

Abstract
In this study we describe the generation of a transgenic mouse model with neuronal overexpression of the human cyclooxygenase-2, h(COX)-2, to explore its role in excitotoxicity. We report that overexpression of neuronal hCOX-2 potentiates the intensity and lethality of kainic acid excitotoxicity in coincidence with potentiation of expression of the immediate early genes c-fos and zif-268. In vitro studies extended the in vivo findings and revealed that glutamate excitotoxicity is potentiated in primary cortico-hippocampal neurons derived from hCOX-2 transgenic mice, possibly through potentiation of mitochondrial impairment. This study is the first to demonstrate a cause-effect relationship between neuronal COX-2 expression and excitotoxicity. This model system will allow the systematic examination of the role of COX-2 in mechanisms of neurodegeneration that involve excitatory amino acid pathways.
Cyclooxygenase (COX) is the rate-limiting enzyme in the production of prostaglandins and, as such, a key target for anti-inflammatory drugs. Indeed, aspirin and other nonsteroidal anti-inflammatory drugs (NSAIDs) are COX inhibitors. 1,2 There are two known isoforms, COX-1 and COX-2, which have quite distinct expression patterns and biological activities. COX-1 is a constitutively expressed protein found in most tissues, whereas COX-2 expression can be induced by a variety of mitogens, including cytokines, hormones, and phorbol esters. 3,4 Inflammatory stimuli have little effect on COX-1 expression, but lead to a rapid rise in COX-2 expression, suggesting a primary role for COX-2 in inflammation. 4,5
Recent studies of the effects of NSAIDs have revealed several possible health benefits of these COX inhibitors. Regular use of NSAIDs appears to reduce the risk of colorectal cancer 6-9 and, more important for the current study, may slow the course of dementia in Alzheimer’s disease (AD). 10 Although it is unclear whether inflammation plays a role in the progression of AD, several lines of evidence indicate that regulation of COX-2 but not COX-1 expression may be involved in some of the neuropathology associated with AD. 11
In rodents, the response to kainic acid (KA)-mediated excitotoxicity, which may be a model for aspects of the hippocampal and cortical neurodegeneration observed in AD, includes a marked up-regulation of COX-2, although COX-1 remains unaffected. 12 The induction of COX-2 expression parallels the appearance of neuronal apoptotic features in cell types affected by KA, and excitotoxic neuronal death in vitro is accompanied by a selective elevation in COX-2 mRNA, indicating that COX-2 may be involved in pathways leading to neuronal death. 12 This information may have important implications for AD in view of the evidence that excitotoxicity may contribute to the widespread pattern of neurodegeneration in the AD brain. 13 Recently we 14 and others 15 found elevation of expression of COX-2, but not COX-1, in neurons of the AD brain.
For the present study, we prepared transgenic mice with neuronal overexpression of the human COX-2 gene, hCOX-2, under the regulation of the rat neuron-specific enolase (NSE) promoter. We used this model to explore the role of neuronal COX-2 expression in KA-mediated excitotoxicity. Moreover, because KA excitotoxicity has profound effects on gene regulation associated with long-lasting cellular responses in rodent brain, 16 we examined the expression of representative immediate early genes (IEG) as an index of cellular response to synaptic activation. We report that overexpression of neuronal COX-2 potentiates the intensity and lethality of KA-mediated seizures in heterozygous hCOX-2 transgenic mice coincident with the potentiation of hippocampal c-fos and zif-268 mRNA expression. Finally, in vitro studies revealed that glutamate excitotoxicity was also potentiated in primary cortico-hippocampal neuron cultures derived from hCOX-2 transgenic mice.
Materials and Methods
Preparation of Transgene Construct
A 1950-bp HindIII cDNA fragment containing the entire coding region for hCOX-2 17 was inserted into a unique HindIII site located within the second exon of the rat neuron-specific enolase (rNSE) gene. This rNSE/hCOX-2 (NHC) transgene contains approximately 2600 bp of rNSE promoter and 5′ flanking region, all of exon 1 (nontranslated sequence), intron 1, and 6 bp of exon 2 (without an ATG starting codon) from the rat NSE gene. 18 In addition, the NHC transgene utilizes a polyadenylation signal from the SV40 late region (see Figure 1A ▶ ).
Figure 1.

NSE/hCOX-2 (NHC) transgene for neuronal overexpression of human COX-2. A: Construction of the NHC transgene is detailed in the Methods section. A schematic representation of the hybrid NHC transgene is presented in which a 4-kb promoter fragment (thin line and dark boxes) from the rat NSE gene was fused to the 1.9-kb hCOX2 cDNA fragment (open box) . The NSE promoter includes 5′ flanking sequences, exon 1, a 1.2-kb intron in the 5′ UTR, and 6 bp of exon 2; the exon sequences are represented by dark boxes. A 930-bp fragment containing the polyadenylation signal (A) from the SV40 late region (cross-hatched box) was fused to the 3′ end of the hCOX-2 cDNA. The location of two HpaI restriction endonuclease sites, which can be used to examine whether or not transgenic mice carry intact copies of the transgene, are indicated. A predicted mRNA of approximately 2500 nucleotides (not including the poly-A tail) is produced from this hybrid gene after splicing of the NSE intron (bottom line of figure). B: Northern analysis of transgenic mice from two independent NHC mouse lines, NHC5 and NHC32. hCOX-2 mRNA expression (top panel) was assessed by northern analysis of total RNA from a WT nontransgenic littermate, a NHC5, and a NHC32 transgenic animal using the entire hCOX-2 cDNA as a probe. The same blot was subsequently reprobed for the expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a control for sample loading (bottom panel). In each of the panels, the positions of 18S and 28S ribosomal RNA are as indicated. LI, liver; KI, kidney; BR, brain; SP, spleen. C: Western blot analysis of hCOX-2 expression in the brain of NHC32 transgenic mice. The expression of hCOX-2 protein in NHC32 mouse brain was assessed by Western blot immunoassay (see Methods). The same blot processed with anti-hCOX-2 was subsequently reacted to anti-β-actin antibody to control for sample loading. The position of molecular weight standards (97, 66, and 46 kd) are as indicated. The anti-hCOX-2 antibody used in this assay recognized an approximately 70-kd protein species from the NHC32 brain homogenate sample.
Purification of Transgene Fragment and Microinjection
A 6.95-kb SalI fragment containing the NHC transgene was purified from plasmid sequences by electrophoresis through low-melting-point agarose. The transgene was further purified by centrifugation through CsCl, followed by dialysis against 10 mmol/L Tris, pH 7.4, and 0.2 mmol/L EDTA. The concentration was adjusted to 5 Fg/ml for microinjection. The purified transgene fragment was used for microinjection of one-cell mouse eggs as previously described. 19 C57Bl/6 × C3H (B6C3) F1 hybrid mice (Taconic Farms, Germantown, NY) were used as the source of fertilized eggs at the pronuclear stage.
Genotyping
Genomic DNA was isolated from tail skin as previously described. 19 Transgenic mice produced from injection of the NHC SalI fragment were identified by dot blot hybridization of tail skin DNA samples with a random-primed, 930-bp EcoRI fragment, which contains the entire SV40 sequence present in the NHC transgene (see Figure 1A ▶ ). The transgenic founder mice identified in this fashion were mated with nontransgenic, B6C3 F1 hybrid mice to produce heterozygous transgenic offspring for establishment of the individual lines, as well as analyses of expression.
Northern Blot Analysis of hCOX-2 Transgene Expression
For the studies of hCOX-2 expression, half of the brain was frozen for extraction of total RNA 20 and the other half was processed for in situ hybridization (ISH) assay as described below. For gel blot Northern hybridization assay, total RNA was resolved electrophoretically through 1.1% agarose, 2.2 mol/L formaldehyde gel as previously described. 21 After transfer to nitrocellulose, expression of the hCOX-2 transgene was determined by hybridization with a random-primed, 1950-bp HindIII fragment containing the entire hCOX-2 cDNA fragment.
ISH Assay
Brain frozen coronal tissue sections (10 μm) were mounted on polylysine-coated slides and stored at −70°C. For ISH, tissue sections were postfixed in PBS containing 4% paraformaldehyde (30 minutes, room temperature), rinsed in phosphate-buffered saline (PBS), and incubated in acetic anhydride (0.25% v/v) in 0.1 mol/L triethanolamine, pH 8.0, for 10 minutes. After acetic anhydride treatment, tissue sections were hybridized with [35S]-cRNA probes (0.3 μg/ml, 2 × 10 9 dpm μg−1); hCOX-2 (1.9 kb hCOX-2 derived from the NHC construct (Figure 1A) ▶ , c-fos, zif-268, 22,23 and mouse (m) COX-2 and mCOX-1 12 were transcribed from linearized cDNA probes. After hybridization (3 hours, 50°C), stringency washes (0.1 × SSC, 60°C), and dehydration, slides were exposed to X-ray film for 5 days for quantification. Slides were then exposed to NTB-2 emulsion (Kodak, Rochester, NY) for approximately 12 days for microscopic analysis of mRNA distribution; after emulsion development, tissue sections were Nissl-counterstained. Hematoxylin and eosin histochemistry was used to assess morphological features of apoptosis on semiadjacent tissue sections. 24 Film autoradiograms were analyzed by computer-assisted video densitometry (Biometrics, Nashville, TN).
Combined ISH and Immunocytochemistry (ICC)
The neuronal expression of hCOX-2 transgene was verified by combining ISH for hCOX-2 mRNA with ICC for NSE on the same brain tissue section as previously described. 25 Following ISH, brain tissue sections were incubated for 30 minutes in PBS containing 10% methanol and 3% H2O2, rinsed, and permeabilized for 30 minutes at 4°C in PBS containing 0.2% Triton X-100, 0.2% normal goat serum, and 20 mg/ml L-lysine (Sigma, St. Louis, MO). Tissue sections were immunoreacted with a rabbit anti-human NSE antibody (1:1000, Dako, Carpinteria, CA). Immunoreactivities were visualized using the ABC method (Vector). Tissue samples were then dehydrated and coated with Kodak NTB2 emulsion as discussed above.
Western Blot Analysis
Brain samples were homogenized in an ice-cold buffer of Tris, pH 7.2, containing 1% NP-40, 0.5% sodium deoxycholate, 0.1% lauryl sulfate, 100 μg/ml phenylmethylsulfonyl fluorid, 3 μg/ml aprotinin, 3 μg/ml pepstatin A, 15 μg/ml propionyl-Leupeptin, and 600 μg/ml Perfabloc (Boehringer Mannheim, Indianapolis, IN), boiled, centrifuged, and electrophoresed on 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) as previously described. 26 Proteins were transferred to nylon Transblot membrane (Biorad, Hercules, CA) and immunoreacted with a polyclonal goat anti-hCOX-2 antibody (1:500 dilution, 3 hours, room temperature) in Superblock blocking solution (Pierce, Rockford, IL). The anti-hCOX-2 antibody was raised against a synthetic peptide derived from the carboxyl region of hCOX-2 sequence (Cayman, Ann Arbor, MI). In control studies we found that anti hCOX-2 antibodies do not cross-react with COX-1 (not shown). Immunoreactivity was visualized autoradiographically using a chemiluminescence detection kit (SuperSignal, Pierce). β-actin immunoreactivity (anti-β-actin, Sigma 1:5000) controlled for selectivity of changes.
KA-Mediated Seizures
KA-induced seizures were induced by intraventricular (ICV) infusion of KA (Sigma) in 4-month-old heterozygous hCOX-2 - NHC32 transgenic mice and control wild-type (WT) littermates, as previously described. 27 KA, 1.15 nmoles/2 μl, or saline was injected in the lateral ventricle of anesthetized mice (Metofan) using a 5-μl Hamilton syringe at a rate of 1μl/minute. Behavioral changes were assessed using a method described by Kondo et al, 28 modified for seizure assessment in mice. Seizures were classified into two groups: mild seizures, in which mice showed staring posture, occasional wet-dog shakes, or automatisms without any myoclonic twitches; and severe seizures, in which mice showed myoclonic twitches of the forelimbs with standing posture or generalized tonic-clonic seizures with standing postures or generalized tonic-clonic seizures with falling or wild seizure jumps.
Because death occurred within 10 minutes after ICV KA infusion in the hCOX-2 transgenic mice (see Results), for comparative analysis of changes in gene expression or neuropathology, the control WT mice were sacrificed at the same time postlesioning. After decapitation, brains were rapidly removed, immediately frozen in methylbutane cooled to −25°C, and then stored at −70°C.
Primary Neuronal Cultures
Cortico-hippocampal primary neuron cultures derived from heterozygous hCOX-2 transgenic and WT mouse embryos (embryonic day 14–16) were prepared as previously described. 26 Briefly, after brain dissection, mechanical trituration, and centrifugation, neurons were seeded onto poly-D-lysine-coated 96-well plates at a density of 2 × 10 5 cells per well. For ISH and/or ICC studies, primary neuron cultures were plated on glass chamber slides precoated with poly-D-lysine. For glutamate neurotoxicity, primary neuron cultures were aged 10 days and then treated with 50 μmol/L glutamate (Sigma) for 24 hours. Neuronal cultures were derived from each independent embryo; treatment and analysis were made in blind. Genotyping was used for identification of hCOX-2 transgenic or WT littermate cultures. Neuronal cultures were treated with glutamate for the indicated time, and parallel cultures derived from the same embryos were simultaneously treated with drug vehicle (control). Glutamate neurotoxicity was assessed by 3(4,5-dimethylthiazol-2-yl)−2,5-diphenyltetrazolium bromide (MTT) colorimetric assay; 14 changes in MTT activity for each independent embryo culture was expressed as a percentage of its own control (identical drug vehicle-treated cultures).
Statistics
Analysis used statistical analysis software (CSS, Statsoft). P values <0.05 were considered to indicate statistically significant differences (t-test). KA-induced seizures studies used n = 6–8 mice per group and the studies were replicated in 3 independent experiments. Fisher’s exact test was used for statistical analysis of mortality rate associated with KA-mediated seizures. ISH studies used n = 4–5 mice per group; changes in mRNA expression were computed from two tissue sections (bilaterally) encompassing the hippocampal formation. Data from in vitro studies were averaged from 3 to 4 independent studies.
Results
Production of Human hCOX-2-Overexpressing Transgenic Mice
Forty-four offspring were born from eggs microinjected with the NSE-hCOX-2 transgene and DNA samples prepared from tail skin biopsies were examined to identify founder transgenic mice. A dot blot containing tail DNA samples from all of the mice was hybridized with an SV40 probe (not shown), resulting in the identification of 5 founder transgenic mice (NHC1, NHC5, NHC30, NHC32, and NHC34) that carry the randomly integrated transgene.
Establishment of Transgenic Lines
All 5 founder transgenics were mated with WT mice to obtain transgenic offspring for establishment of the transgenic lines and for analysis of hCOX-2 expression. Offspring from these matings were genotyped by tail DNA dot blot analysis using an SV40 probe to detect the NHC transgene. Only the NHC5 and NHC32 founders transmitted the transgene to multiple offspring. In contrast, transmission from the NHC1 and NHC30 founders was much lower than expected for mendelian transmission of the transgene sequences (not shown).
Tissue-Specific Expression of hCOX-2 Transgene
Total RNA was prepared from various organs isolated from NHC5 and NHC32 transgenic offspring, as well as a WT nontransgenic littermate. Northern blot hybridization of total RNA indicated that, as expected, NHC5 and NHC32 transgenic mice express the hCOX-2 transgene mRNA only in the brain (Figure 1B) ▶ . In addition, the hCOX-2 hybridization signal indicates that the size of the major transcript in both lines is as predicted for the NHC gene (approximately 2500 nucleotides). A second, higher molecular weight transcript is also observed in both lines (Figure 1B) ▶ . Because this analysis was performed on total cellular RNA, the most likely interpretation is that the larger RNA represents hnRNA in which the 1200-bp NSE intron has not been spliced out, whereas the smaller major transcript represents the mature mRNA.
Expression of hCOX-2 protein in the NHC32−hCOX-2 mouse brain was assessed by Western blot immunoassay (Figure 1C) ▶ . The anti-hCOX-2 antibody used in this assay recognized an approximately 70-kd protein species as assessed by Western blot assay of whole NHC32-hCOX-2 brain homogenate (Figure 1C ▶ , top panel). β-actin immunoreactivity controlled for equal loading (Figure 1C ▶ , bottom panel). This result indicates that the NHC transgene is specifically expressed in the brain and that the mRNA transcribed from the transgene is of sufficient length to encode the entire hCOX-2 protein.
Cell Type Expression of hCOX-2
The regional distribution of hCOX-2 mRNA expression in the mouse brain of the NHC32-hCOX-2 transgenic mice was assessed by ISH using a [35S]-hCOX-2 antisense cRNA probe containing the entire hCOX-2 cDNA fragment and visualized by X-ray film autoradiography (Figure 2A) ▶ . ISH for hCOX-2 mRNA combined with NSE ICC on the same tissue section confirmed the neuronal expression of hCOX-2 mRNA in the brain of hCOX-2 transgenic mice (parietal cortex, NHC32 line; Figure 2C ▶ ); no detectable hCOX-2 signal was found in NSE-immunopositive neurons of WT mouse brain (Figure 2D) ▶ .
Figure 2.

In situ hybridization (ISH) analysis of human COX-2 in transgenic and WT mice. A: Regional distribution of hCOX-2 in the brain of NHC32-hCOX-2 transgenic mouse. B: WT control littermate (4 months old), as assessed by ISH assay for hCOX-2 mRNA and visualized by X-ray autoradiography, respectively. C: Colocalization of hCOX-2 mRNA hybridization signal with NSE-immunopositive neurons in hCOX-2 transgenic mouse brain. D: Lack of hCOX-2 mRNA hybridization signal in NSE-positive neurons of WT control brain. E: Optical densities were quantified by computerized video densitometry from film autoradiograms using a Bioquant image analysis system (Biometrics). Values are expressed as mean ± SE, *P < 0.001, hCOX-2 (NHC32) versus WT controls, n = 6–8 per group. CTX, parietal cortex; PYR, pyramidal layer of the hippocampal formation; DG, dentate gyrus of the hippocampal formation; WM, white matter; THL, thalamus; PKK, cerebellar Purkinje’s layer. Scale bars, 1 mm (A and B) and 8 μm C and D).
Quantitative ISH autoradiography in the brain of NHC32-hCOX-2 transgenic mice (Figure 2E) ▶ revealed greater than eightfold hCOX-2 mRNA expression in parietal cortex (CTX), the pyramidal CA3-CA1 (PYR) and dentate gyrus (DG) granule neuron layers of the hippocampal formation, thalamus (THL), cerebellar Purkinje cell layer (PRK) of NHC32-hCOX2 transgenic mice (Figure 2A) ▶ , relative to hCOX-2 hybridization signal in the brain of WT control littermates (Figure 2B) ▶ . No detectable expression of hCOX-2 was found in corpus callosum white matter (Figure 2A ▶ ; n = 6–8 per group, 4-month-old mice; P < 0.001). The intensity of hCOX-2 mRNA hybridization signal in the brain of WT control littermates (Figure 2B) ▶ approximated the intensity of the [35S]-hCOX-2 sense-strand hybridization signal on brain tissue sections of NHC32-hCOX-2 transgenics and WT controls (not shown).
hCOX-2 mRNA Expression Relative to Endogenous COX-2 mRNA and Expression of Endogenous COX-1 mRNA
Because of the constitutive expression of COX-2 mRNA in rodent brain, we examined the relative levels of hCOX-2 mRNA expression over endogenous murine (m) COX-2 mRNA; mCOX-2 mRNA expression was assessed by ISH using a murine [35S]-COX-2 antisense cRNA probe. Quantitative ISH revealed greater than three- to fourfold hCOX-2 mRNA expression in the DG granule and pyramidal (CA3-CA1) neuron layers of the hippocampal formation, parietal cortex, and thalamus over the mCOX-2 mRNA hybridization signal (Figure 3A) ▶ . In control studies, we found that mCOX-2 mRNA expression did not differ in control WT littermates and NHC32-hCOX-2 transgenics (not shown).
Figure 3.

hCOX-2 mRNA expression relative to mouse COX-2 mRNA in hCOX-2 transgenic mice and endogenous mCOX-1 mRNA expression. A: hCOX-2 mRNA is expressed as a percentage of the endogenous mCOX-2 mRNA as assessed by ISH assay in adjacent brain tissue sections of the same brain (NHC32). Under the stringency conditions used in this study, we found negligible cross-hybridization of the hCOX-2 [35S]-cRNA probe with the endogenous mCOX-2 mRNA (see Figure 2B ▶ ). B: mCOX-1 mRNA expression in brain of NHC32-hCOX-2 transgenics relative to WT controls. Optical densities were quantified by computerized video densitometry from film autoradiograms using a Bioquant image analysis system (Biometrics). Values are expressed as mean ± SE, in A *P < 0.01, n = 5–6 per group.
In parallel control studies, we also explored the regulation of the endogenous mCOX-1 mRNA and found no detectable change of expression in any brain region examined of the NHC32-hCOX-2 transgenics compared to WT controls (Figure 3B) ▶ .
Neuronal Overexpression of hCOX-2 Potentiates KA-Induced Seizures in NHC32-hCOX-2 Transgenic Mice
Neuronal hCOX-2 overexpression potentiated seizures and increased mortality during response to ICV (lateral ventricle) KA infusion. There was marked increase in the intensity of KA-mediated seizures in NHC32 hCOX-2 transgenic mice compared to WT control littermates (Table 1) ▶ . Vigorous status epilepticus, leading to accelerated death within 10 minutes after ICV infusion of 1.25 nmoles of KA, occurred in each of the hCOX-2 transgenic mice. Mild seizures but no death occurred in WT control littermates within 10 minutes after ICV infusion of an equal dose of KA (n = 6–8 per group, P < 0.004, Fisher’s exact test; Table 1 ▶ ). For further comparative analysis of changes in gene expression or neuropathology, the control WT mice were sacrificed within 10 minutes after lesioning. No detectable seizures were found in either hCOX-2 transgenics or WT controls following ICV infusion of an equal volume of vehicle (2 μl, saline).
Table 1.
Potentiation of KA-Induced Seizures in hCOX-2 Transgenic Mice Compared to Wild-Type Littermates
| n | Mortality | Remarks | |
|---|---|---|---|
| Wild-type littermates | 6 | 0% | Seizures were associated with staring posture and mild wet-dog shake behavior without any myoclonic twitches. |
| hCOX-2 transgenics | 8 | 100% | Seizures characterized by intense myoclonic twitches of the forelimbs associated with generalized tonic-clonic seizures with wild seizure jumps. |
Fisher’s exact test, P < 0.004.
Severe seizures in NHC32-hCOX-2 transgenic mice were characterized by intense myoclonic twitches of the forelimbs associated with generalized tonic-clonic seizures with wild seizure jumps. In contrast, seizures in WT littermates ICV injected with the same dose of KA were in general associated with staring posture and wet-dog shake behavior without any myoclonic twitches (Table 1) ▶ .
In pilot titration studies, potentiation of KA-mediated seizures in NHC32-hCOX-2 transgenic mice was found after ICV infusion of KA at doses between 0.75 and 3 nmoles. Mild or no detectable seizures were found in both hCOX-2 transgenics and WT control littermates after ICV infusion of KA at doses below 0.75 nmoles (not shown). Similar potentiation of KA-induced seizures occurred in NHC5-hCOX-2 transgenic mice (data not shown).
Potentiation of KA-Mediated Induction of c-fos and zif-268 mRNA Expression in NHC32-hCOX-2 Transgenic Mice
In NHC32-hCOX-2 transgenic mice, generalized status epilepticus, characterized by intense tonic-clonic seizures and death by 10 minutes after KA (1.25 nmoles), coincided with potentiation of c-fos mRNA expression in neuronal layers the hippocampal formation, and parietal cortex relative to KA-injected control WT littermates. Quantitative ISH autoradiography revealed greater than three- to fourfold (n = 4, P < 0.05) potentiation of c-fos mRNA expression in the granule neuron layer of the DG ipsilateral to the lesion side compared to WT controls (Figure 4, A and B ▶ ; Figure 5A ▶ , left panel). Similarly, there was >1.5-fold potentiation (P < 0.05) of c-fos mRNA expression in the CA3-CA1 pyramidal layer of the hippocampal formation (Figure 4, A and B ▶ ; Figure 5B ▶ , left panel) and in the parietal cortex (Figure 4, A and B ▶ Figure 5C ▶ , left panel), ipsilateral to the lesion side, compared to WT controls.
Figure 4.
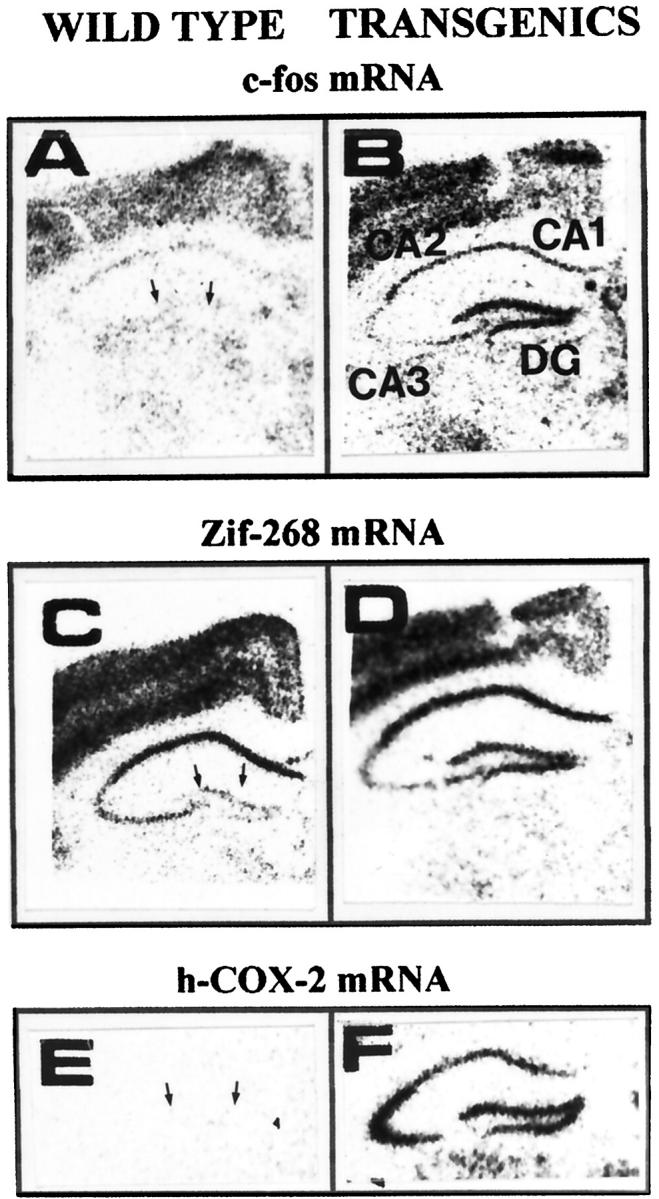
Potentiation of hippocampal c-fos and zif-268 mRNA expression in hCOX-2 transgenic mice compared to WT littermates in response to KA-induced seizures (10 minutes after lesioning). A and B: c-fos mRNA. C and D: zif-268 mRNA expression in WT controls (A, C) and in hCOX-2 transgenic mice (B, D) 10 minutes after ICV KA infusion, respectively. E: hCOX-2 mRNA expression in the hippocampal formation of WT (negative control). F: hCOX-2 transgenics, respectively. Hybridization signals from mouse brain sections are visualized by X-ray film autoradiography. In A, C, and E, arrows point toward the granule neuron layer of the hippocampal dentate gyrus (DG). In B, CA1 CA2, CA3 pyramidal layers of the hippocampal formation.
Figure 5.
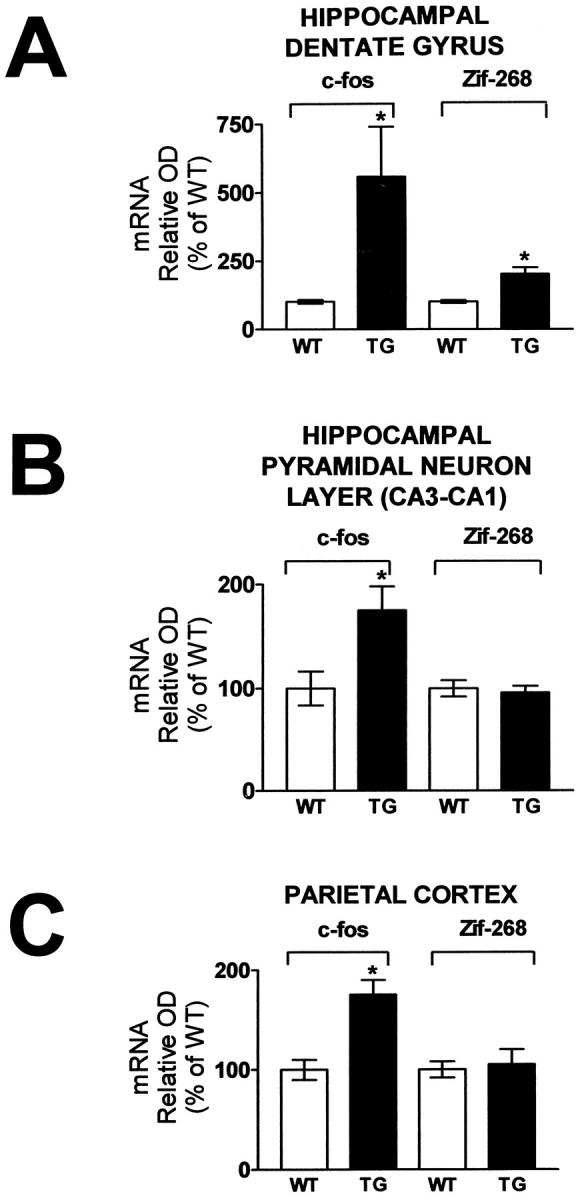
Potentiation of c- fos and zif-268 mRNA expression in the hippocampal dentate gyrus, pyramidal neuron layer, and parietal cortex of hCOX-2 transgenic mice compared to WT littermates in response to KA-induced seizures. Changes in c-fos and zif-268 mRNA expression in the granule cell layer of the dentate gyrus (A), in the pyramidal neuron layer of the hippocampal formation (B), and in the parietal cortex (C), respectively. Relative optical densities were quantified by computerized video densitometry from film autoradiograms using a Bioquant image analysis system (Biometrics). Control studies confirmed that the hippocampal level of c-fos and zif-268 mRNA expression did not differ between control WT littermates and hCOX-2 transgenics following ICV infusion of saline (not shown). Values are expressed as mean ± SE, n = 4 per group, *P < 0.05, WT versus transgenic.
Similar potentiation of zif-268 mRNA expression (approximately twofold, P < 0.05) in NHC32-hCOX-2 transgenic mice versus WT littermates was found in the granule neuron layer of the DG (Figure 4, C and D ▶ ; Figure 5A ▶ , right panel). On the contrary, no detectable potentiation of zif-268 mRNA expression was found in the CA3-CA1 pyramidal neuron of the hippocampal formation, compared to WT control littermates (Figure 4, C and D ▶ ; Figure 5B ▶ , right panel), as assessed by ISH assay on tissue sections adjacent to those used for c-fos mRNA. No detectable potentiation of zif-268 mRNA hybridization signal was found in parietal cortex of hCOX-2-NHC32 transgenic mice compared to control WT littermates (Figure 4, C and D ▶ ; Figure 5C ▶ , right panel).
In response to ICV infusion of drug vehicle (saline) in NHC-32-hCOX-2 transgenic mice, we found no detectable potentiation of hippocampal and cortical c-fos and zif-268 mRNA hybridization signal relative to control WT littermates (not shown). Moreover, hematoxylin and eosin histochemistry was used to identify neurons with pyknotic condensed nuclei in neuronal layer of the hippocampal formation and cerebral cortex. No overt sign of neurodegeneration was found in either hCOX-2 transgenics or WT control littermates within 10 minutes after ICV KA (not shown).
Neuronal hCOX-2 Overexpression Potentiates Glutamate-Mediated Excitotoxicity in Vitro
Ten-day-old primary cortico-hippocampal neuron cultures generated from control WT embryos resulted in 31 ± 2% impairment of redox activity after exposure to glutamate (50 μmol/L, 24 hours) relative to vehicle-treated control cultures, as assessed by MTT assay. However, glutamate treatment in parallel cortico-hippocampal neuronal cultures generated from hCOX-2 (NHC32) transgenic embryos resulted in 46 ± 3% impairment of redox impairment relative to vehicle-treated control cultures. Thus, overexpression of COX-2 in cortico-hippocampal neurons potentiated glutamate-mediated redox impairment by approximately 50% relative to WT controls (n = 5–6 independent cultures per group, P < 0.004; Figure 6A ▶ )
Figure 6.
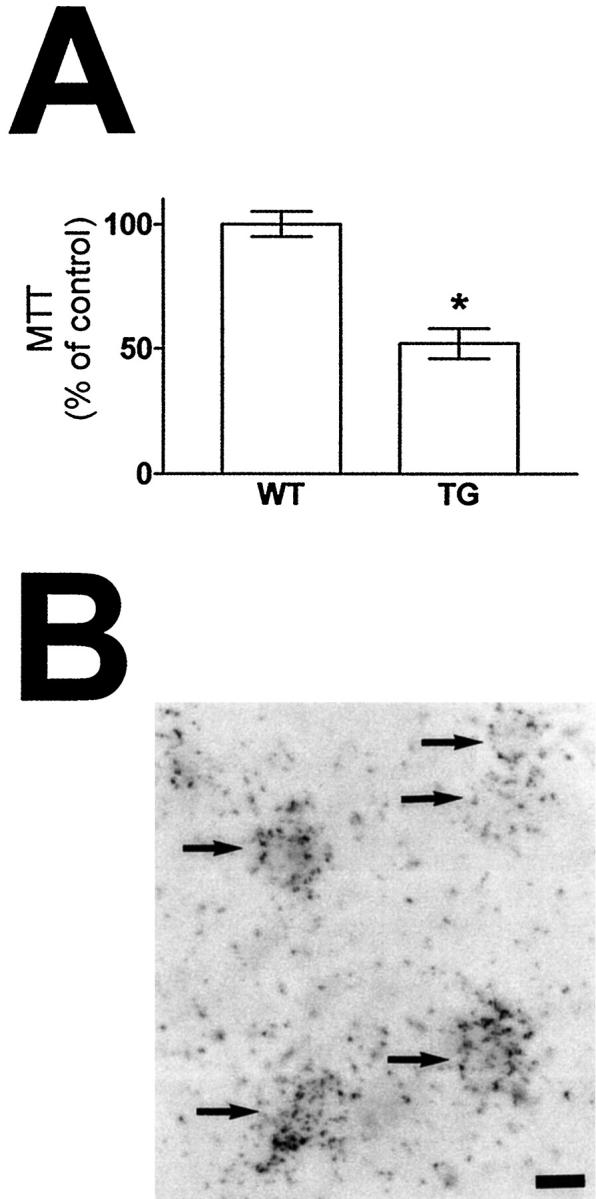
Potentiation of glutamate-mediated impairment of redox activity in primary cortico-hippocampal neurons derived from hCOX-2 transgenic mice compared to WT control mice. A: Primary cortical-hippocampal neuron cultures were derived from 14-embryonic-day-old transgenic NHC32-hCOX-2 or control WT embryos. Ten days after plating, neurons were treated with 50 μmol/L glutamate or vehicle for 24 hours’ treatment and glutamate toxicity was assessed by MTT assay. Changes in MTT are expressed as percentage of identical cultures treated with drug vehicle. B: hCOX-2 mRNA expression was visualized by combined ISH and NSE ICC using bright field illumination. Values are expressed as mean ± SE, n = 4–8 per group in n = 5 independent studies (*P < 0.05) transgenic versus WT. Scale bar in C, 10 μm. Arrows point toward NSE-immunopositive neurons expressing hCOX-2 (silver grains).
In control studies, ISH for hCOX-2 mRNA combined with NSE ICC confirmed neuronal hCOX-2 expression in the cortico-hippocampal neuronal cultures generated from hCOX-2 (NHC32) transgenic embryos (Figure 6B) ▶ .
Discussion
In this study we report that overexpression of neuronal hCOX-2 in NHC32 transgenic mice potentiated the intensity of KA-mediated seizures, leading to increased mortality coincident to increased expression of the transcription factors c-fos and zif-268. Moreover, glutamate neurotoxicity in vitro, as assessed by MTT assay, was potentiated in primary cortico-hippocampal neuron cultures derived from NHC32 transgenic mice compared to cultures derived from WT littermates. Our conclusion that potentiation of KA-induced excitotoxicity resulted from overexpression of hCOX-2 in neurons, rather than from disruption of another gene in the NHC32 transgenic line, is supported by similar results obtained with NHC5-hCOX-2 transgenics, a second line with neuronal overexpression of hCOX-2.
To generate our transgenic mice, a modified rat NSE promoter was used to regulate expression of hCOX-2 in neurons. The rat NSE promoter has been successfully used by multiple groups to selectively direct the overexpression of heterologous proteins in neurons of transgenic mice. 18,29-37 Previous studies reported the use of a rat NSE promoter consisting only of a 1.8-kb sequence located 5′ end to the first exon; this 1.8-kb promoter fragment has been found to regulate selective neuron-specific expression, but also with some minor expression in other tissues. 29-35
In this study, in addition to a longer segment of the promoter and 5′ flanking region (2.6 kb), the rat NSE regulatory fragment also included exon 1, intron 1, and 6 bp of exon 2 (Figure 1A) ▶ . Intron 1 contains a putative neuronal regulatory element, 38 and this larger rat NSE promoter construct was previously used to direct specific expression of heterologous proteins to neurons in transgenic mice. 18,36,37 The results presented in this study confirm the cell type specificity determined by the larger NSE promoter, which includes intron 1; transgene expression in the NHC32-hCOX-2 line examined was restricted to the brain, whereas ISH revealed that the detectable brain expression is essentially only neuronal.
One possible mechanism by which neuronal COX-2 overexpression may influence the neuronal response to KA-induced seizures and glutamate excitotoxicity in vitro may involve the control of transcription factor DNA-binding activities. We found that neuronal hCOX-2 overexpression in NHC32-hCOX-2 transgenic mice potentiate the expression of the IEG transcription factors c-fos and zif-268 in subsets of hippocampal neurons in response to KA, compared to WT littermates. This regulation of gene expression might occur through paracrine and autocrine signaling mediated by prostaglandins (PGs) through PG receptors, which are members of the G-protein-coupled family of receptors. In non-neuronal cells, the signals depend on the specific agonist and the receptor, but include changes in cyclic AMP, turnover of phosphatidylinositol, and activation of both protein kinase C (PKC) and mitogen-activated protein (MAP) kinase. 39 Thus, it is possible that overexpression of hCOX-2 mRNA in neurons may increase sensitivity or lower the threshold level of excitation to KA through changes in gene expression. We also note that there is evidence suggesting a direct link between COX-2 and c-fos and zif-268 expression in excitotoxicity. All three are rapidly transcribed during the response to KA-induced seizures 40 and inhibition of intracellular platelet activating factor (PAF), 41 whereas limiting KA-mediated induction of zif-268 expression also attenuates COX-2 induction. 40
The lack of potentiation of zif-268 mRNA expression in the hippocampal CA3-CA1 pyramidal layers and parietal cortex of hCOX-2 transgenic mice versus WT littermates in response to KA-induced seizures is puzzling; it may indicate a regional variation in the time course of changes in gene expression in this model system. In support of this hypothesis we note that a common feature of these responses to excitotoxic insult is the concerted induction of IEG with a distinct time course of activation. 42,43
COX-1 and COX-2 are key enzymes in the generation of prostanoids including PGs. Both enzymes catalyze two functional reactions: a cyclooxygenase reaction to form the endohydroperoxide PGG2, and an iron heme-mediated peroxidase activity to reduce PGG2 to PGH2. 11 PGH2 is the precursor to all other PG. The primary source of arachidonate for COX is thought to be membrane phospholipid; arachidonic acid (AA) is released from esterified phospholipid by the action of phospholipase A2. Substances that increase intracellular Ca2+, such as glutamate, 44 also activate phospholipase A2 45 to make available the substrate AA for COX reactions. Thus, agents increasing neuronal intracellular free Ca2+ in presence of high levels of COX-2 may lead to higher levels of prostanoid synthesis. Because several of the prostanoid products of AA metabolism generated by COX may also control the activity of excitatory and inhibitory amino acid pathways, 46 it is plausible that overexpression of neuronal COX-2 might also modulate glutamate mediated responses through excitotoxic mechanisms. In support of this hypothesis there is evidence that (n-methyl-d-aspartate (NMDA)-mediated neuronal death is diminished in a dose-dependent manner by COX-2 inhibitors in primary neuronal cultures. 47 However, we note that there is also evidence that PGs may protect from glutamate toxicity in cortical neurons under certain conditions. 48
Free radicals are intermediate products in COX-mediated PG synthesis, and lipid peroxides influence the activity of COX. 49 Moreover, the role of free radicals in NMDA receptor-mediated glutamate excitotoxicity is well established in various experimental conditions. 50,51 Thus, given that oxidative stress seems to be involved in mechanisms of excitotoxicity, 52,53 it is not unexpected that neuronal COX-2 activity may contribute to neurodegeneration via oxidative mechanisms 11 ; this COX-2 influence may not necessarily be closely linked to PG production. 54 Oxidative stress resulting from COX-2 activity may have important implications for neurodegenerative diseases such as AD, in which an elevated glutamatergic tone, also defined as disinhibition syndrome, may be responsible for a widespread pattern of neurodegeneration. 13 The evidence of potentiation of glutamate-mediated redox impairment in primary neuron cultures from the hCOX-2 transgenic mice suggests that COX-2 overexpression indeed may be involved in mechanisms of oxidative stress in vitro. Future studies will clarify the role of neuronal COX-2 expression in neurodegenerative pathways such as caspase activation and apoptosis in vitro and in vivo.
In conclusion, this study demonstrated a possible causative role for neuronal COX-2 expression in excitotoxicity. This model system will allow a systematic examination of the role of COX-2 in mechanisms of neurodegeneration that involve excitatory amino acid pathways. Ongoing laboratory studies exploring the role of selective and nonselective COX-2 inhibitors on KA-mediated responses in NHC32-hCOX-2 transgenics will clarify the causative role of COX-2 in mechanisms of excitotoxicity.
Footnotes
Address reprint requests to Dr. Giulio Maria Pasinetti, Neuroinflammation Research Laboratories, Mount Sinai School of Medicine, Department of Psychiatry, Box 1229, One Gustave L. Levy Place, New York, New York 10029. E-mail: gp2@doc.mssm.edu.
Supported by National Institute on Aging grants AG13799, AG14239, and AG14766 to G. M. P.
References
- 1.Marnett L: Aspirin and the potential role of prostaglandins in colon cancer. Cancer Res 1992, 52:5575-5589 [PubMed] [Google Scholar]
- 2.Meade EA, Smith WL, DeWitt DL: Differential inhibition of prostaglandin endoperoxide synthase (cyclooxygenase) isozymes by aspirin and other non-steroidal anti-inflammatory drugs. J Biol Chem 1993, 268:6610-6614 [PubMed] [Google Scholar]
- 3.Kujubu DA, Fletcher BS, Varnum BC, Lim RW, Herschman HR: TIS10, a phorbol ester promoter-inducible mRNA from Swiss 3T3 cells, encodes a novel prostaglandin synthase/cyclooxygenase homologue. J Biol Chem 1991, 266:12866-12872 [PubMed] [Google Scholar]
- 4.O’Banion MK, Winn VD, Young DA: CDNA cloning and functional activity of a glucocorticoid-regulated inflammatory cyclooxygenase. Proc Natl Acad Sci USA 1991, 89:4888-4892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cao C, Matsumura K, Yamagata K, Watanabe Y: Induction by lipopolysaccharide of cyclooxygenase-2 mRNA in rat brain: Its possible role in the febrile response. Brain Res 1995, 697:187-196 [DOI] [PubMed] [Google Scholar]
- 6.Giovanucci E, Rimm EB, Stampfer MJ, Colditz GA, Ascherio A, Willett WC: Aspirin use and the risk for colorectal cancer and adenoma in male health professionals. Ann Intern Med 1994, 121:241-246 [DOI] [PubMed] [Google Scholar]
- 7.Greenberg ER, Baron JA, Freeman DHJ, Mandel JS, Haile R: Reduced risk of large-bowel adenomas among aspirin users: the polyp prevention study group. J Natl Cancer Inst 1993, 85:912-916 [DOI] [PubMed] [Google Scholar]
- 8.Thun MJ, Namboodiri MM, Heath CW, Jr: Aspirin use and reduced risk of fatal colon cancer. N Engl J Med 1991, 325:1593-1596 [DOI] [PubMed] [Google Scholar]
- 9.Thun MJ, Namboodiri MM, Calle E, Flanders W, Heath CJ: Aspirin use and risk of fatal cancer. Cancer Res 1993, 53:1322-1327 [PubMed] [Google Scholar]
- 10.McGeer PL, Schulzer M, McGeer EG: Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer’s disease: a review of 17 epidemiologic studies. Neurology 1996, 47:425-443 [DOI] [PubMed] [Google Scholar]
- 11.Pasinetti GM: Cyclooxygenase and inflammation in Alzheimer’s disease: experimental approaches and clinical interventions. J Neurosci Res 1998, 54:1-6 [DOI] [PubMed] [Google Scholar]
- 12.Tocco G, Freire-Moar J, Schreiber SS, Sakhi SH, Aisen PS, Pasinetti GM: Maturational regulation, and regional induction of cyclooxygenase-2 in rat brain: Implications for Alzheimer’s disease. Exp Neurol 1997, 144:399-349 [DOI] [PubMed] [Google Scholar]
- 13.Olney JW, Wozniak DF, Farber NB: Excitotoxic neurodegeneration in Alzheimer disease: new hypothesis and new therapeutic strategies. Arch Neurol 1997, 54:1234-1240 [DOI] [PubMed] [Google Scholar]
- 14.Pasinetti GM, Aisen PS: Cyclooxygenase-2 expression is increased in frontal cortex of Alzheimer’s disease brain. Neuroscience 1998, 87:319-324 [DOI] [PubMed] [Google Scholar]
- 15.Oka A, Takashima S: Induction of cyclo-oxygenase 2 in brains of patients with Down’s syndrome and dementia of Alzheimer type: specific localization in affected neurones and axons. Neuroreport 1997, 8:1161-1164 [DOI] [PubMed] [Google Scholar]
- 16.Adams J, Collaco-Moraes Y, de Belleroche J: Cyclooxygenase-2 induction in cerebral cortex: an intracellular response to synaptic excitation. J Neurochem 1996, 66:6-12 [DOI] [PubMed] [Google Scholar]
- 17.Hla T, Nielsen N: Human cyclooxygenase-2 cDNA. Proc Natl Acad Sci USA 1992, 89:7384-7388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mucke L, Masliah E, Johnson WB, Ruppe MD, Alford M, Rockenstein EM, Forss-Petter S, Pietropaolo M, Mallory M, Abraham CR: Synaptotrophin effects of human amyloid β protein precursors in the cortex of transgenic mice. Brain Res 1994, 666:151-167 [DOI] [PubMed] [Google Scholar]
- 19.Hogan B, Costantini F, Lacy E: Manipulating the Mouse Embryo: A Laboratory Handbook. 1986. Cold Spring Harbor Laboratory Cold Spring Harbor, NY
- 20.Chirgwin JM, Przybyla AE, MacDonald RJ, Rutter WJ: Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochem 1979, 18:5294-5301 [DOI] [PubMed] [Google Scholar]
- 21.Lehrach H, Diamond D, Wozney JM, Boedtker H: RNA molecular weight determinations by gel electrophoresis under denaturing conditions, a critical examination. Biochemistry 1977, 16:4743-4751 [DOI] [PubMed] [Google Scholar]
- 22.Schreiber SS, Maren S, Tocco G, Shors TJ, Thompson RF: A negative correlation between the induction of long-term potentiation and activation of immediate early genes. Mol Brain Res 1991, 11:89-91 [DOI] [PubMed] [Google Scholar]
- 23.Schreiber SS, Tocco G, Najm I, Thompson RF, Baudry M: Cycloheximide prevents kainate-induced neuronal death and c-fos expression in adult rat brain. J Mol Neurosci 1993, 4:149-159 [DOI] [PubMed] [Google Scholar]
- 24.Ho L, Osaka H, Aisen PS, Pasinetti GM: Induction of cyclooxygenase (COX)-2 but not COX-1 gene expression in apoptotic cell death. J Neuroimmunol 1998, 89:142-149 [DOI] [PubMed] [Google Scholar]
- 25.Pasinetti GM: In situ hybridization to brain tissue sections using labeled single-strand complememtary RNA probes. Methods in Molecular Biology, vol. 13: Protocols in Molecular Neurobiology. Edited by A Longstaff and P Revest. Totowa, NJ, Humana Press, 1992, pp 155–165
- 26.Pasinetti GM, Johnson SA, Oda T, Rozovsky I, Finch CE: Clusterin (SGP-2): a multifunctional glycoprotein with regional expression in astrocytes and neurons of the adult rat brain. J Comp Neurol 1994, 339:387-400 [DOI] [PubMed] [Google Scholar]
- 27.Tocco G, Musleh W, Sakhi S, Schreiber SS, Baudry M, Pasinetti GM: Complement and glutamate neurotoxicty. Genotypic influences of C5 in a mouse model of hippocampal neurodegeneration. Mol Chem Neuropathol 1997, 31:289–299 [DOI] [PubMed]
- 28.Kondo T, Sharp FR, Honkaniemi J, Mikawa S, Epstein CJ, Chan PH: DNA fragmentation and prolonged expression of c-fos, c-jun, and hsp-70 in kainic acid- induced neuronal cell death in transgenic mice overexpressing human CuZn-superoxide dismutase. J Cereb Blood Flow Metab 1997, 17:241-256 [DOI] [PubMed] [Google Scholar]
- 29.Castillo MB, Celio MR, Andressen C, Gotzos V, Rulicke T, Berger MC, Weber J, Berchtold MW: Production and analysis of transgenic mice with ectopic expression of parvalbumin. Arch Biochem Biophys 1995, 317:292-298 [DOI] [PubMed] [Google Scholar]
- 30.Colucci-D’Amato GL, Santelli G, D’Alessio A, Chiappetta G, Mineo A, Manzo G, Vecchio G, DeFranciscis V: Dbl expression driven by the neuron-specific enolase promoter induces tumor formation in transgenic mice with a p53(±) background. Biochem Biophys Res Commun 1995, 216:762-770 [DOI] [PubMed] [Google Scholar]
- 31.Farlie PG, Dringen R, Rees SM, Kannourakis G, Bernard O: Bcl-2 transgene expression can protect neurons against developmental and induced cell death. Proc Natl Acad Sci USA 1995, 92:4397-4401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fattori E, Lazzaro D, Musiani P, Modesti A, Alonzi T, Ciliberto G: IL-6 expression in neurons of transgenic mice causes reactive astrocytosis and increase in ramified microglial cells but no neuronal damage. Eur J Neurosci 1995, 7:2441-2449 [DOI] [PubMed] [Google Scholar]
- 33.Forss-Petter S, Danielson PE, Catsicas S, Battenberg E, Price J, Nerenberg M, Sutcliffe JG: Transgenic mice expressing β-galactosidase in mature neurons under neuron-specific enolase promoter control. Neuron 1990, 5:187-197 [DOI] [PubMed] [Google Scholar]
- 34.Malherbe P, Richards JG, Martin JR, Bluethmann H, Maggio J, Huber G: Lack of β-amyloidosis in transgenic mice expressing low levels of familial Alzheimer’s disease missense mutations. Neurobiol Aging 1996, 17:205-214 [DOI] [PubMed] [Google Scholar]
- 35.Martinou JC, Dubois-Dauphin M, Staple JK, Rodriguez I, Frankowski H, Missotten M, Albertini P, Talabot D, Catsicas S, Pietra C: Overexpression of BCL-2 in transgenic mice protects neurons from naturally occurring cell death and experimental ischemia. Neuron 1994, 13:1017-1030 [DOI] [PubMed] [Google Scholar]
- 36.Rall GF, Mucke L, Oldstone MB: Consequences of cytotoxic T lymphocyte interaction with major histocompatibility complex class I-expressing neurons in vivo. J Exp Med 1995, 182:1201-1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao J, Paganini L, Mucke L, Gordon M, Refolo L, Carman M, Sinha S, Oltersdorf T, Lieberburg I, McConlogue L: β-secretase processing of the β-amyloid precursor protein in transgenic mice is efficient in neurons but inefficient in astrocytes. J Biol Chem 1996, 271:31407-31411 [DOI] [PubMed] [Google Scholar]
- 38.Sakimura K, Kushiya E, Ogura A, Kudo Y, Katagiri T, Takahashi Y: Upstream and intron regulatory regions for expression of the rat neuron-specific enolase gene. Mol Brain Res 1995, 28:19-28 [DOI] [PubMed] [Google Scholar]
- 39.Prescott SM, White R: Self promotion? Intimate connections between APC and prostaglandin H synthase-2. Cell 1996, 87:783-786 [DOI] [PubMed] [Google Scholar]
- 40.Marcheselli V, Bazan NG: Sustained induction of prostaglandin endoperoxide synthase-2 by seizures in hippocampus- Inhibition by a platelet-activating factor antagonist. J Biol Chem 1996, 271:24794-24799 [DOI] [PubMed] [Google Scholar]
- 41.Bazan NG: A signal terminator. Nature 1995, 374:501-502 [DOI] [PubMed] [Google Scholar]
- 42.Janssen-Timmen V, Lemaire P, Mattei M, Relevant O, Charnay P: Structure chromosome mapping and regulation of the mouse zinc finger gene krox-24: evidence for a common regulatory pathway for immediate early serum response genes. Gene 1989, 80:325-336 [DOI] [PubMed] [Google Scholar]
- 43.Simonato M, Hosford D, Labiner D, Shin C, Mansbach HH, McNamara J: Differential expression of immediate early genes in the hippocampus in the kindling model of epilepsy. Mol Brain Res 1991, 11:115-124 [DOI] [PubMed] [Google Scholar]
- 44.Mattson M, Lovell MA, Furukawa K, Markesbery WR: Neurotrophic factors attenuate glumate-induced accumulation of peroxides, elvation of intracellular Ca++ concentration, and neurotoxicity and increase antioxidant enzyme activities in hippocampal neurons. J Neurochem 1995, 65:1740-1751 [DOI] [PubMed] [Google Scholar]
- 45.Murakami M, Nakatani Y, Atsumi G, Inoue K, Kudo I: Regulatory functions of phospholipase A2. Crit Rev Immunol 1997, 217:225-283 [DOI] [PubMed] [Google Scholar]
- 46.Kimura H, Okamoto K, Sakai Y: Modulatory effects of prostaglandin D2, E2 and F2 on the postsynaptic action of inhibithory and excitatory amino acids in cerebellar Purkinje cells dendrites in vitro. Brain Res 1985, 330:235-244 [DOI] [PubMed] [Google Scholar]
- 47.Hewett SJ, Hewett JA: COX-2 contributes to NMDA-induced neuronal death in cortical cell cultures. Soc Neurosci 1997, 23:1666(meeting abstr) [Google Scholar]
- 48.Cazevieille C, Muller A, Meynier F, Dutrait N, Bonne C: Protection by prostaglandin from glutamate toxicity in cortical neurons. Neurochem Int 1994, 24:395-398 [DOI] [PubMed] [Google Scholar]
- 49.Halliwell B, Gutteridge JMC: Free Radical in Biology and Medicine, 3rd edition. Oxford, Clarendon Press, 1995
- 50.Dawson VL, Dawson TM, London E, Bredt DS, Snyder SH: Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc Natl Acad Sci USA 1991, 88:6368-6371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cazevieille C, Muller A, Meynier F, Bonne C: Superoxide and nitric oxide cooperation in hypoxia reoxygenation-induced neuron injury. Free Rad Biol Med 1993, 14:389-395 [DOI] [PubMed] [Google Scholar]
- 52.Patel M, Day BJ, Crapo JD, Fridovich I, McNamara JO: Requirement for superoxide in excitotoxic cell death. Neuron 1996, 16:345-355 [DOI] [PubMed] [Google Scholar]
- 53.Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT: Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron 1989, 2:1547-1558 [DOI] [PubMed] [Google Scholar]
- 54.Shoami E, Rosenthal J, Lavy S: The effect of incomplete cerebral ischemia on prostaglandin levels in rat brain. Stroke 1982, 13:494-499 [DOI] [PubMed] [Google Scholar]