of β-actin in transfected HeLa cells overexpressing the death-domain-containing protein MyD88, but not in nontransfected cells. Colocalization of this protein with the β-actin of apoptotic cells, like that of the “apoptosis-specific protein” reported in several examples of apoptosis, 46 may indicate a direct, rather than passive role for actin in cell death.
The release of cellular fragments containing aggregations of actin may thus be one feature of a common process involving the redistribution of the cytoskeleton during apoptosis. The phagocytosis of such fragments by neighboring cells could result in the subsequent proteolysis of actin in secondary lysosomes. Variations in the extent of this process, particularly between facultative and obligate phagocytes, may provide an explanation for the diversity of observations regarding actin proteolysis in cultures of apoptotic cells.
This study has shown that the release of mitochondrial cytochrome c into the cytosol of apoptotic THP.1 cells is normally followed by striking morphological changes in the cytoskeleton, Golgi apparatus, and mitochondria. All of these changes, unlike the release of cytochrome c, are prevented by the inhibition of caspase activity. Our results indicate that cytochrome c release is not the result of the onset of the mitochondrial permeability transition or the loss of the inner mitochondrial membrane potential. The development of ultracondensed mitochondria, which occurs after the release of cytochrome c, is currently the earliest morphologically detected hallmark of apoptosis in THP.1 cells.
Acknowledgments
We are grateful for the invaluable assistance of Huijun Zhu for treatment of cells, Roger Snowden for flow cytometry, and Ray Gilbert for the preparation of samples for electron microscopy.
Footnotes
Address reprint requests to Dr. David Dinsdale, MRC Toxicology Unit, Hodgkin Building, P.O. Box 138, Lancaster Road, Leicester LEI 9HN, UK. E-mail: dd5@le.ac.uk.
References
- 1.Kerr JFR, Wyllie AH, Currie AR: Apoptosis: a basic biological phenomenon with wide ranging implications in tissue kinetics. Br J Cancer 1972, 26:239-257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Earnshaw WC: Nuclear changes in apoptosis. Curr Opin Cell Biol 1995, 7:337-343 [DOI] [PubMed] [Google Scholar]
- 3.Cohen GM: Caspases: the executioners of apoptosis. Biochem J 1997, 326:1-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Majno G, Joris I: Apoptosis, oncosis, and necrosis. Am J Pathol 1995, 146:3-15 [PMC free article] [PubMed] [Google Scholar]
- 5.Wyllie AH: Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature 1980, 284:555-556 [DOI] [PubMed] [Google Scholar]
- 6.Cohen GM, Sun X-M, Snowden RT, Dinsdale D, Skilleter DN: Key morphological features of apoptosis may occur in the absence of internucleosomal DNA fragmentation. Biochem J 1992, 286:331-334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Green DR: Apoptotic pathways: the roads to ruin. Cell 1998, 94:695-698 [DOI] [PubMed] [Google Scholar]
- 8.Zamzami N, Marchetti P, Castedo M, Zanin C, Vayssiere J-L, Petit PX, Kroemer G: Reduction in mitochondrial potential constitutes an early irreversible step of programmed lymphocyte death in vivo. J Exp Med 1995, 181:1661-1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petit PX, Susin S-A, Zamzami N, Mignotte B, Kroemer G: Mitochondria and programmed cell death: back to the future. FEBS Lett 1996, 396:7-13 [DOI] [PubMed] [Google Scholar]
- 10.Reed JC: Cytochrome c: can’t live with it—can’t live without it. Cell 1997, 91:559-562 [DOI] [PubMed] [Google Scholar]
- 11.Vander Heiden MG, Chandel NS, Williamson EK, Schumacker PT, Thompson CB: Bcl-xl regulates the membrane potential and volume homeostasis of mitochondria. Cell 1997, 91:627-637 [DOI] [PubMed] [Google Scholar]
- 12.Van Engeland M, Kuijpers HJH, Ramaekers FCS, Reutelingsperger CPM, Schutte B: Plasma membrane alterations and cytoskeletal changes in apoptosis. Exp Cell Res 1997, 235:421-430 [DOI] [PubMed] [Google Scholar]
- 13.Brancolini C, Lazarevic D, Rodriguez J, Schneider C: Dismantling cell-cell contacts during apoptosis is coupled to a caspase-dependent proteolytic cleavage of α-catenin. J Cell Biol 1997, 139:759-771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jaunin F, Burns K, Tschopp J, Martin TE, Fakan S: Ultrastructural distribution of the death-domain-containing MyD88 protein in HeLa cells. Exp Cell Res 1998, 243:67-75 [DOI] [PubMed] [Google Scholar]
- 15.Kayalar C, Ord T, Testa MP, Zhong L-T, Bredesen DE: Cleavage of actin by interleukin 1B-converting enzyme to reverse DNase I inhibition. Proc Natl Acad Sci USA 1996, 93:2234-2238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown SB, Bailey K, Savill J: Actin is cleaved during constitutive apoptosis. Biochem J 1997, 323:233-237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kamada S, Kusano H, Fujita H, Ohtsu M, Koya RC, Kuzumaki N, Tsujimoto Y: A cloning method for caspase substrates that uses the yeast two-hybrid system: cloning of the antiapoptotic gene gelsolin. Proc Natl Acad Sci USA 1998, 95:8532-8537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhuang J, Dinsdale D, Cohen GM: Apoptosis, in human monocytic THP.1 cells, results in the release of cytochrome c from mitochondria prior to their ultracondensation, formation of outer membrane discontinuities, and reduction in inner membrane potential. Cell Death Differ 1998, 5:953-962 [DOI] [PubMed] [Google Scholar]
- 19.Zhu H, Fearnhead HO, Cohen GM: An ICE-like protease is a common mediator of apoptosis induced by diverse stimuli in human monocytic THP.1 cells. FEBS Lett 1995, 374:303-308 [DOI] [PubMed] [Google Scholar]
- 20.Zhu HJ, Dinsdale D, Alnemri ES, Cohen GM: Apoptosis in human monocytic THP.1 cells involves several distinct targets of N-tosyl-l-phenylalanyl chloromethyl ketone (TPCK). Cell Death Differ 1997, 4:590-599 [DOI] [PubMed] [Google Scholar]
- 21.MacFarlane M, Cain K, Sun X-M, Alnemri ES, Cohen GM: Processing/activation of at least four interleukin-1B converting enzyme-like proteases occurs during the execution phase of apoptosis in human monocyte tumor cells. J Cell Biol 1997, 137:469-479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hackenbrock CR: Ultrastructural bases for metabolically linked mechanical activity in mitochondria. J Cell Biol 1968, 37:345-369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laiho KU, Trump BF: Studies on the pathogenesis of cell injury: effects of inhibitors of metabolism and membrane function on the mitochondria of Ehrlich ascites tumor cells. Lab Invest 1975, 32:163-182 [PubMed] [Google Scholar]
- 24.Papadimitriou JC, Drachenberg CB, Shin ML, Trump BF: Ultrastructural studies of complement mediated cell death: a biological reaction model to plasma membrane injury. Virchows Arch 1994, 424:677-685 [DOI] [PubMed] [Google Scholar]
- 25.James TN, Terasaki F, Pavlovich ER, Vikhert AM: Apoptosis and pleomorphic micromitochondriosis in the sinus nodes surgically excised from five patients with the long QT syndrome. J Lab Clin Med 1993, 122:309-323 [PubMed] [Google Scholar]
- 26.Mancini M, Anderson BO, Caldwell E, Sedghinasab M, Paty PB, Hockenbery DM: Mitochondrial proliferation and paradoxical membrane depolarization during terminal differentiation and apoptosis in a human colon carcinoma cell line. J Cell Biol 1997, 138:449-469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jia L, Dourmashkin RR, Newland AC, Kelsey SM: Mitochondrial ultracondensation, but not swelling, is involved in TNFalpha-induced apoptosis in human T-lymphoblastic leukaemic cells. Leuk Res 1997, 21:973-983 [DOI] [PubMed] [Google Scholar]
- 28.Jia L, Dourmashkin RR, Allen PD, Gray PD, Newland AC, Kelsey SM: Inhibition of autophagy abrogates tumour nectrosis factor a induced apoptosis in human T-lymphobastic leukaemic cells. Br J Haematol 1997, 98:673-685 [DOI] [PubMed] [Google Scholar]
- 29.Sun X-M, MacFarlane M, Zhuang J, Wolf BB, Green DR, Cohen GM: Distinct caspase cascades are initiated in receptor-mediated and chemical-induced apoptosis. J Biol Chem 1999, 274:5053-5060 [DOI] [PubMed] [Google Scholar]
- 30.Liu X, Kim CN, Yang J, Jemmerson R, Wang X: Induction of apoptotic program in cell-free extracts: requirements for dATP and cytochrome c. Cell 1996, 86:147-157 [DOI] [PubMed] [Google Scholar]
- 31.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X: Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91:479-489 [DOI] [PubMed] [Google Scholar]
- 32.Lucocq JM, Pryde JG, Berger EG, Warren G: A mitotic form of the Golgi apparatus in HeLa cells. J Cell Biol 1987, 104:865-874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pypaert M, Nilsson T, Berger EG, Warren G: Mitotic Golgi clusters are not tubular endosomes. J Cell Sci 1993, 104:811-818 [DOI] [PubMed] [Google Scholar]
- 34.Villa PG, Henzel WJ, Sensenbrenner M, Henderson CE, Pettmann B: Calpain inhibitors, but not caspase inhibitors, prevent actin proteolysis and DNA fragmentation during apoptosis. J Cell Sci 1998, 111:713-722 [DOI] [PubMed] [Google Scholar]
- 35.Mashima T, Fujita N, Noguchi K, Tsuruo T: Identification of actin as a substrate of ICE and an ICE-like protease and involvement of an ICE-like protease but not ICE in VP-16-induced U937 apoptosis. Biochem Biophys Res Commun 1995, 217:1185-1192 [DOI] [PubMed] [Google Scholar]
- 36.Mashima T, Naito M, Noguchi K, Miller DK, Nicholson DW, Tsuruo T: Actin cleavage by CPP-32/apopain during the development of apoptosis. Oncogene 1997, 14:1007-1012 [DOI] [PubMed] [Google Scholar]
- 37.Song Q, Wei T, Lees-Miller S, Alnemri E, Watters D, Lavin MF: Resistance of actin to cleavage during apoptosis. Proc Natl Acad Sci USA 1997, 94:157-162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang F, Sun X, Beech W, Teter B, Wu S, Sigel J, Vinters HV, Frautschy SA, Cole GM: Antibody to caspase-cleaved actin detects apoptosis in differentiated neuroblastoma and plaque-associated neurons and microglia in Alzheimer’s disease. Am J Pathol 1998, 152:379-389 [PMC free article] [PubMed] [Google Scholar]
- 39.Guenal I, Risler Y, Mignotte B: Down-regulation of actin genes precedes microfilament network disruption and actin cleavage during p53-mediated apoptosis. J Cell Sci 1997, 110:489-495 [DOI] [PubMed] [Google Scholar]
- 40.Kondo T, Takeuchi K, Doi Y, Yonemura S, Nagata S, Tsukita T, Tsukita S: ERM (ezrin/radexin/moesin)-based molecular mechanism of microvillar breakdown at an early stage of apoptosis. J Cell Biol 1997, 139:749-758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rappaport L, Oliviero P, Samuel JL: Cytoskeleton and mitochondrial morphology and function. Mol Cell Biochem 1998, 184:101-105 [PubMed] [Google Scholar]
- 42.Chow SC, Orrenius S: Rapid cytoskeleton modification in thymocytes induced by the immunotoxicant tributyltin. Toxicol Appl Pharmacol 1994, 127:19-26 [DOI] [PubMed] [Google Scholar]
- 43.Endresen PC, Prytz PS, Aarbakke J: A new flow cytometric method for discrimination of apoptotic cells and detection of their cell cycle specificity through staining of F-actin and DNA. Cytometry 1995, 20:162-171 [DOI] [PubMed] [Google Scholar]
- 44.Levee MG, Dabrowska MI, Lelli JL, Hinshaw DB: Actin polymerization and depolymerization during apoptosis in HL-60 cells. Am J Physiol 1996, 271:C1981-C1992 [DOI] [PubMed] [Google Scholar]
- 45.Rubtsova SN, Kondratov RV, Kopnin PB, Chumakov PM, Kopnin BP, Vasiliev JM: Disruption of actin microfilaments by cytochalasin D leads to activation of p53. FEBS Lett 1998, 430:353-357 [DOI] [PubMed] [Google Scholar]
- 46.Grand RJA, Milner AE, Mustoe T, Johnson GD, Owen D, Grant ML: A novel protein expressed in mammalian cells undergoing apoptosis. Exp Cell Res 1995, 218:439-451 [DOI] [PubMed] [Google Scholar]