

Abstract
Transforming growth factor-β-1 (TGF-β1) is secreted by cells in a latent form (L-TGF-β1) noncovalently bound to a latency-associated peptide. Activated alveolar macrophages obtained from rat lungs after bleomycin-induced pulmonary injury released increased amounts of active TGF-β1 as well as plasmin, a protease, and thrombospondin-1 (TSP-1), a trimeric glycoprotein. Previously we had demonstrated that plasmin was critical to the activation of L-TGF- β1. In the present study we demonstrated that TSP-1 is also important for the activation of L-TGF- β1 because the activation can be inhibited by anti-TSP-1 monoclonal antibody. Proteins obtained from alveolar macrophage cell lysates immunoprecipitated with antibodies specific for TSP-1 were identified on immunoblots as LAP and TGF-β1, indicating that TSP-1/L-TGF-β1 complexes are present on alveolar macrophages. However, in the presence of plasmin both latency-associated peptide and TGF-β1 were decreased in the same cell lysates, indicating that L-TGF-β1 associated with TSP-1 is released by plasmin. Using immunofluorescence and antibodies to TGF-β1 and CD36, a receptor for TSP-1, there was colocalization of TGF-β1 with CD36. Because TSP-1 but not TGF-β1 is a natural ligand for CD36, these findings suggest that the L-TGF-β1 in a complex with TSP-1 localizes to the macrophage cell surface when TSP-1 interacts with its receptor, CD36. Furthermore, the association of TSP-1/L-TGF-β1 complex with CD36 is necessary to the activation of L-TGF-β1 because antibodies to CD36 prevent the colocalization of TGF-β1 with CD36 as observed by immunofluorescence and inhibit activation of the L-TGF-β1 by explanted alveolar macrophages. These findings suggest that activation of L-TGF-β1 by plasmin occurs at the cell surface of activated alveolar macrophages and requires a TSP-1/CD36 interaction.
At sites of lung injury before connective tissue synthesis there is an influx of activated macrophages. 1,2 When activated, macrophages secrete a number of pro-inflammatory and fibrogenic cytokines. 1,2 Of these cytokines, transforming growth factor-β-1 (TGF-β1), a multifunctional peptide, is one of the most potent regulators of inflammation and connective tissue synthesis. 3 TGF-β1 is synthesized as a large 390-amino acid precursor that undergoes a number of intracellular processing steps, including cleavage of the latent-associated peptide (LAP), to produce the mature 25-kd TGF-β1 protein. 4 However, with rare exceptions, when TGF-β1 is secreted by cells it remains noncovalently associated in a 1:1 ratio with its LAP. 4 The noncovalent association of TGF-β1 with its LAP renders the TGF-β1 unable to interact with its receptor and, therefore, biologically inactive. 4 TGF- β1 and its receptors are ubiquitously expressed, and, because TGF-β1 has numerous biological effects, the ability of a cell to activate L-TGF-β1 on secretion may be an important regulatory mechanism of TGF- β1 action in vivo.
We had previously demonstrated that after pulmonary injury induced by the antineoplastic antibiotic bleomycin, explanted alveolar macrophages generated maximal quantities of biologically active TGF-β1 and plasmin 7 days after bleomycin induced pulmonary injury. 5 Furthermore, the secretion of active TGF-β1 was totally inhibited by the presence of α2-antiplasmin, a naturally occurring inhibitor of plasmin. 5 When large quantities of plasmin were added to activated alveolar macrophages, there was further activation of the L-TGF-β1. 5 However, when plasmin was added to the L-TGF-β1 in cell-free CM, no 9further activation of L-TGF-β1 occurred. 5 Our findings suggested that the generation of plasmin is important in the posttranslational activation of alveolar macrophage-derived L-TGF- β1 during an inflammatory pulmonary injury response and that the activation requires the presence of intact macrophages. 5
In this paper, we demonstrate that alveolar macrophages also secrete increased quantities of TSP-1, a glycoprotein previously reported to activate L-TGF-β1 both in the presence of cells and in cell-free solution. 6 When alveolar macrophages were cultured in the presence of neutralizing antibodies to TSP-1, the posttranslational activation of L-TGF-β1 was abrogated. Furthermore, antibodies to the TSP-1 receptor, CD36, also abrogated activation of alveolar macrophage-derived L-TGF-β1. Our findings support a model in which L-TGF-β1 is held at the cell surface by a TSP-1/CD36 interaction and is processed by plasmin generated by activated alveolar macrophages.
Materials and Methods
Animals
Female Sprague-Dawley rats, which were free of respiratory disease and weighed between 250 and 300 g, were obtained from the University of Manitoba vivarium. In each experiment, all rats were matched for age and weight.
Reagents
Bleomycin (Blenoxane) was a gift from Bristol-Myers Squibb (Evansville, IN). Neutralizing antibody to TGF-β1–3 was obtained from Genzyme (Cambridge, MA). Antibody to TGF-β1 used for Western blot analysis was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The recombinant anti-human LAP antibody was obtained from R&D Systems (Minneapolis, MN). TSP-1 depleted of TGF-β activity (sTSP-1) and monoclonal antibodies to human platelet TSP-1 depleted of associated TGF- β1 (mAb 133) used in the enzyme-linked immunosorbent assay (ELISA) and TSP-1 immunoprecipitates were either used by or obtained from Dr. Murphy-Ullrich. 6,7 Anti-TSP-1 antibody used in experiments to neutralize TSP-1 from activated alveolar macrophages was obtained from Sigma (St. Louis, MO). The CD36 antibody, 5F1, was provided by the Fifth International Workshop on Leukocyte Differentiation Antigens. 8
Bleomycin Administration
This procedure is described in detail elsewhere. 5,9,10 For some experiments rats were sacrificed at several time intervals after bleomycin or normal saline treatment, 5,9,10 whereas for other experiments alveolar macrophages were harvested 7 days after bleomycin administration. The latter time point was used based on our findings that alveolar macrophages are maximally stimulated at this time to secrete active TGF-β1. 5
Macrophage Cultures
This procedure is described in detail elsewhere. 5,10 The lungs were lavaged to obtain cells for culture of alveolar macrophages. Alveolar macrophages were maintained in serum-free media containing Gentamicin (4 mg/100 ml; Roussel, Montreal, PQ), Fungizone (100 μl/100 ml; Gibco BRL, Grand Island, NY) and 0.2% clotted bovine calf plasma (BCP; National Biological Laboratory Limited, Dugald, MB). The macrophages were cultured in the absence or presence of a number of reagents consisting of anti-TSP-1 antibody, 5F1 (anti-CD36 antibody), CD36 synthetic peptide (aa 93–110), or sTSP-1. In experiments to determine the effects of exogenous sTSP-1, alveolar macrophages were cultured with varying quantities of sTSP-1 for 2 hours before the collection of conditioned media (CM). In addition, CM from the same alveolar macrophages cultured in parallel was incubated in a cell-free solution with sTSP-1 for 2 hours. Incubation of CM with sTSP-1 for 2 hours was chosen based on our previous findings, that sTSP-1 can activate L-TGF-β1 within 2 hours. 11 In some experiments the cells were cultured in the absence or presence of 5F1, the CD36 antibody, before the addition of sTSP-1. In experiments to determine whether or not both plasmin and TSP-1 are required to activate alveolar macrophage-derived L-TGF-β1, the alveolar macrophages were cultured with sTSP-1 in the absence or presence of aprotinin, an inhibitor of plasmin activity. 12 After 20 hours of incubation in 5% CO2 at 37°C, the media was collected in the presence of protease inhibitors (leupeptin 0.5 μg/ml, Amersham, Poole, UK; aprotinin 1 μg/ml, and pepstatin 1 μg/ml, both from Sigma, Oakville, ON), and frozen at −80°C until ready for TGF-β quantitation. 5,10
CCL-64 Mink Lung Epithelial Growth Inhibition Assay for TGF-β
The CCL-64 growth inhibition assay to identity and quantitate TGF-β has been described. 5,9,10,13 . Briefly, to subconfluent cells in 0.2% BCP and resuspended in α-MEM, 0.2% BCP, 10 mmol/L Hepes at pH 7.4, penicillin (25 μg/ml) and streptomycin (25 μg/ml), and cultured as 5 × 10 4 cells per 0.5 ml in 24-well costar dishes (Flow Laboratories, Inc., Mississauga, ON) was added neutral CM or CM that was acidified and subsequently neutralized. After 22 hours the cells were pulsed with 0.25 Ci of 5-[125I] iodo 2′-deoxyuridine (ICN Pharmaceutical, Costa Mesa, CA) for 2 to 3 hours at 37°C and eventually lysed with 1 ml of 1 N NaOH for 30 minutes at room temperature and the 125I-UdR was counted in a γ counter (LKB Instruments, Gaithersburg, MD). A standard curve using porcine TGF-β1 was included in each assay. For confirmation of TGF-β activity, neutralizing monoclonal antibody to TGF-β1–3 (Genzyme) was added before the addition of the CM 5,9,10,13 and resulted in abrogation of all TGF-β activity.
Detection and Quantitation of TSP-1 by Direct ELISA
The wells of 96-well plates (Falcon tissue culture plates) were coated with 200 ng/well of the mAb 133 (anti-sTSP-1). For each experiment, a standard curve containing wells with 200 μl of CM and several concentrations of sTSP-1, ranging from 15–150 ng/well, was included. To quantitate TSP-1 in CM, 200 μl of alveolar macrophage derived CM in carbonate buffer, pH 9.6, was incubated overnight at 4°C. The next day, the wells were washed three times with PBS, 0.05%, Tween −20. Nonspecific binding sites were blocked by incubating with 250 μl/well of 1% BSA for 1 hour at 37°C. The wells were then washed with PBS and incubated with 200 μl/well of mAb 133 (7.5 μg/ml) in PBS-Tween for 90 minutes at 37°C. The wells were then washed and incubated with 30 ng/ml of alkaline phosphatase-labeled goat anti-mouse lgG for 90 minutes at 37°C, and then assayed for color development using the Sigma 104 AP substrate. Color development was stopped by adding 50 μl of 2N NaOH, and absorbency at 405 nm was read using a Bio-Tek ELISA reader.
Preparation of Synthetic CD36 Peptides
The CD36 peptide, YRVRFLAKENVTQDAEDNC(93–110) was synthesized, based on the work of Leung et al. 14 The peptide was synthesized with an Applied Biosystems model 431A peptide synthesizer using Fmoc (N-(9-Fluoreny D-methoxycarbonyl) chemistry and purified by reverse high pressure liquid chromatography, using a C18 column.
Localization of CD36 and TGF-β1 by Immunofluorescence
Alveolar macrophages were obtained by bronchoalveolar lavage 7 days after intratracheal normal saline or bleomycin administration, and were adjusted to 1 × 10 6 cells/ml. For some experiments, alveolar macrophages obtained after bleomycin administration were cultured in the presence of anti-CD36 antibody (20 μg per 10 6 macrophages) for 30 minutes before the immunofluoresence procedure. The immunofluorescence analysis was performed as previously described. 15 Briefly, cytospin smears of 1 × 10 5 cells suspension were fixed with 3.7% formaldehyde for 10 minutes and washed with PBS. Nonspecific binding was blocked by 100% lamb serum (Gibco BRL) for 5 minutes. The cells were then incubated with anti-TGF-β1 (Santa Cruz Biotechnology) and anti-CD36 antibody, both at a concentration of 1 μg/ml, for 45 minutes. After washing with PBS the cells were incubated for 30 minutes with polyclonal anti-rabbit antibody conjugated to tetramethylrhodamine-isothiocyanate (TRITC) or conjugated with fluorescein isothiocyanate (FITC) monoclonal anti-mouse IgM as secondary antibodies for the detection of TGF-β1 and CD36 antibodies, respectively. Both secondary antibodies were used as 1 μg/ml. The slides were washed three times with PBS. Nuclear staining was done using 4′, 6′ Diamidino-2-phenylindole (DAPI) at a concentration of 1 μg/ml for 5 minutes. The slides were mounted in anti-bleach (12% glycerol, 4.8% Mowiol 4–88 (Hoechst), 2.4% DABCO (1, 4 Diazabicylo [2.2.2]-octane (Fluka) in 0.2 Mol/L Tris/HCl, pH = 8.5). Image analysis was performed using a Zeiss Axiophot microscope, equipped with a cooled CCD camera CH 250/a (Optikon/Photometrics), driven by IPLabs Spectrum software version 3.1, Signal Analytics). For TRITC-conjugated antibodies, the filter combination was BP540/FT580/LP590, resulting in a red emission identifying the location of TGF-β1on the macrophage surface. For the FITC-conjugated antibody, the filter combination was 450–490/FT510/515–565, resulting in a green emission identifying the location of CD36 on the macrophage surface. For DAPI, the filter combination was G365/FT395/LP420, resulting in a blue emission. To determine whether TGF-β1 and CD36 were colocalized, the images were obtained by a pixel overlap, which was achieved with IPLab Spectrum/Multiprobe (V.3.1, Signal Analytics, Fairfax, VA). In areas where the TGF-β1 was localized to the same region as CD36, the emission was yellow.
Protein Extraction and Immunoprecipitation
Alveolar macrophages obtained from rats that had been treated with normal saline or bleomycin 7 days earlier were lysed by incubating the cells for 20 minutes on ice in a RIPA lysis buffer (50 mmol/L Tris-Cl, pH 7.5; 150 mmol/L NaCl, 1% Nonidet P-40, 0.1% sodium deoxycholate and a cocktail of protease inhibitors; phenylmethylsulfonyl fluoride 1 mmol/L, leupeptin 1 μg/ml, and aprotinin 0.1 μg/ml, all from Sigma). In some experiments the alveolar macrophages were cultured overnight in the absence or presence of α2-antiplasmin or aprotinin (Sigma), both inhibitors of plasmin activity. 12 The lysate was centrifuged for 20 minutes at 12,000 × g at 4°C, the supernatant collected, and the total protein content determined by the Bradford dye-binding assay (Bio-Rad, Mississauga, ON). The total protein extract (300 μg) and 10 μg of anti-sTSP-1 antibody (mAb 133) or IgG as an isotype control for the anti-sTSP-1 antibody was incubated overnight at 4°C. After a further incubation of 2 hours with 30 μl of Protein G Plus/Protein A-Agarose (Calbiochem, San Diego, CA) the immune complexed beads were collected. The beads were washed four times with RIPA buffer and placed in a final suspension with 25 μl of Laemmli buffer and boiled for 10 minutes. The supernatant containing the precipitated proteins was then used for Western blot analysis.
Western Blot Analysis
The protein samples (25 μl) were electrophoresed on 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) in a MiniPROTEAN II Electrophoresis Cell (Bio-Rad, Hercules, CA). Rainbow colored protein molecular weight markers (Amersham) were run parallel to each blot as an indicator of the molecular weight. Equality in loading of protein was evaluated using silver staining (not shown). The separated proteins were transferred at 50 V overnight onto nitrocellulose membrane (Gibco BRL) in a Mini trans-Blot chamber with transfer buffer (25 mmol/L Tris Cl, 192 mmol/L glycine, and 20% methanol). The nitrocellulose membrane was blocked for 1 hour by using 5% instant skim milk powder in TBS. For detection of LAP a 1:500 dilution of anti-recombinant human LAP antibody (anti-rh LAP; R&D Systems) in 1% instant skim milk powder was used. After washing, the nitrocellulose membrane was incubated with horseradish peroxidase linked with the secondary antibody (Goat anti-mouse IgG; Bio-Rad) as recommended by the manufacturer. Finally the washed blots were exposed to enhanced chemiluminescence detection system (Amersham) and recorded on an autoradiograph (Kodak X-Omat film). Before reprobing, the nitrocellulose membrane was incubated at 50°C for 30 minutes with a stripping buffer (100 mmol/L 2-mercaptoethanol, 2% sodium dodecyl sulfate, and 62.5 mmol/L Tris-HCl, pH 6.7). The blots were rinsed twice with TBS. To ensure the removal of antibodies, membranes were incubated with the enhanced chemiluminescence detection reagents and exposed to film (Kodak). No band was detected, confirming that all antibodies were stripped off the membrane. The same nitrocellulose membrane was blocked using 5% instant skim milk powder in TBS. For detection of TGF-β1, 0.5 μg/ml of rabbit polyclonal IgG TGF-β1 antibody (Santa Cruz Biotechnology) was used as above.
Statistical Analysis
Statistical analysis using analysis of variance was done by Dr. Bob Tate, Biostatistical Unit, University of Manitoba.
Results
Activation of Alveolar Macrophage-Derived L-TGF-β1 by Thrombospondin (TSP)-1
In addition to plasmin, TSP-1, a large trimeric glycoprotein, 16 had also been demonstrated to be a physiological substance that can activate L-TGF-β1. 6,17 TSP-1 was constitutively secreted by alveolar macrophages, but after bleomycin injury, TSP-1 secretion was further increased and reached maximal levels 7 days after bleomycin administration (Figure 1A) ▶ . The secretion of TSP-1 declined rapidly thereafter, and by 28 days after bleomycin administration, the secretion returned to that of control levels of alveolar macrophages from normal saline-treated rats. To determine whether the presence of TSP-1 in the CM was necessary for the activation of L-TGF-β1, alveolar macrophages were cultured in the absence or presence of anti-TSP-1 monoclonal antibody. Anti-TSP-1 antibody inhibited the activation of L-TGF- β1 but had no effect on the secretion of the latent form of TGF-β1 (Figure 1B) ▶ by the alveolar macrophages. Neutral CM of alveolar macrophages activated by in vivo bleomycin injury contained 67.6 ± 7.9 pg of TGF-β1 per 10 6 cells. After the addition of 50 μg/ml of the isotype control for anti-TSP-1, 50.13 ± 13.4 pg of TGF-β1 per 10 6 was present in neutral CM (P < 0.934). Because TSP-1 has been reported to activate L-TGF-β1 in solution, 6 we next determined if alveolar macrophage-derived L-TGF-β1 could be directly activated in CM by sTSP-1. After 20 hours in culture, activated alveolar macrophages secreted large quantities of L-TGF-β1. 5 The addition of sTSP-1 to this cell-free L-TGF-β1 unexpectedly diminished the quantity of active TGF-β1 that could be detected by our bioassay (Figure 1C) ▶ . In contrast, sTSP-1 added directly to the same CM but, in the presence of alveolar macrophages, further activated L-TGF-β1 (Figure 1C) ▶ . These findings demonstrate that TSP-1 is effective in promoting the activation of alveolar macrophage-derived L-TGF-β1 in the presence of intact macrophages.
Figure 1.

Activation of alveolar macrophage derived L-TGF-β1 by sTSP-1. A: Quantity of TSP-1 secreted by explanted alveolar macrophages obtained from rats at varying lengths of time after bleomycin administration. Each point is the mean of samples from 3 to 6 rats. The P value is <0.0003 when the quantity of TSP-1 produced by alveolar macrophages from normal saline-treated rats is compared to TSP-1 produced by alveolar macrophages from rats treated with bleomycin 7 days earlier. B: The effects of anti-TSP-1 antibody on the activation of alveolar macrophage-derived L-TGF-β1. Alveolar macrophages obtained 7 days after bleomycin administration were cultured in the absence of anti-TSP-1 antibody or in the presence of varying concentrations. The TGF-β1 present in neutral CM (□) represents bioactive TGF-β1; that present in acidified then neutralized CM (▪) represents the total TGF-β1 in the same sample. Each point is the mean of samples from 6 rats. The quantity of TGF-β1 in neutral CM compared to the quantity of TGF-β1 in acidified CM has a P ≤ 0.001 when the anti-TSP-1 antibody used was 50 μg/ml/10 6 macrophages, and P ≤ 0.003 when the anti-TSP-1 antibody used was 5.0 or 0.5 μg/ml/10 6 macrophages. There was no statistical difference in the quantity of TGF-β1 in neutral and acidified CM when no anti-TSP-1 antibody was present. C: Induction of activation of L-TGF-β1 by addition of sTSP-1. Alveolar macrophages were obtained 7 days after bleomycin administration and cultured for 20 hours before the addition of sTSP-1 (0–4 μg/ml) to alveolar macrophages (▪) or the CM (□) from parallel cultures of alveolar macrophages. After a 2-hour incubation with sTSP-1, the TGF- β1 in neutral and acidified CM (not shown) of the same sample was determined. The -fold increase in the quantity of TGF-β1 present was calculated by using the quantity of active TGF-β1 present in the absence of sTSP-1 as the denominator and the quantity of TGF-β1 present after the addition of sTSP-1 as the numerator. Each point is the mean of samples from 3 rats. The -fold increase in the quantity of TGF-β1 in neutral CM when alveolar macrophages were treated with 0.4 or 4.0 μg/ml of sTSP-1, compared to TGF-β1 in CM when no sTSP-1 was present, has P value ≤ 0.05.
Cell Surface Association of L-TGF- β1 to CD36 Is Required for Activation of L-TGF-β1
TSP-1 not only complexes with L-TGF-β1, 6,7,11 but it also binds to cell surface receptors such as CD36 18,19 which are prominently expressed on macrophages. 18,19 We next determined if the CD36 binding of TSP-1 is required for the posttranslational processing and activation of L-TGF- β1. At the highest concentration, the presence of antibodies specific to CD36 totally abrogated the activation of L-TGF-β1 (Figure 2A) ▶ . Neutral CM of alveolar macrophages activated by in vivo bleomycin injury contained 70 ± 10 pg of TGF-β1 per 10 6 cells, while after the addition of 20 μg/ml of the isotype control for anti-CD36, 55 ± 1 pg of TGF-β1 per 10 6 cells was present (P < 0.949). There was an induction of L-TGF- β1 in the presence of CD36 antibody but the mechanism of induction is not clear. The interaction of the CD36 with its antibody has been reported to lead to an oxidative burst in monocytes 20 and induction of the protein tyrosine kinases fyn, lyn, and yes. 21 We are presently determining if either of these events results in increased production and secretion of L-TGF-β1 by macrophages after anti-CD36 antibody interaction. Alternatively it is also possible that the L-TGF-β1 is sequestered on the cell surface via the CD36 receptor. The presence of the CD36 antibody may then displace the L-TGF-β1 from the cell surface and increase the quantity of L-TGF-β1 present in the CM.
Figure 2.

Effects of anti-CD36 antibody on the activation of L-TGF-β1. Alveolar macrophages obtained 7 days after bleomycin administration were cultured in the absence or presence of different concentrations of anti-CD36 antibody or the CD36 synthetic peptide 93–110. A: The TGF-β1 present in neutral CM □ and acidified then neutralized CM ▪ from the same sample was quantitated. Each point is the mean of samples from 4 rats. The quantity of TGF-β1 present in neutral CM compared to TGF-β1 in acidified CM has a P value ≤0.0001 when the antibody was 20 μg/ml/10 6 macrophages, and P ≤ 0.01 when the antibody was 10 μg/ml/10 6 macrophages. The other comparisons between TGF-β1 in neutral and acidified CM were not significant. B: The effects of CD36 synthetic peptide aa 93–110 on the activation of L-TGF-β1. Alveolar macrophages were cultured in the absence or presence of several concentrations of the synthetic peptide of CD36 aa 93–110. TGF-β1 in neutral □ and acidified, then neutralized CM ▪ was quantitated. All points are the mean of 4 to 6 animals. The quantity of TGF-β1 in neutral CM, compared to the TGF-β1 in acidified CM of the same sample, had a P value ≤0.0006 only when CD36 peptide 93–110 was present. C: Effects of anti-CD36 antibody on the activation of L-TGF-β1 in the presence of sTSP-1. Alveolar macrophages were cultured in the absence or presence of 20 μg/ml/10 6 macrophages of anti-CD36 antibody, and in the absence or presence of 0.4 μg/ml/10 6 macrophages of sTSP-1. The TGF- β1 present in neutral CM was quantitated. Each point is the mean of samples from 4–6 rats. TGF-β1 in neutral CM of alveolar macrophages from bleomycin (BLM)-treated rats compared to normal saline (N/S)-treated rats has a P value ≤0.07. TGF-β1 in neutral CM of alveolar macrophages from BLM-treated rats cultured in the absence of sTSP-1, compared to when sTSP-1 was present, has a P value ≤0.02. TGF-β1 in neutral CM of alveolar macrophages from BLM treated rats cultured with sTSP-1, compared to when sTSP-1 and anti-CD36 antibody was also present, has a P value ≤0.006.
The binding of TSP-1 to the extracellular domain of CD36 is blocked by a CD36 synthetic peptide from amino acids 93–110 (CD3693–110). 14 When activated alveolar macrophages were incubated with the synthetic peptide CD3693–110 mimicking the TSP-1 binding domain, the activation of L-TGF-β1 was abrogated (Figure 2B) ▶ . Although the CD36 peptide 93–110 decreased activation of L-TGF-β1, unlike the CD36 antibody, the presence of the peptide did not induce L-TGF-β1 generation. The reason for this is not clear, but it could be that the peptide does not interact with the CD36 receptor in the same manner as the antibody 20,21 and, therefore, not cause further activation of alveolar macrophages to release L-TGF-β1. The synthetic peptide corresponding to an area unrelated to CD36-TSP-1 interaction, CD36204–288, had no effect on the activation of L-TGF- β1(data not shown). The addition of sTSP-1 to alveolar macrophages increased the activity of TGF-β1 present in neutral CM (Figures 1C and 2C) ▶ ▶ . Further confirmation that the association of TSP-1 with CD36 on alveolar macrophages was important in activation of L-TGF- β1 was the finding that, when the CD36 antibody was added to activated alveolar macrophages in the presence of sTSP-1, there was a marked diminution of the effects of sTSP-1 in generating mature TGF-β1 (Figure 2C) ▶ .
Using indirect immunofluorescent staining, alveolar macrophages obtained from rats previously treated with normal saline had detectable TGF-β1 (red emission; Figure 3A ▶ ) and CD36 (green emission; Figure 3B ▶ ) on the cell surface. In addition, both CD36 and TGF-β1 were colocalized (yellow emission) to a number of regions on the same cells (Figure 3C) ▶ . However, alveolar macrophages obtained 7 days after in vivo bleomycin injury had a marked increase in the number of cells with cell surface localization of TGF-β1 (Figure 3D) ▶ and CD36 (Figure 3E) ▶ , and the colocalization of TGF-β1 and CD36 was also increased (Figure 3F) ▶ . When alveolar macrophages obtained 7 days after bleomycin injury were cultured in the presence of anti-CD36 antibody before the immunofluorescent staining the presence of TGF-β1 (Figure 3G) ▶ and CD36 (Figure 3H) ▶ was markedly reduced and was not detected consistently on the same cells (Figure 3, G and H) ▶ as observed in Figure 3, A–F ▶ . Furthermore, the colocalization of TGF-β1 and CD36 was either not present or markedly decreased (Figure 3I) ▶ . Although alveolar macrophages from normal saline-treated rats expressed little TGF-β1 and CD36 there was almost complete colocalization with CD36, whereas after anti-CD36 pretreatment of alveolar macrophages there was markedly less colocalization of CD36 and TGF-β1. These findings suggest that the association of TGF-β1 with CD36 occurs in a specific manner rather than randomly.
Figure 3.
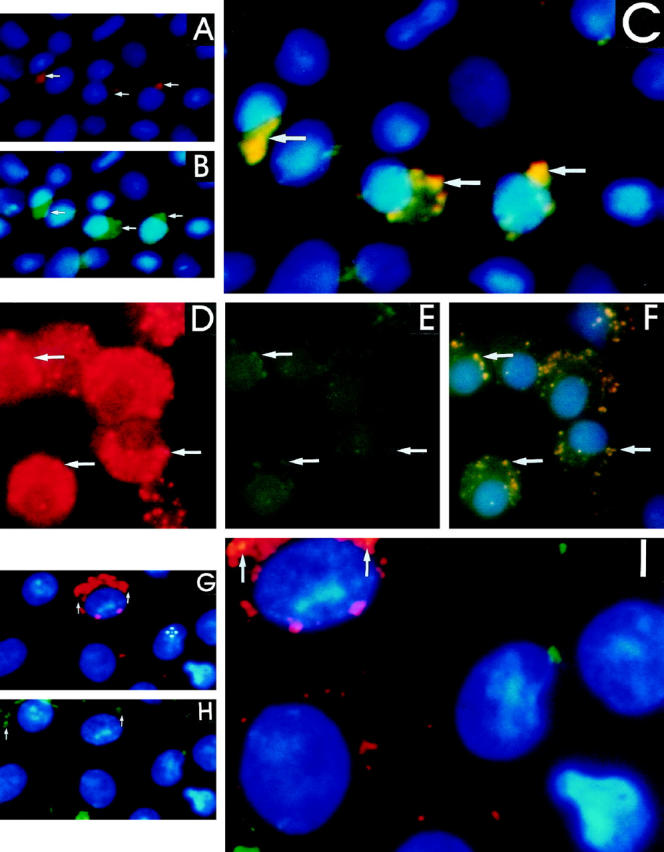
Localization of TGF-β1 and CD36 on the alveolar macrophage using immunofluoresence. Alveolar macrophages obtained 7 days after normal saline administration treated with (A) polyclonal anti-rabbit lgG antibody conjugated with TRITC reacts with cell surface-associated anti-TGF-β1 primary antibody, and the location is seen as a red coloration (arrow). B: Monoclonal anti-mouse lgM antibody conjugated with FITC reacts with cell surface-associated anti-CD36 primary antibody; the location is seen as a green coloration (arrow). C: Immunofluorescent images obtained from the above observations were used to generate a pixel overlay of the images resulting in a yellow coloration (arrow) and demonstrating the regions of colocalization of TGF-β1 and CD36. Alveolar macrophages obtained 7 after bleomycin administration and treated as described above identify (D) TGF-β1 seen as a red coloration (arrow); (E) CD36 seen as a green coloration (arrow). F: Immunofluorescent images obtained from the above observations were used to generate a pixel overlay of images resulting in a yellow coloration (arrow). Alveolar macrophages obtained 7 days after bleomycin administration treated in the same manner as described above but incubated with anti-CD36 antibody before cell surface localization of (G) TGF-β1 and (H) CD36. A pixel overlay of the images demonstrates either no areas of colocalization of TGF-β1 and CD36 or markedly decreased areas of colocalization (arrows) on a few cells (I).
Proteins obtained from the immunoprecipitates using alveolar macrophage cell lysates and anti-sTSP-1 antibodies were identified by Western blot analysis using anti-LAP-1 and revealed bands at 68 and 34 kd (Figure 4) ▶ . The location of bands using anti-LAP-1 antibody at 68 kd has previously been confirmed to correspond to the dimer of LAP-1, whereas the 34-kd band on the same blot has been confirmed to correspond to the monomer of LAP-1. 22 When the same nitrocellulose membrane was immunoblotted with anti-TGF-β1 antibodies a band was detected at 25 kd, corresponding to the dimer of TGF-β1, and another band detected at 13 kd, corresponding to the monomer of TGF- β1. Differences of the LAP-1 and TGF-β1 band intensity between alveolar macrophages obtained after normal saline or bleomycin treatment could be ascertained in the bands corresponding to the monomer and dimer of TGF-β1, which were decreased in samples from bleomycin-treated rats. The decrease was most apparent for the band corresponding to the monomer of TGF-β1. Alveolar macrophages obtained after bleomycin injury but not normal saline administration release active TGF-β1 by the action of plasmin. 5 In the absence of plasmin inhibitors before protein extraction, less TGF-β1 is likely to be associated with the macrophages. Thus, proteins obtained from alveolar macrophages after immunoprecipitation with TSP-1 contain less TGF-β1 than that obtained from alveolar macrophages after normal saline administration. The effects of plasmin on cell-associated TGF-β1 was further confirmed in the findings reported in Figure 5A ▶ . The results in Figure 4 ▶ indicate that resting macrophages as well as those obtained after bleomycin injury response have TSP-1/L-TGF-β1 complexes. Because the anti-sTSP-1 antibody (mAb 133) used in our work only recognizes TSP-1, 6,7 it follows that the TGF-β1 and LAP-1 obtained from immunoprecipitates using the anti-sTSP-1 antibody (mAb 133) must have been complexed with TSP-1, and the TSP-1/L-TGF-β1 complex was present on alveolar macrophages.
Figure 4.

Immunoblot analysis of LAP and TGF-β1 antigens obtained from immunoprecipitates using anti-sTSP-1 antibody (mAb 133) and lysates of alveolar macrophages. Alveolar macrophages obtained 7 days after treatment with either normal saline (lane 1) or bleomycin (lane 2) were immunoblotted using anti-LAP antibodies and demonstrate protein bands compatible with the dimer (68 kd) and monomer (34 kd) of LAP. The same nitrocellulose filter was reprobed for immunoblotting with anti-TGF-β1 antibodies and demonstrate the presence of TGF-β1 protein as a dimer (25 kd) and monomer in samples obtained after normal saline (lane 1) and primarily as a dimer after bleomycin treatment (lane 2). The immunoblots are representative of experiments done 6 times. The numbers on the left denote the molecular weight of the bands in kilodaltons.
Figure 5.
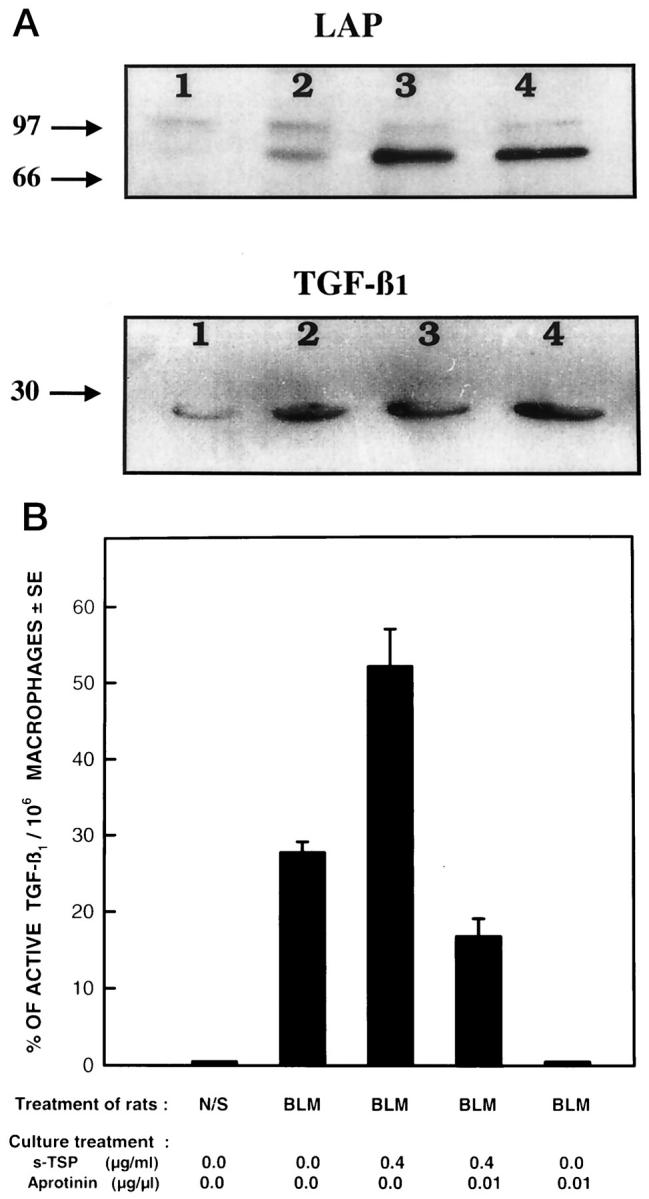
The effects of plasmin inhibitors on the expression of LAP, TGF-β1 and activation of L-TGF-β1. Immunoblot analysis of LAP and TGF-β1 antigens obtained from immunoprecipitates using anti-TSP-1 antibody (mAb133) and lysates of alveolar macrophages cultured in the absence or presence of α2-antiplasmin or aprotinin. A, top: Alveolar macrophages obtained 7 days after bleomycin administration cultured in the absence (lane 1) or presence of α2-antiplasmin at 10−4 units/ml (lane 2), α2-antiplasmin 10−1 unit/ml (lane 3), or aprotinin 0.01 μg/μm l (lane 4) were immunoblotted with anti-LAP antibodies and demonstrate protein bands compatible with L-TGF-β (100 kd) and the dimer (68 kd) of LAP. A, bottom: The same nitrocellulose filter was reprobed for immunoblotting with anti-TGF-β1 antibodies and demonstrates the dimer (25 kd) of TGF-β1. The culture conditions of alveolar macrophages used for lanes 1–4 are described above. The immunoblots are representative of experiments done 3 times. The numbers on the left denote the molecular weight of the bands in kilodaltons. B: Effects of aprotinin on the activation of L-TGF-β1 in the presence of sTSP-1. Alveolar macrophages obtained 7 days after bleomycin administration were cultured in the absence or presence of 0.01 μg/μm l/10 6 macrophages of aprotinin and/or in the absence or presence of 0.4 μg/ml/10 6 macrophages of sTSP-1. The TGF-β1 present in neutral and acidified then neutralized CM was quantitated and the percentage of active TGF-β1 was calculated. Each point is the mean of 3 to 7 rats. Percentage of active TGF-β1 in CM of alveolar macrophages from BLM-treated rats compared to normal saline N/S-treated rats or the CM of alveolar macrophages obtained after BLM treatment cultured with aprotinin have a P value ≤0.0001. Percentage of active TGF-β1 in CM of alveolar macrophages cultured in the presence of sTSP-1 compared to when no TSP-1 or aprotinin were present has a P value ≤0.0004. Percentage of active TGF-β1 in CM of alveolar macrophages obtained after BLM treatment and no in vitro treatment, compared to when sTSP-1 and aprotinin was also present, has a P value ≤0.0976. Percentage of active TGF-β1 in CM of alveolar macrophages cultured with sTSP-1 compared to when sTSP-1 and aprotinin were present has a P value <0.0001.
It is of note that the immunofluorescent localization of TGF-β1 on alveolar macrophages obtained after bleomycin administration compared to normal saline treatment demonstrates that TGF-β1 is extensively and diffusely present on the cell surface (Figure 3D) ▶ . Alveolar macrophages obtained for these experiments were immediately placed in the fixative (3.7% formaldehyde), permitting little time for plasmin-mediated release of TGF-β1. Furthermore, the immunofluorescent procedure detects TGF-β1 present on the cell by its association with TSP-1 and CD36 as well as with its own binding proteins, such as the TGF-β receptor type II. The colocalization of TGF-β1 with CD36 (Figure 3F) ▶ represents TGF-β1 associated with TSP-1, which is not as extensive in its distribution as TGF-β1 (Figure 3D) ▶ or CD36 (Figure 3E) ▶ . For the experiments shown in Figure 4 ▶ , to obtain enough alveolar macrophages for immunoprecipitation studies, the lavage procedure was prolonged and may have resulted in the release of TGF-β1 by plasmin generated by the same cells. 5 Unlike the immunofluorescent detection of all cell surface-associated TGF-β1 in these experiments, the immunodetection of TGF-β1 was restricted to that associated with TSP-1 and susceptible to plasmin-mediated release. For these reasons, after bleomycin injury compared to normal saline treatment the TGF-β1 detected by immunofluorescence on alveolar macrophages is extensive (Figure 3D) ▶ , whereas that detected by Western blot analysis after immunoprecipitation with TSP-1 is decreased (Figure 4) ▶ .
The Role of Plasmin in Activation of Alveolar Macrophage-Derived L-TGF- β1
In our previous work we had demonstrated that the posttranslational activation of alveolar macrophage-derived L-TGF-β1 was dependent on the generation of plasmin by the same cells. 5 Plasmin activates L-TGF- β1 by releasing the mature form of TGF- β1 from its noncovalent association with its LAP. 4 The effects of plasmin can be inhibited by α2-antiplasmin and aprotinin. 12,23 α2-Antiplasmin is a natural inhibitor of plasmin, whereas aprotinin inhibits the effects of plasmin as well as trypsin, α-chymotrypsin, and kallikrein. 23 The L-TGF-β1 associated with TSP-1 is also released by plasmin because when plasmin was inactivated by the presence of α2-antiplasmin (lanes 2 and 3) or aprotinin (lane 3), LAP-1 and TGF-β1 remained complexed with TSP-1 (Figure 5A) ▶ . However, when plasmin activity was not neutralized by α2-antiplasmin or aprotinin, there was a decreased quantity of LAP-1 and TGF-β1 associated with TSP-1 (Figure 5A ▶ , lane 1). In addition we found that in lanes 1–4 using anti-LAP-1 antibodies a band was visible at 100 kd corresponding to bands observed on Western blot analysis to the presence of LAP-1 associated with TGF-β1. 24-26 The detection of a band using anti-LAP-1 antibody at 68 kd has previously been confirmed to correspond to the dimer of LAP-1. 22 Although our findings confirm that plasmin can release TGF-β1 and thus diminish the quantity of TGF-β1 detected in association with TSP-1 (lane 1), we have also demonstrated that the prolonged presence of plasmin in overnight cultures of alveolar macrophages can release or degrade the LAP associated with TSP-1 (lane 1). The mechanism by which plasmin may regulate the association of LAP with TSP-1 is currently under investigation. Because TSP-1 can itself activate L-TGF-β1, we next determined if the activation of alveolar macrophage derived L-TGF-β1 by sTSP-1 also required the presence of plasmin. sTSP-1 added to CM in the presence of alveolar macrophages further increased the amount of active TGF-β1 generated (Figures 1C, 2C, and 5B) ▶ ▶ ▶ . It is of interest that the effects of α2-antiplasmin on the reduction of TGF-β activity in the presence of TSP-1 (not shown) was the same as that of aprotinin. Although, in addition to plasmin, aprotinin also inhibits the effects of trypsin, α-chymotrypsin, and kallikrein, plasmin is the only protease consistently reported to be generated by macrophages. Therefore, the effects of aprotinin in these experiments most likely results from inhibition of plasmin activity. In the presence of aprotinin or α2-antiplasmin (not shown), which inhibit plasmin activity, the induction of activation of L-TGF-β1 by sTSP-1 was significantly diminished but not totally abolished. It then follows that if the activation of L-TGF-β1 was totally dependent on sTSP-1, then the absence or presence of a plasmin inhibitor would not affect the activation of L-TGF-β1 by sTSP-1. These findings confirm that TSP-1, as well as plasmin, is required to activate alveolar macrophage-derived L-TGF- β1. These findings also demonstrate that the LAP-1 and TGF-β1 associated with TSP-1, which is complexed with its receptor, CD36, are the targets for plasmin activity. Because sTSP-1, anti-TSP-1, and CD36 peptide 93–110 have no effect on the generation of plasmin (D Xu, T Yehualaeshet, and N Khalil, manuscript in preparation) the regulation of activation of L-TGF-β1 by sTSP-1, anti-TSP-1, or CD36 peptide is not mediated by their effects on plasmin. Collectively, these findings support a model for the posttranslational activation of L-TGF- β1 where L-TGF- β1 is complexed with TSP-1 and held at the cell surface when TSP-1 interacts with its TSP-1 receptor, CD36. Cell surface localization of L-TGF-β1 favors plasmin-mediated release of mature TGF-β1 (Figure 6) ▶ .
Figure 6.

Proposed model for the activation of alveolar macrophage-derived L-TGF-β1. Resting alveolar macrophages secrete small amounts of L-TGF-β1 and TSP-1 but no plasmin. CD36 and TGF-β1 are present in small quantities on the cell surface, but no active TGF-β1 is produced. After bleomycin-induced lung injury, the alveolar macrophages are activated to secrete increased quantities of L-TGF-β1 and TSP-1 and generate increased quantities of plasmin. TSP-1 associates with the alveolar macrophage-derived L-TGF-β1 released in the immediate vicinity of the macrophage. The TSP-1/L-TGF-β1 complex then associates with the cell surface of the alveolar macrophage by the CD36 receptor. After association of CD36 with the TSP-1/L-TGF-β1 complex, the plasmin generated by the macrophages releases the TGF-β1 from the LAP. TGF-β1 is then available to react with its receptor and have a biological effect. Plgn, plasminogen; pm, plasmin; Plgn-R, plasminogen receptor.
Discussion
We have examined the posttranslational activation of alveolar macrophage-derived L-TGF-β1 in a well recognized model of lung injury and fibrosis induced by the antineoplastic antibiotic, bleomycin. 5,9,10 After bleomycin induced lung injury there was an increase in total lung TGF-β content, which was due to overproduction of TGF-β1 primarily synthesized by the alveolar macrophages. 5,9,10 Furthermore, these macrophages not only were induced to produce latent TGF- β1, but also were able to convert it to an active form. 5,10 In the present study we have demonstrated that the increased secretion of TSP-1 by alveolar macrophages is important for the activation of L-TGF- β1.
Although the thrombospondins exist in five isoforms, 16 the activation of L-TGF-β1 is a property unique to the TSP-1 isoform. 27 In earlier work, it was demonstrated that L-TGF-β1 present in CM from bovine aortic endothelial cells was activated in solution by the addition of sTSP-1. 11,27 The WSXW motif present in the type 1 repeats of TSP-1 were involved in the recognition of the mature form of TGF-β1. 11,27 After binding the WSXW region, the sequence of RFK, which is unique to TSP-1, interacted with a site at the amino terminus of L-TGF-β1, resulting in generation of bioactive TGF-β1 11,27 (and S Schultz-Cherry and J Murphy-Ullrich, unpublished data). This activation was independent of cell surface association and plasmin. 11,27 Similar to the findings of Arnoletti 28 and Tuszynski, 29 our study demonstrates that although TSP-1 is important in the activation of L-TGF-β1 from activated alveolar macrophages, the process requires the presence of cells and plasmin. 5 In contrast to our earlier work, 11,27 the addition of sTSP-1 to alveolar macrophage-derived CM diminished the TGF-β1 detected. The difference in the effects of sTSP-1 on L-TGF-β1 in solution may be related in part to the properties of the CM in the two studies. Unlike bovine aortic endothelial cells, after bleomycin injury, alveolar macrophages release a number of substances that could either degrade the sTSP-1 and TGF-β1 or interfere with the association of sTSP-1 to L-TGF-β1. For example, activated macrophages release plasmin, 5 cathepsin G, 30 and elastase. 31 These proteases are likely to be present in the CM from activated macrophages and may degrade the TGF-β1 that is present in the CM. We know from previous experiments 5 that in the absence of protease inhibitors leupeptin, pepstatin, and aprotinin, the TGF-β1 present in CM is not stable and the TGF-β1 activity deteriorates rapidly in CM (data not shown). Furthermore, the ubiquitous antiprotease α2-macroglobulin, which is released by activated macrophages, 32 has been demonstrated to complex with both active and latent TGF-β1. 33 The active TGF-β1 associated with α2-macroglobulin is not detectable in neutral CM by the CCL-64 assay. 33
It is also possible that substances in the CM from activated alveolar macrophages affect TSP-1. For example, TSP-1 binds a number of proteases such as plasmin, 34 cathepsin G, 35 and elastase, 36 which may be present in our CM. Soon after binding these proteases, TSP-1 undergoes proteolytic degradation, 34-36 making less of it available to activate L-TGF-β1. Furthermore, there may be substances in the CM that could interfere with the association of TSP-1 to the L-TGF-β1. For example, the WSXW sequence in the type-1 repeats of TSP-1 that mediate binding of the TSP-1 to TGF-β1 is close to the heparin 37 and fibronectin 38 binding regions. Both molecules are released from activated macrophages 39,40 and would be expected to be present in the alveolar macrophage CM. Binding of heparin or fibronectin to sTSP-1 could possibly interfere with the association of sTSP-1 with L-TGF-β1 and thus with activation of L-TGF-β1. In addition, TSP-1 can also bind to α2-macroglobulin 33 that is released by activated macrophages. The binding of sTSP-1 to α2-macroglobulin would diminish the quantity of TSP-1 available to activate L-TGF-β1. Association of TSP-1 or TGF-β1 with α2-macroglobulin and the proteolytic affects of proteases could explain the diminished quantity of TGF-β1 detected in cell-free CM after sTSP-1 was added. Lastly, although both L-TGF-β1 and sTSP-1 are highly conserved proteins, it is possible that there may be conformational differences between bovine and rat L-TGF-β1 and TSP-1, leading to differences in the association of sTSP-1 with L-TGF-β1 and in the activation process.
It has been demonstrated that the efficiency of protease-substrate interactions is increased if the substrate is anchored to the surface of the cell producing the protease. 41 In our model the presence of a cell surface is important for processing of alveolar macrophage-derived L-TGF- β1 by plasmin. However the mechanism of localization of L-TGF-β1 to the cell surface is unique and requires that TSP-1 complexed with L-TGF-β1 interact at the cell surface. TSP-1 not only forms complexes with L-TGF- β1, but binds to the cell surface proteins CD36 18,19 and CD51. 42 Unlike CD51, CD36, which is an 88-kd membrane glycoprotein, is consistently found on the surfaces of monocytes and macrophages. 18,19 No previous report has demonstrated that CD36 binds TGF-β1, but CD36 expression functions as a receptor for TSP-1, 43 collagen, 43 and a ligand exposed on the surface of erythrocytes infected with the parasite Plasmodium falciparum. 43 The CD36-TSP-1 interaction has been reported to be involved in platelet-monocyte adhesion, 44 platelet-tumor-cell adhesion, 45 platelet aggregation, 46 and macrophage uptake of apoptotic cells. 47 The findings in this study demonstrate for the first time that the CD36-TSP-1 interaction is important in the activation of alveolar macrophage-derived L-TGF- β1. This is because the presence of an antibody to CD36 in cultures of activated alveolar macrophages diminishes TGF-β1 localization to the cell surface and totally abrogates the activation of L-TGF-β1. Because CD36 has not been reported to bind L-TGF- β1, the colocalization of TGF-β1 with CD36 must occur when TSP-1 that is complexed with L-TGF- β1 interacts with its receptor, CD36. There was further confirmation that the interaction of TSP-1 to CD36 is important for activation of L-TGF-β1, in that the presence of the CD36 antibody in cultures of alveolar macrophages incubated with sTSP-1 resulted in inhibition of the enhanced activation of L-TGF- β1 observed when sTSP-1 alone was added to alveolar macrophages.
It is of interest that a peptide mimicking the region of CD36 between amino acids 93 and 110, previously described as critical for the interaction of TSP-1 with CD36, 14 also diminished the generation of active TGF-β1 in vitro. In addition, when the CD36 peptide 93–110 is administered to rats concomitantly with bleomycin, alveolar macrophages do not generate active TGF-β1, 48 and there is less inflammation and fibrosis in the lungs 48 (T Yehualaeshet, R O’Connor, A Begleiter, J Murphy-Ullrich, R Silverstein, N Khalil, manuscript in preparation). These observations suggest that the interaction of TSP-1 with L-TGF-β1 is important for activation of alveolar macrophage-derived L-TGF- β1 in vivo and may subsequently regulate pulmonary inflammation and fibrosis. Even though the interaction of TSP-1 and CD36 is necessary for the activation of L-TGF-β1, the plasmin generated by the same macrophages is also important for activation of L-TGF-β1, because plasmin releases LAP-1 and TGF-β1complexed with TSP-1. Although TSP-1 can itself activate L-TGF-β1, our findings suggest that after a pulmonary injury, activated alveolar macrophages generate TSP-1 that functions to localize L-TGF- β1 to their cell surfaces. Localization of L-TGF-β1 to the alveolar macrophages’ cell surface must lead to optimization of conditions that favor activation of L-TGF-β1 by plasmin that is released into the vicinity of the alveolar macrophages. 5 Furthermore, because plasmin can also be bound to the cell surface, 41 the association of L-TGF-β1 to the cell surface would lead to localizing L-TGF-β1 in close proximity to the cell surface-bound plasmin and thus facilitate the release of L-TGF-β1 by the actions of plasmin.
Several types of cells in the lung, such as endothelial cells, 49 epithelial cells, 50 and fibroblasts, 51 can be sources of TGF-β after bleomycin injury. It is of interest that recently Munger et al 52 described a protease- and TSP-1-independent activation of L-TGF-β1 in bleomycin-induced lung injury in mice. 52 Their findings suggest that the arginine-glycine-aspartic acid sequences in the LAP-1 of TGF-β1 associates with the integrin αvβ6 on alveolar epithelial cells. Munger et al have proposed a model of activation of L-TGF-β1 wherein the interaction of L-TGF-β1 with αvβ6 is followed by the association of αvβ6 with the actin cytoskeleton, leading to a conformational change in L-TGF-β1 attached to the cell surface. The conformational change in L-TGF-β1 leads to the mature TGF-β1 interacting with its receptor, TGF-β receptor type II. 52 It is not known if bleomycin administration to rats, a different species of rodent, regulates αvβ6 expression on alveolar epithelial cells, nor whether αvβ6 is important in the activation of L-TGF-β1 in our model. Nonetheless, because the presence of active TGF-β1 parallels the inflammatory changes seen in this model, 5,9,10 our findings suggest that alveolar macrophages may be an additional source of active TGF-β1. In conclusion, our findings demonstrate that plasmin 5 and TSP-1, which are diminished early in the bleomycin injury response, may result in terminating the activation of alveolar macrophage-derived L-TGF- β1, as well as its inflammatory and fibrotic effects. This then suggests that the regulation of inflammation that is mediated by TGF-β1 is dependent on the posttranslational activation of alveolar macrophage-derived L-TGF-β1 by both plasmin and TSP-1.
Acknowledgments
We thank Arnold H. Greenberg for many helpful discussions and review of the manuscript; Carol Whitman, Angela Kemp, and Antonio Pallero for technical assistance; Dr. Bob Tate for the statistical analysis of the data; and Arlene Klassen and Heather Buhler for typing the manuscript.
Footnotes
Address reprint requests to Dr. Nasreen Khalil, B.C. Centre for Disease Control, University of British Columbia, 655 West 12th Avenue, Vancouver, British Columbia, V5Z 4R4 Canada.
Supported by the Medical Research Council of Canada and the Respiratory Health Network of Centers of Excellence (to N. K.) and National Institutes of Health HL50061 (to J. E. M.-U.).
References
- 1.Rappolee DA, Werb Z: Macrophage derived growth factors. Curr Topics Microbiol Immumol 1992, 181:87-140 [DOI] [PubMed] [Google Scholar]
- 2.Stein M, Keshav A: The versatility of macrophages. Clin Exp Allergy 1992, 22:19-27 [DOI] [PubMed] [Google Scholar]
- 3.Wahl SM: Transforming growth factor β: the good, the bad and the ugly. J Exp Med 1994, 108:1587-1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gleizes PE, Munger JS, Nunes I, Harpel JG, Mazzieri R, Noguera S, Rifkin DB: TGF-β latency: biological significance and mechanism of activation. Stem Cells 1997, 15:190-197 [DOI] [PubMed] [Google Scholar]
- 5.Khalil N, Corne S, Whitman C, Yacyshyn H: Plasmin regulates the activation of cell associated latent TGF-β1 secreted by rat alveolar macrophages after in vivo bleomycin injury. Am J Resp Mol Cell Biol 1996, 15:252-259 [DOI] [PubMed] [Google Scholar]
- 6.Schultz-Cherry S, Murphy-Ullrich JE: Thrombospondin causes activation of latent transforming growth factor-β secreted by endothelial cells by a novel mechanism. J Cell Biol 1993, 122:923-932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murphy-Ullrich JC, Schultz-Cherry S, Hook M: Transforming growth factor-β complexes with thrombospondin. Mol Biol Cell 1992, 3:181-188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Silverstein RL, La Salla J, Pearce SE: CD36 cluster report. Leucocyte Typing V: White Cell Differentiation Antigens. Edited by Schlossman SF, Boumsell L, Gilks W, Harlan JM, Kishimoto T, Morimoto C, Ritz O, Silverstein RL, Springer TA, Tedder TF, and Todd RF. Oxford, Oxford University Press, 1995, 1271:1274–1275
- 9.Khalil N, Bereznay O, Sporn MB, Greenberg AH: Macrophage production of transforming growth factor-β and fibroblast collagen synthesis in chronic pulmonary inflammation. J Exp Med 1989, 170:727-737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khalil N, Whitman C, Zuo L, Danielpour D, Greenberg AH: Regulation of alveolar macrophage transforming growth factor-β secretion by corticosteroids in bleomycin-induced pulmonary inflammation in the rat. J Clin Invest 1993, 92:1812-1818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schultz-Cherry S, Lawler J, Murphy-Ullrich JE: The type-1 repeats of thrombospondin-1 activates latent transforming growth factor-β. J Biol Chem 1994, 269:26783-26788 [PubMed] [Google Scholar]
- 12.Witman B: On the reaction of plasmin or plasmin-streptokinase complex with aprotinin or α2-antiplasmin. Thromb Res 1980, 17:143-152 [DOI] [PubMed] [Google Scholar]
- 13.Danielpour D, Hart LL, Flanders KC, Roberts AB, Sporn MB: Immunodetection and quantitation of the two forms of transforming growth factor-β (TGF-β1 and TGF-β2) secreted by cells in culture. J Cell Physiol 1989, 138:78-86 [DOI] [PubMed] [Google Scholar]
- 14.Leung LL, Wei-Xing L, McGregor JL, Albrecht G, Howard RJ: CD36 peptides enhance or inhibit CD36 thrombospondin binding: a two-step process of ligand receptor interaction. J Biol Chem 1992, 267:18244-18250 [PubMed] [Google Scholar]
- 15.Mai S: Overexpression of C-myc precedes amplification of gene encoding dihydrofolate reductase. Gene 1994, 148:253-260 [DOI] [PubMed] [Google Scholar]
- 16.Bornstein P: Thrombospondins: structure and regulation of expression. FASEB J 1992, 6:3290-3299 [DOI] [PubMed] [Google Scholar]
- 17.Yee JA, Yan L, Dominguez JC, Allan EH, Martin TJ: Plasminogen-dependent activation of latent transforming growth factor β (TGF-β) by growing cultures of osteoblast-like cells. J Cell Physiol 1993, 157:528-534 [DOI] [PubMed] [Google Scholar]
- 18.Huh YH, Pearch SF, Yesner LM, Schindler JL, Silverstein RL: Regulating expression of CD36 during monocyte-to-macrophage differentiation: potential role of CD36 in foam cell formation. Blood 1996, 87:2020-2028 [PubMed] [Google Scholar]
- 19.Yesner LM, Huh HY, Pearch SF, Silverstein RL: Regulation of monocyte CD36 and thrombospondin-1 expression by soluble mediators. Arterioscler Thromb Vasc Biol 1996, 16:1019-1025 [DOI] [PubMed] [Google Scholar]
- 20.Trezzini C, Jungi TW, Spycher MD, Maly FE, Rao P: Human monocytes CD36 and CD16 are signalling molecules: evidence from studies using antibody-induced chemiluminescence as a tool to probe signal transduction. Immunology 1990, 71:29-37 [PMC free article] [PubMed] [Google Scholar]
- 21.Huang MM, Bolen JB, Barnewell JW, Shattil SJ, Brugge JS: Membrance glycoprotein IV (CD36) is physically associated with Fyn, Lyn and Yes protein-tyrosine kinases in human platelets. Proc Natl Acad Sci USA 1991, 88:7844-7848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang Y, Dignam JD, Gentry LE: Role of carbohydrate structures in the binding of β1-latency-associated peptide to ligands. Biochem 1997, 36:11923-11932 [DOI] [PubMed] [Google Scholar]
- 23.Huber D, Philipp J, Fontana A: Protease Inhibitors interfere with the transforming growth factor-β-dependent but not the transforming growth factor-β-independent pathway of tumor cell-mediated immunosuppression. J Immunol 1992, 148:277-284 [PubMed] [Google Scholar]
- 24.Olofsson A, Miyazono K, Kanzaki T, Coloselti P, Engstrom U, Heldin C-H: Transforming growth factor-β1, -β2, and -β3 secreted by a human glioblastoma cell line: identification of small and different forms of large latent complexes. J Biol Chem 1992, 267:1948-1958 [PubMed] [Google Scholar]
- 25.Mcmahon GA, Dignam JD, Gentry LE: Structural characterization of the latent complex between transforming growth factor β1 and β1-latency-associated peptide. Biochem J 1996, 313:343-351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Okada F, Yamaguchi K, Ichihara A, Nakamura T: Purification and structural analysis of the latent form of transforming growth factor-β from rat platelets. J Biochem 1989, 106:304-310 [DOI] [PubMed] [Google Scholar]
- 27.Schultz-Cherry S, Chen H, Mosher DF, Misenheimer TM, Krutzsch HC, Roberts DD, Murphy-Ullrich JE: Regulation of transforming growth factor-β activation by discrete sequences of thrombospondin-1. J Biol Chem 1995, 270:7304-7310 [DOI] [PubMed] [Google Scholar]
- 28.Arnoletti JP, Albo D, Granick MS, Solomon MP, Rothman NL, Tuszynski AB: Thrombospondin and transforming growth factor β-1 increase expression of urokinase-type plasminogen activator and plasminogen activator inhibitor 1 in human MDA-MB-231 breast cancer cells. Cancer 1995, 76:998-1005 [DOI] [PubMed] [Google Scholar]
- 29.Tuszynski GP, Nicosia RF: The role of thrombospondin-1 in tumor progression and angiogenesis. BioEssays 1996, 18:71-75 [DOI] [PubMed] [Google Scholar]
- 30.Welgus HG, Connolly NL, Senior RM: 12-0-Tetradecanoyl-phorbol-13-acetate-differentiated U937 cells express a macrophage-like profile of neutral proteinases: high levels of secreted collagenase and collagenase inhibitors accompany low levels of intracellular elastase and cathepsin-G. J Clin Invest 1986, 77:1675-1681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Valentine R, Fisher GL: Characteristics of bovine alveolar macrophage elastase. J Leukocyte Biol 1984, 35:449-457 [DOI] [PubMed] [Google Scholar]
- 32.Bonner JC, Hoffman M, Brody AR: Alpha2-macroglobulin secreted by alveolar macrophages serves as a binding protein for a macrophage-derived homologue of platelet-derived growth factor. Am J Respir Cell Mol Biol 1989, 1:171-179 [DOI] [PubMed] [Google Scholar]
- 33.Souchelnitskiy S, Chambaz EM, Feige JJ: Thrombospondins selectively active one of the two latent forms of transforming growth factor-β present in adrenocortical cell-conditioned medium. Endocrine 1995, 136:5118-5126 [DOI] [PubMed] [Google Scholar]
- 34.Anonick PK, Yoo JK, Webb DJ, Gonias SL: Characterization of the antiplasmin activity of human thrombospondin-1 in solution. Biochem J 1993, 289:903-909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rabhi-Sabile S, Pidard D, Lawler J, Renesto P, Chignard M, Legrand C: Proeolysis of thrombospondin during cathepsin-G induced platelet aggregation: functional role of the 165-kd carboxy-terminal fragment. FEBS Lett 1996, 386:82-88 [DOI] [PubMed] [Google Scholar]
- 36.Hogg PJ, Owensby DA, Mosher DF, Misenheimer TM, Chesterman CN: Thrombospondin is a tight-binding, competitive inhibitor of neutrophil elastase. J Biol Chem 1993, 268:7139-7146 [PubMed] [Google Scholar]
- 37.Murphy-Ullrich JE, Giurusiddappa S, Franzier WA, Hook M: Heparin-binding peptides from thrombospondins 1 and 2 contain focal adhesion-labilizing activity. J Biol Chem 1993, 268:26784-26789 [PubMed] [Google Scholar]
- 38.Sipes JM, Giuo N, Negre E, Yogel T, Krutzsch HC, Roberts DD: Inhibition of fibronectin binding and fibronectin-mediated cell adhesion to collagen by a peptide from the second type I repeats of thrombospondin. J Cell Biol 1993, 121:469-477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kolset SO, Larsen T: Sulphur-containing macromolecules in cultured monocyte-like cells. Acta Histochem 1988, 84:67-75 [DOI] [PubMed] [Google Scholar]
- 40.Driscoll KE, Maurer JK, Higgins J, Poynter J: Alveolar macrophage cytokine and growth factor production in a rat model of crocidolite-induced pulmonary inflammation and fibrosis. J Toxicol Environ Health 1995, 46:155-169 [DOI] [PubMed] [Google Scholar]
- 41.Vassalli JD, Wohlwend A, Belin D: Urokinase-catalyzed plasminogen activation at the monocyte/macrophage cell surface: a localized and regulated proteolylic system. Curr Topics Microbiol Immunol 1992, 181:65-86 [DOI] [PubMed] [Google Scholar]
- 42.Clezardin P, Frappart L, Clerget M, Pechoun C, Delmas PD: Expression of thrombospondin (TSP-1) and its receptors (CD36 and 51) in normal, hyperplastic and neoplastic human breast. Cancer Res 1993, 53:1421-1430 [PubMed] [Google Scholar]
- 43.Daviet L, Mcgregor JT: Vascular biology of CD36: role of this new adhesion molecule family in different disease states. Thromb Haemost 1997, 78:65-69 [PubMed] [Google Scholar]
- 44.Silverstein RL, Asch AS, Nachman RL: GPIV mediates thrombospondin-dependent platelet-monocyte adhesion. J Clin Invest 1989, 84:546-552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Silverstein RL, Baird M, Lo SK, Yesner LM: Sense and antisense cDNA transfection of CD36 (glycoprotein IV) in melanoma cells. J Biol Chem 1992, 267:16607-16612 [PubMed] [Google Scholar]
- 46.Leung LL: Role of thrombospondin in platelet aggregation. J Clin Invest 1984, 74:1764-1772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ren Y, Silverstein RL, Allen J, Savill J: CD36 gene transfer confers capacity for phagocytosis of cells undergoing apoptosis: J Exp Med 1995, 181:1857-1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yehaulaeshet T, Douglas D, O’Connor R, Khalil N: In vitro and in vivo inhibition of activation of alveolar macrophage-derived latent transforming growth factor-β 1 (TGF-β1) after bleomycin lung injury. Am J Respir Crit Care Med 1998, 157:A265(abstr.) [Google Scholar]
- 49.Phan SH, Gharaee-Kermani M, Wolber F, Ryan US: Stimulation of rat endothelial cell transforming growth factor-β production by bleomycin. J Clin Invest 1991, 87:148-154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Khalil N, O’Connor R, Flanders K, Shing W, Whitman C: Autocrine regulation of type-2 epithelial cell proliferation by transforming growth factor-β during bleomycin induced lung injury in the rat. Am J Physiol 1994, 77:L498-507 [DOI] [PubMed] [Google Scholar]
- 51.Cutroneo K, Breen E, Absher M, Phan S: Bleomycin regulation of transforming growth factor-β mRNA in rat lung fibroblasts and subpopulations. Chest 1991, 99:65(suppl) [DOI] [PubMed] [Google Scholar]
- 52.Munger J, Huang X, Kawakatsu H, Griffiths MJD, Datton SL, Wu J, Pittet J-F, Kaminski N, Garat C, Mathay MA, Rifkin DB, Sheppard D: The integrin αvβ6 binds and activates TGF-β1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999, 96:319-328 [DOI] [PubMed] [Google Scholar]