

Abstract
Inflammation of the arterial wall in giant cell arteritis induces a series of structural changes, including the formation of new vasa vasorum. To study the regulation of neoangiogenesis in giant cell arteritis, temporal arteries were examined for the extent and localization of microvessel generation and for the production of angiogenic factors. In normal arteries, vasa vasorum were restricted to the adventitia, but in inflamed arteries, capillaries emerged in the media and the intima. These capillaries displayed a distinct topography with a circumferential arrangement in the external one-third of the intima. Neovascularization was closely correlated with the formation of lumen-obstructing intima, the fragmentation of the internal elastic lamina, and the presence of multinucleated giant cells. Comparison of tissue cytokine transcription in temporal arteries of giant cell arteritis patients with and without up-regulated neoangiogenesis identified interferon-γ and vascular endothelial growth factor but not fibroblast growth factor-2 as mediators associated with vasa vasorum proliferation. Giant cells and CD68-positive macrophages at the media-intima junction were found to be the major cellular sources of vascular endothelial growth factor. These data demonstrate that formation of new vasa vasorum in vasculitis is regulated by inflammatory cells and not by arterial wall cells, raising the possibility that it represents a primary disease mechanism and not a secondary hypoxia-induced event. Increased neovascularization in interferon-γ-rich arteries suggests that the formation of new vasa vasorum is determined by the nature of the immune response in the arterial wall, possibly resulting from the generation and functional activity of multinucleated giant cells.
Giant cell arteritis (GCA) is an inflammatory vasculopathy that preferentially affects medium-sized and large arteries. 1,2 Inflammatory infiltrates that penetrate all layers of the arterial wall are composed of CD4 T cells and macrophages, and they can include multinucleated giant cells and can be arranged as granulomas. The disease displays a tropism for arteries of extracranial distribution but may also involve the aorta and its major branches. The vascular morbidity of this syndrome is related either to luminal stenosis of inflamed arteries 3 with subsequent tissue infarction or to arterial wall destruction and aneurysm formation. 4 Pathogenetic studies of GCA have indicated that the syndrome is the sequela of an antigen-driven immune response. 5 This concept has been substantiated by in vivo studies, demonstrating that GCA can be perpetuated in temporal arteries engrafted into SCID mice. 6 Depletion of T cells from artery grafts in SCID mouse chimeras was sufficient to paralyze the functional activity of cytokine-producing macrophages and to abrogate the vasculitic lesions. Selected CD4 T cells undergo clonal expansion in the arterial wall and produce interferon (IFN)-γ. 7 IFN-γ-releasing CD4 T cells have been mapped to the adventitial layer of inflamed arteries, giving rise to the hypothesis that the site of the immunological injury in this vasculopathy is the adventitia. 8
Inflammation of the arterial wall in GCA is followed by a series of structural changes that are partially the result of tissue-destroying mechanisms. A typical finding in GCA is the degradation of the internal elastic laminae (IEL). Giant cells, which have a tendency to accumulate along the IEL, have been implicated in removing fragments of digested elastic tissue. However, arterial wall destruction with hemorrhage is not a typical manifestation of GCA. Rather, patients are threatened by ischemia resulting from arterial stenosis. Stenotic lesions are due to the concentric growth of the intima with little evidence of thrombosis. The degree of luminal stenosis has been closely correlated with the production of platelet-derived growth factors (PDGF)-AA and -BB. 3 Macrophages and giant cells in the media and along the media-intima junction have been identified as the major cellular sources of PDGF. These findings suggest that the growth of the intima, not the destruction of arterial wall layers, is the major factor determining the pathological consequences of GCA.
In addition to the fragmented elastic lamina and the hyperplastic intima, inflamed arteries are characterized by the emergence of new intramural capillaries. The normal arterial wall is a relatively avascular tissue, comparable only to cartilage. Vasa vasorum are restricted to the adventitial layer, and the media and intima are supplied via diffusion from either the lumen or the adventitia. 9 Angiogenesis is a physiological process in embryonic development and wound repair, but it can also contribute to pathological events. 10,11 It can be a primary disease mechanism, such as in diabetic retinopathy, 12 or can play an indirect role by supporting the growth of pathological tissue, such as in tumors and in rheumatoid arthritis. 13 Growth factors previously reported as driving microvessel formation include vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF), whereas inflammatory cytokines locally produced in GCA arteries, such as IFN-γ, interleukin (IL)-1, and IL-6, do not appear to play a direct role. 12,14-17
The current study was designed to explore whether neoangiogenesis is regulated by infiltrating inflammatory cells or by arterial wall stromal cells. In temporal arteries of patients with GCA, new vasa vasorum appeared primarily in the media and the intima, thereby supplying blood to formerly avascular areas of the arterial wall. The degree of neoangiogenesis in the inflamed arteries was closely correlated with the hyperplastic reaction of the intima and the fragmentation of the elastic membranes. Tissue transcription of VEGF and of the T cell product IFN-γ but not of FGF-2 correlated with the extent of neovascularization. The major source of VEGF was multinucleated giant cells and macrophages accumulating at the media intima junction, suggesting that inflammatory cells and not stromal cells or endothelial cells regulate vasa vasorum formation in GCA.
Materials and Methods
Patients
Temporal artery specimens were obtained from 40 patients undergoing biopsy for diagnostic purposes. The diagnosis of GCA was confirmed by histopathology. All patients fulfilled the American College of Rheumatology criteria for the diagnosis of GCA. 18 None of the patients was on glucocorticoids or other anti-inflammatory medication.
Antibodies
Endothelial cells were identified with a polyclonal rabbit Ab against von Willebrand’s factor (vWF), 1:750 (Dako, Carpinteria, CA). The expression of VEGF was visualized with a polyclonal rabbit Ab, 1:100 (Calbiochem, La Jolla, CA). FGF-2 was detected with a polyclonal rabbit Ab, 1:100 (Collaborative Biomedical Products, Bedford, MA). Mouse mAb against CD68 (PG-M1), 1:150, as well as all biotinylated secondary antibodies were obtained from Dako. Streptavidin-conjugated alkaline phosphatase and Vector Red substrate kits were purchased from Vector Laboratories (Burlingame, CA).
Immunohistochemistry
Four-micron sections of paraffin-embedded temporal artery biopsy specimens from 28 GCA patients and 10 normal controls were deparaffinized and endogenous peroxidase was blocked. Slides were steamed in 0.1% citrate buffer for 30 minutes to facilitate antigen recovery. After blocking of non-specific binding with 5% normal goat serum, adjacent sections were incubated overnight (4°C) with antibodies specific for human vWF, FGF-2, and VEGF or 30 minutes at room temperature with anti-CD68 Ab. After incubation with the corresponding secondary Ab for 30 minutes, slides stained for FGF-2 and VEGF were developed with the Vectastain alkaline phosphatase kit and Vector Red. vWF and CD68 stains were visualized with Cy-2 or FITC-labeled goat-anti-rabbit or rabbit-anti-mouse Abs (1:200, Jackson Laboratories, Bar Harbor, ME). Sections were counterstained with DAPI for 10 minutes (Sigma, St. Louis, MO) and coverslipped with Cytoseal (Stephens Scientific, Riverdale, NY) or Vectashield (Vector Laboratories). Parallel sections were stained with hematoxylin/eosin (H&E). Negative controls were included in each series and were incubated with 1% normal goat serum instead of the primary Abs.
vWF and VEGF-stains were visualized by using a confocal Axiphot fluorescent laser-scanning microscope (Carl Zeiss, Inc., Oberkochen, Germany). The total number of lumina was counted for the three layers of the vessel wall over the entire cross-section of the artery.
Quantification of Neointima Formation and Destruction of the Internal Elastic Lamina
For quantification of IEL destruction, digital imaging software (KS400, Kontron Electronics, Eching, Germany) was used. H&E-stained paraffin sections of temporal artery sections were scanned using a confocal laser scanning microscope (Axiphot) and the resulting 256 gray-scale images were analyzed with the KS400 software. The total cross section of the temporal artery, the area of the neointima (lumen to IEL), and the area of the lumen were measured. The extent of IEL-destruction was determined as the percentage of the total circumference of the IEL.
Polymerase Chain Reaction (PCR) Primers and Biotinylated Probes
The following primers and probes were used: β-actin (5′: ATG GCC ACG GCT GCT TCC AGC; 3′: CAT GGT GGT GCC GCC AGA CAG; probe: TTC CTT CCT GGG CAT GGA GT), IFN-γ (5′: ACC TTA AGA AAT ATT TTA ATG C; 3′: ACC GAA TAA TTA GTC AGC TT; probe: ATT TGG CTC TGC ATT ATT TTT CTG T), FGF-2 (5′: GCA GAA GAG AGA GGA GTT G; 3′: TAG CAG AGA TTG GGA GAA A, probe: TGT GCT AAC CGT TAC CTG GC), and VEGF (5′: ATG AAC TTT CTG CTG TCT TGG; 3′: TCA CCG CCT CGG CTT GTC ACA; probe: CAC CAT GCA GAT TAT GCG GA).
Reverse Transcription (RT)-PCR and Cytokine Semiquantification
Total RNA was extracted from 18 GCA temporal artery samples by using a commercially available reagent (TRIZOL, Life Technologies, Grand Island, NY). cDNA from temporal artery tissue was analyzed for β-actin transcripts by semiquantitative PCR and then adjusted to contain equal numbers. 19 The adjusted cDNA was amplified under nonsaturating conditions with cytokine-specific primers for 30 cycles.
Each PCR amplification cycle consisted of denaturation at 94°C for 30 seconds, annealing at either 52°C (FGF-2), 55°C (β-actin, IFN-γ), or 58°C (VEGF) for 1 minute, and polymerization at 72°C for 1 minute with a final 10-minute extension at 72°C. Each PCR included a set of serial dilutions of cytokine cDNA with a known number of copies.
Amplified products were labeled with digoxygenin-11-dUTP and then semiquantified in a liquid hybridization assay with biotinylated internal probes using a PCR enzyme-linked immunosorbent assay kit (both Roche Molecular Biochemicals-Boehringer Mannheim, Indianapolis, IN). In these assays, the labeled PCR products were hybridized for 2 hours with 200 ng/ml probe at 42°C for β-actin and IFN-γ and at 55°C for VEGF and FGF-2. Hybrids were immobilized on streptavidin-coated microtiter plates and, after washing, were detected with a peroxidase-labeled anti-digoxigenin antibody. The plates were developed by a color reaction using ABTS (2,2′-azino-di[3-ethylbenzthiazoline sulfonate] diammonium salt) substrate and quantified using a kinetic microplate reader (Molecular Devices, Sunnyvale, CA). The number of cytokine-specific sequences was determined by interpolation onto a standard curve and was expressed as the number of cytokine sequences per 2 × 10 5 β-actin sequences. 19
Statistical Analysis
Statistical analysis was carried out using Sigma Stat software (SPSS, Chicago). The number of blood vessels was correlated with elastic lamina destruction and in situ cytokine production by exponential regression analysis.
Results
Neovascularization in the Arterial Wall Correlates with Intimal Hyperplasia
Microvessels in the wall of temporal arteries were detected by staining for vWF. Normal temporal arteries contained a network of vasa vasorum that was restricted to the adventitial layer (data not shown). In arteries affected by GCA, microvessels were found in all three layers of the arterial wall (Figure 1) ▶ . The extent of neovascularization varied markedly among patients with pronounced microvessel formation in some cases and minimally increased vascularization in others. In temporal arteries with minimal intimal hyperplasia, vasa vasorum were found almost exclusively in the adventitia (Figure 1A) ▶ . Compared to normal temporal arteries, the diameter of intramural blood vessels was smaller and they were increased in number. In contrast, in inflamed arteries with marked intimal hyperplasia, new vasa vasorum were found throughout the arterial wall, including the media and the intima (Figure 1B) ▶ . In the media, clusters of newly formed vessels colocalized with dense mononuclear infiltrates. In the intima, vasa vasorum displayed a distinct topography. They were arranged in a circumferential distribution in the outer one-third of the intima, distant from the intima-media junction. Vasa vasorum were distinctly absent from the base of the hyperplastic intima and were not found in the direct vicinity of the IEL. Also, with loss of arterial lumen, vWF was often found diffusely under the macroendothelial layer but remained highly localized to endothelial cells in the newly formed vessels.
Figure 1.
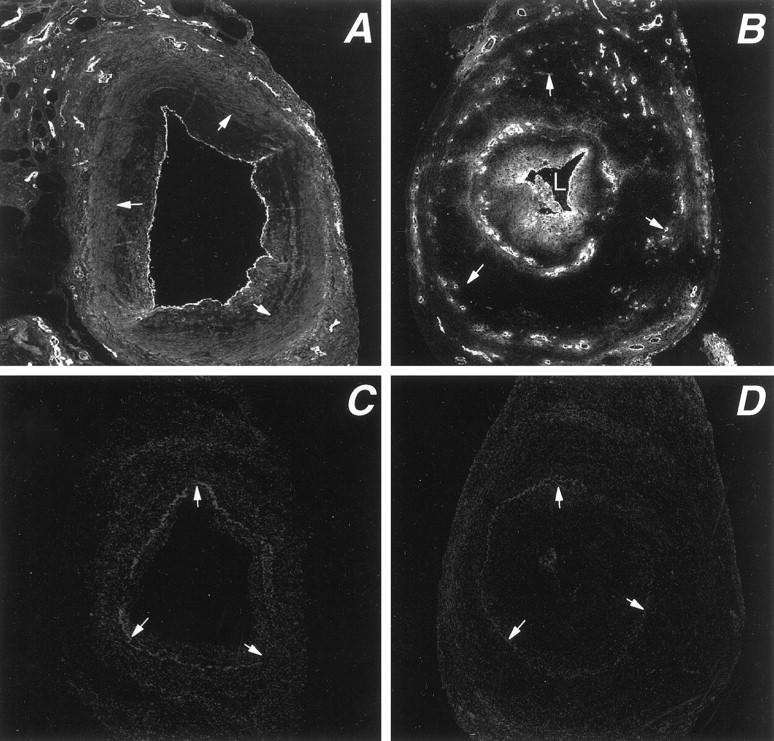
Distribution of vasa vasorum in temporal arteries affected by GCA. Tissue sections from temporal arteries of GCA patients were stained with antibodies specific for von Willebrand’s factor and a Cy-2-labeled secondary antibody to visualize capillary lumina. Stained sections were scanned by confocal microscopy. In temporal arteries with minimal intimal thickening, vasa vasorum are essentially restricted to the adventitial layer (A). In temporal arteries with marked luminal stenosis due to intimal hyperplasia, microvessels emerge in the media and form a circumferential ring in the outer intima (B). Arrows delineate the internal elastic lamina. Negative control stains are shown as C and D. Original magnification, ×100.
To explore the relationship between vasa vasorum formation and remodeling of the inflamed arterial wall, temporal artery sections from 28 GCA patients were examined (Table 1) ▶ . The number of microvessels in the entire circumference of the arterial wall ranged from 13 to 284. Correlation between the area of arterial lumen and the area of the intimal layer demonstrated a close association between intimal hyperplasia and vasa vasorum formation (P = 0.004). The number of newly formed vasa vasorum was highest in cases with marked hyperplasia of the intima, leading to luminal stenosis. Not only did arterial sections with intimal hyperplasia contain more microvessels, the absence or presence of a thickened intimal layer was also correlated with the emergence of capillaries in the otherwise avascular media and intima. Results of 19 patients who had marked intimal hyperplasia (stenosis >50%) and of 9 patients with minimal to moderate stenosis (<50%) are summarized in Figure 2 ▶ . In the absence of hyperplastic intima and luminal stenosis, microvessels were only found in the adventitia. Arteries with intimal hyperplasia had a mean of 22 vasa vasorum per section in the media (P = 0.001, compared to arteries lacking luminal stenosis) and a mean of 50 vasa vasorum in the intima (P < 0.0001), confirming that the hyperplastic intima was a preferred site of neoangiogenesis.
Table 1.
Neoangiogenesis and Intimal Hyperplasia in GCA
| Patient | Microvessels no/section | Stenosis % area | Neointima % area |
|---|---|---|---|
| 1 | 13 | 16.56 | 1.07 |
| 2 | 38 | 25.97 | 8.15 |
| 3 | 40 | 27.43 | 3.29 |
| 4 | 43 | 34.37 | 4.42 |
| 5 | 48 | 42.07 | 11.46 |
| 6 | 73 | 48.03 | 9.00 |
| 7 | 77 | 79.55 | 47.73 |
| 8 | 85 | 24.24 | 3.22 |
| 9 | 91 | 97.67 | 19.78 |
| 10 | 95 | 60.35 | 11.58 |
| 11 | 96 | 72.33 | 21.87 |
| 12 | 101 | 99.96 | 38.77 |
| 13 | 104 | 17.84 | 3.14 |
| 14 | 114 | 97.54 | 27.02 |
| 15 | 115 | 99.78 | 35.16 |
| 16 | 129 | 92.35 | 23.79 |
| 17 | 132 | 79.13 | 23.61 |
| 18 | 136 | 99.54 | 47.93 |
| 19 | 138 | 89.48 | 33.93 |
| 20 | 145 | 24.86 | 7.44 |
| 21 | 173 | 99.89 | 30.60 |
| 22 | 182 | 93.77 | 40.00 |
| 23 | 209 | 94.56 | 34.41 |
| 24 | 218 | 64.79 | 21.77 |
| 25 | 225 | 77.99 | 24.97 |
| 26 | 228 | 93.49 | 34.80 |
| 27 | 256 | 93.86 | 10.75 |
| 28 | 284 | 84.63 | 37.00 |
Figure 2.

Layer-specific distribution of vasa vasorum in temporal arteries with minimal and marked intimal hyperplasia. Temporal arteries from a cohort of 28 patients with GCA were stained for von Willebrand’s factor to visualize microvessels. Based upon the thickening of the intimal layer, patients were divided into two categories with minimal to moderate intimal hyperplasia (<50%) or marked luminal stenosis (>50%), designated − and ++, respectively. Capillary lumen were counted in the entire arterial circumference and assigned to the adventitia, media, or intima. Results are given as means ± SD. Neovascularization in the media and intima occurred only in cases with intimal hyperplasia. The number of microvessels in the adventitia did not correlate with luminal stenosis.
Neovascularization Correlates with Fragmentation of the Internal Elastic Lamina
To identify parameters distinguishing patients with and without upregulated neoangiogenesis, temporal arteries were analyzed for characteristic features besides the presence of a lumen-compromising intimal layer. Comparison of inflamed temporal arteries with and without angiogenesis in the inner arterial layers indicated that neovascularization correlated with a high number of multinucleated giant cells (MGCs). MGCs occur in 50–60% of patients with GCA and have been implicated in the fragmentation of the IEL, a typical finding of GCA. 20,21 Therefore, we explored whether the extent of IEL degradation was directly correlated with neoangiogenesis. In Figure 3 ▶ , two temporal artery tissues are compared, one with neoangiogenesis limited to the adventitia and one with vasa vasorum throughout the entire arterial wall. Giant cell formation, neovascularization, and disruption of the IEL were closely correlated. In cases of advanced degradation, new vasa vasorum emerged in the intimal layer. In temporal arteries with an intact IEL, vasa vasorum were restricted to the adventitia and were only slightly increased in number.
Figure 3.
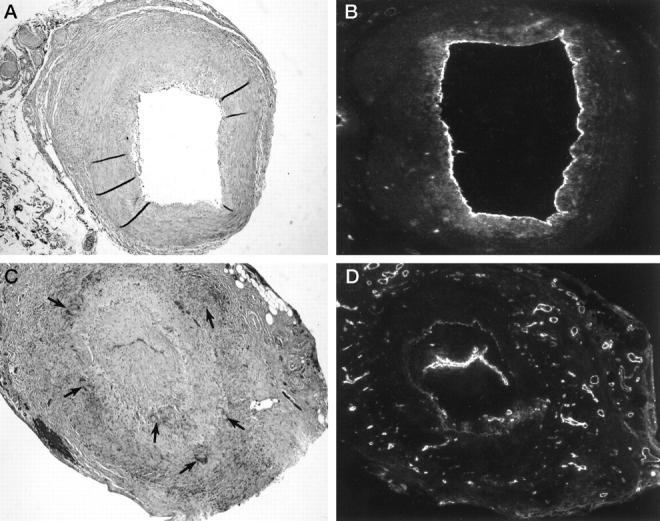
Giant cell formation, fragmentation of the internal elastic lamina, and neoangiogenesis in GCA. Temporal arteries from GCA patients with and without marked neoangiogenesis were compared. Hematoxylin/eosin (A, C) and von Willebrand’s factor staining (B, D) of two representative specimens are shown. Original magnification, ×100. B and C: In the temporal artery with vasa vasorum confined to the adventitia, multinucleated giant cells were absent and the internal elastic lamina (IEL), was well maintained. C and D: In the temporal artery with numerous microvessels in the media and the hyperplastic intima, a garland of multinucleated giant cells (arrows) was arranged along the disrupted IEL.
To investigate the relationship between elastic tissue digestion and microvessel proliferation, the perimeter of the IEL and the fraction of the IEL that had undergone destruction were determined by digital image analysis in 28 GCA patients (Figure 4) ▶ . In cases with almost circumferential destruction of the IEL, a high number of new blood vessels was formed. Conversely, if more than 70% of the elastic lamina was still intact, the number of microvessels was low. Nine of the 28 patients had fragmentation of less than 30% of the lamina perimeter. Most of these patients had fewer than 100 microvessels per arterial circumference. In the two individuals in this group with 104 and 142 microvessels, all of the vasa vasorum were found in the adventitia and none in the media and intima. Patients with more than 70% destruction of the IEL had 100 to 300 microvessels. From the data shown in Figure 4 ▶ , it appears that even with almost complete disruption of the lamina, the number of capillaries still increased. This observation raised the possibility that a second factor, besides the destruction of the elastic membrane, was involved in regulating angiogenesis. Comparison of the patients in the subset with almost complete lamina destruction did not reveal a relationship between the duration of vasculitis and the extent of neovascularization (data not shown).
Figure 4.

Correlation of the destruction of the internal elastic lamina (IEL) and the formation of new vasa vasorum in GCA. Cross-sections of temporal arteries from 28 patients with GCA were stained for von Willebrand’s factor to identify vasa vasorum. The number of capillary lumina in all layers of the arterial wall were counted. The perimeter of the IEL and the fraction of disrupted IEL were determined by digital image analysis. The percentage of fragmented IEL is shown in correlation to the number of microvessels in the arterial wall. Vasa vasorum formation was low in patients with a large proportion of the IEL intact but was high in patients with almost complete disruption of the IEL (R2 = 0.53).
Tissue Expression of VEGF Correlates with Neovascularization
To explore which cytokines and growth factors had a role in supporting neoangiogenesis, tissue extracts from temporal artery sections were analyzed for the expression of cytokine and growth factor transcripts. IFN-γ has been identified as a key cytokine in GCA and its tissue expression has been correlated with variations in clinical disease pattern. 8,21 VEGF and FGF-2 are cytokines that have been implicated in inducing the outgrowth of new capillaries. 14-17 IFN-γ-, FGF-2-, and VEGF-specific sequences were quantified in tissue extracts of 18 consecutive temporal artery biopsies from GCA patients. Results are presented in Figure 5 ▶ . The transcription of VEGF varied markedly among patients and ranged from 100 to 10,000 VEGF-specific sequences/2 × 10 5 β-actin sequences. High copy numbers correlated with increased neovascularization (R2 = 0.78). In contrast, no correlation was seen for FGF-2 production (R2 = 0.11). Although IFN-γ is not directly angiogenic, copy numbers of tissue IFN-γ mRNA were also closely correlated with the number of newly formed microvessels in the media and intima (R2 = 0.53). Possibly, IFN-γ has an indirect effect on angiogenesis by influencing the production of angiogenic growth factors in the arterial tissue. FGF-2 did not appear to have a role in neovascularization in GCA whereas VEGF emerged as a prime candidate.
Figure 5.

Expression of cytokine gene transcripts in temporal arteries with minimal and marked neoangiogenesis. Total RNA was obtained from 18 consecutive temporal artery biopsies from patients with GCA. cDNAs were adjusted for the number of β-actin transcripts and amplified with primer sets specific for IFN-γ, FGF-2, and VEGF. Results are given as the number of gene transcripts per 2 × 10 5 copies of β-actin. In parallel, sections of the temporal artery specimens were stained with antibodies to von Willebrand’s factor and microvessels per section were enumerated as described in Figure 1 ▶ . Transcription of IFN-γ and VEGF, but not of FGF-2 (βFGF), correlated with neoangiogenesis.
VEGF Derives from Multinucleated Giant Cells and from Macrophages at the Media-Intima Junction
The close correlation between tissue expression of VEGF and neovascularization raised the questions whence this growth factor derived and whether the origin of VEGF could provide information on why new vasa vasorum displayed a distinct topography in the intimal layer. Immunohistochemical analysis of tissue sections demonstrated that VEGF protein was easily detected in the arterial wall. By two-color staining, more than 90% of the VEGF-positive cells expressed the CD68 marker. Among these, the most intense staining was in MGCs (Figure 6) ▶ . Not all CD68-positive macrophages in the arterial wall contributed to VEGF production. Expression of VEGF protein in adventitial or intimal macrophages was the rare exception. Characteristically, it was found in the MGCs and macrophages aligned along the media-intima junction. Occasionally, very few smooth muscle cells and endothelial cells stained weakly positive with anti-VEGF antibodies. VEGF-positive macrophages and MGCs were arranged along the medial aspect of the IEL. These findings implicated MGCs in the regulation of neovascularization in the inflamed arterial wall in GCA.
Figure 6.
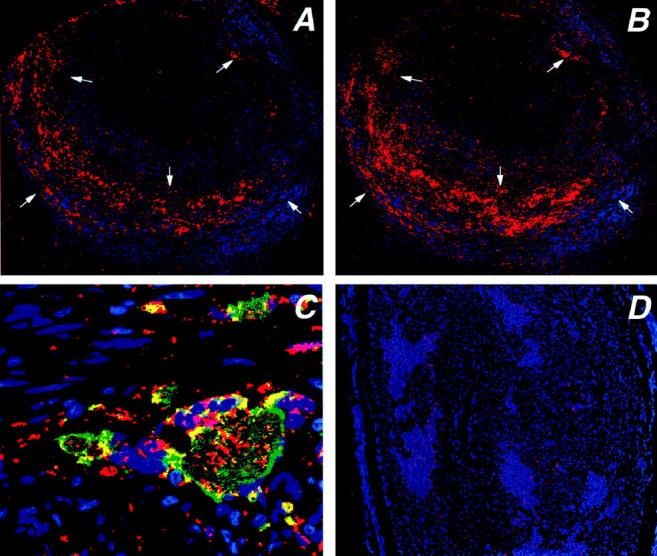
Production of vascular endothelial growth factor (VEGF) in temporal arteries with GCA. Tissue sections from temporal arteries were stained with anti-VEGF and anti-CD68 Abs. Anti-CD68 was visualized by using an FITC-labeled secondary antibody, anti-VEGF was developed by using a secondary biotinylated antibody, streptavidine-alkaline phosphatase and vector red. Stained sections were scanned by confocal microscopy and composite pictures were generated. Stainings for VEGF (A) and CD68 (B) are shown at an original magnification of ×100. VEGF production in the arterial wall was unevenly distributed with VEGF+ cells (red) accumulating at the media-intima junction. A small population of VEGF-producing cells was detected along the external elastic lamina. Cell nuclei are stained blue. More than 90% of the VEGF-producing cells expressed the CD68 marker. C: Strong expression of VEGF was typical for multinucleated giant cells. Blue, cell nuclei; red, VEGF; green, CD68; yellow, co-expression of VEGF and CD68. Original magnification, ×630. D: Control stainings without primary antibodies only showed minimal autofluorescence.
Discussion
Infiltration of the arterial wall by inflammatory cells in vasculitis induces a series of structural changes, only some of them destructive in nature. How these events in the artery relate to clinical disease is only partially understood. The purpose of the current study was to investigate the formation of new vasa vasorum in order to understand how the arterial wall responds to the inflammatory attack and how this translates into clinical manifestations of arteritis. The results of our study provide evidence that inflammatory cells, but not stromal cells of the arterial wall or endothelial cells, are the critical cell population in regulating the outgrowth of new capillaries. This observation contrasts with findings in atherosclerotic disease where smooth muscle cells and CD45RO-positive cells, but not tissue-infiltrating macrophages, are producers of VEGF. 22 Importantly, the multinucleated giant cell, a unique form of fused macrophages, emerged as a central player in modulating neovascularization in GCA. These findings suggest that neoangiogenesis in the inflamed arterial wall is not simply a consequence of tissue hypoxia, but, rather, is a primary mechanism introduced by the inflammatory infiltrate. The leading role of inflammatory cells in supporting neovascularization raises the question whether vasa vasorum formation could precede other critical events in the arterial wall, such as intimal hyperplasia, and could thus represent a primary disease mechanism. Recognition that MGCs, due to their potential to produce growth factors, hold a central position in the process of arterial stenosis should renew interest in this cell population. Understanding the precise role of MGCs in blood vessel wall inflammation is important because MGCs may provide ideal targets for novel therapeutic interventions.
The arterial wall itself is an unusual site for the formation of microvessels. In normal temporal arteries, vasa vasorum were restricted to the adventitial layer, confirming that the media and the intima are avascular. It is currently believed that the artery wall tissue receives nutrients by diffusion outward from the main lumen and inward from the adventitia. 9 Thickening of the arterial wall could therefore be suspected to generate hypoxia-induced angiogenesis. The tendency of new capillaries to form a circular arrangement in the intimal tissue but not at the intima-media junction could be interpreted as indicating a watershed with increased oxygen demands of the expanding intima. Hypoxia-initiated angiogenesis was suggested in a recent study describing abundant development of new vasa vasorum in porcine coronary arteries following experimental hypercholesterolemia. 23 Alterations in the arterial wall vascularization preceded the emergence of classical atherosclerotic lesions but were closely correlated to an increase in wall thickness. Data presented here indicate that hypoxia is very unlikely to be the triggering factor inducing neovascularization in vasculitis. Rather, VEGF was supplied by MGCs, which are not a reaction to hypoxic stress but represent a tissue-infiltrating cell. Studies describing an increased number of adventitial vasa vasorum in advanced coronary atherosclerosis 24,25 might also indicate that mechanisms other than hypoxia regulate the emergence of new microvessels in the arterial wall. 26-29
Variations in the degree of vasa vasorum neovascularization between patients provided an important clue and an opportunity to search for parameters associated with intramural angiogenesis. Therefore, we began to investigate on a molecular level how the outgrowth of new capillaries was supported. Semiquantification of cytokines and growth factors led to the identification of VEGF as an angiogenic factor that was closely correlated with the degree of microvessel formation. More interestingly, the tissue cytokine expression pattern demonstrated that the in situ activity of the IFN-γ gene was associated with MGC formation/up-regulated neovascularization/intimal hyperplasia. These results were in line with earlier reports correlating the local production of IFN-γ with MGC formation in GCA patients. 21 Full-blown vasculitis with vascular morbidity requires the transcription of IFN-γ in the arteries. In the absence of IFN-γ, only subclinical vessel wall inflammation is generated, presenting clinically as polymyalgia rheumatica. 30 High levels of tissue IFN-γ have been associated with a particular disease pattern characterized by the development of blindness, stroke, and jaw claudication. 21 Vice versa, low tissue transcription of IFN-γ has been encountered in patients with predominantly systemic manifestations, often lacking localized vascular manifestations. Tissue production of IFN-γ has also been associated with the emergence of MGCs, which are a typical but not a necessary component of vascular infiltrates in GCA. The questions of why some patients form MGCs and other do not and how that relates to the clinical picture of GCA has remained unanswered.
Data presented here allow us to propose a disease model partially addressing this conundrum (Figure 7) ▶ . The model predicts that IFN-γ derives from antigen-specific activation of CD4+ T cells in the adventitia. This event transmits signals to other cells, particularly CD68+ macrophages recruited to the arterial wall. Specialized macrophages in the media and along the media-intima junction undergo fusion and form MGCs. MGCs produce VEGF and thus facilitate the growth of new vasa vasorum. Because the site of the immunological injury is the adventitia and the new microvessels must sprout from vasa vasorum in the adventitia, additional signals from the adventitia might come into play. The molecular basis for the spatial distance between IFN-γ production and MGC formation remains to be elucidated. Support for a critical contribution of IFN-γ in MGC generation also comes from in vitro studies. Although many different cytokines have been suspected to induce monocyte fusion, strong evidence has pointed to IFN-γ as the fusogenic mediator. 31,32 In this model, induction of neoangiogenesis would reflect a certain type of T-cell-mediated immune response and would not be a consequence of tissue hypoxia resulting from the proliferation of the intimal layer. A fascinating aspect of this model is that IFN-γ-producing cells have characteristic features of antigen-activated T cells. 8 The nature and concentration of disease-relevant antigens in GCA may therefore influence the expression of the disease with its variant forms of vascular and systemic morbidity.
Figure 7.

Schematic model summarizing the role of multinucleated giant cells in vasculitis. A model is proposed that the generation, differentiation, and functional activity of multinucleated giant cells and macrophages at the media intima junction are regulated through T lymphocytes stimulated by antigen in the adventitia. Giant cells and specialized macrophages have a role in several pathways, including regulation of neoangiogenesis, intimal hyperplasia, and matrix degradation.
Formation of new microvessels may precede or accompany other structural changes that are also driven by MGCs. These include hyperplasia of the intima and digestion of the elastic tissue. Molecules critically involved in these processes can all be traced to the MGCs and, to some extent, to the macrophages sitting along the IEL. 3,33 As a rule, VEGF production in macrophages and giant cells does not involve cells localized on the intimal side of the IEL. Tsurumi et al have recently shown a reciprocal relation between NO and VEGF in a balloon arterial injury model. 34 Intimal macrophages in GCA preferentially express NOS-2. 33 It is therefore possible that locally produced NO down-regulates VEGF.
The precise function of MGCs in the vascular infiltrates in GCA has not been understood. It has been suggested that MGCs derive from macrophages to provide improved effector function in phagocytosis, intracellular killing of microorganisms, and sequestration of pathogens that are difficult to remove. Conclusive evidence for an infectious etiopathogenesis of GCA has not been forthcoming. Also, in vitro studies have shown that the ability to phagocytose is not maintained in macrophages after cell fusion. 32 Two lines of evidence support the hypothesis that MGCs are primarily tissue-destroying cells. First, they can produce matrix metalloproteinase (MMP)-2, an enzyme with substrate specificity for elastin. 33,35 MGCs characteristically lie next to fragmented pieces of IEL and could thus be instrumental in digesting the elastic membranes. 36 Second, MGCs have been found to be surrounded by lipid peroxidation products, as evidenced by the presence of the toxic aldehyde 4-hydroxynonenal (4-HNE). 37 The current study emphasizes a different aspect of the functional profile of MGCs, namely their ability to secrete VEGF. MGCs have been reported to also synthesize other growth factors involved in intimal proliferation, such as PDGF-AA and PDGF-BB. 3 Taken together, these data indicate that these peculiar cells have secretory abilities and a dual function in tissue destruction and attempted tissue repair.
Although our results directly implicate MGCs as the critical provider of the angiogenic factor VEGF and provide indirect evidence for a potential role of T cells in regulating the pattern and extent of vasa vasorum formation in the arterial wall, the mechanism(s) by which MGCs are induced and activated and how their function is regulated remain to be elucidated. The experiments reported here did not address the time sequence of neoangiogenesis, intimal hyperplasia, and IEL degradation. The co-occurrence of all three events may reflect shared regulatory pathways or interdependence of these pathological reactions. All of them are part of the arterial wall remodeling process that finally leads to luminal stenosis and eventual ischemia. Because all three tissue response patterns can be linked to MGCs, these unique cells emerge as critical cellular players in GCA. The findings are relevant to the potential for eliminating MGCs or disrupting their generation as novel therapeutic approaches to arteritis.
Acknowledgments
We thank Toni L. Higgins for secretarial support.
Footnotes
Address reprint requests to Cornelia M. Weyand, M.D., Mayo Clinic, 401 Guggenheim Building, 200 First Street, S.W., Rochester, MN 55905. E-mail: weyand.cornelia@mayo.edu.
Supported in part by the Mayo Foundation and by a grant from the National Institutes of Health (RO1 EY11916).
References
- 1.Hunder GG: Giant cell arteritis and polymyalgia rheumatica. Med Clin North Am 1997, 81:195-219 [DOI] [PubMed] [Google Scholar]
- 2.Nordborg E, Nordborg C, Malmvall BE, Andersson R, Bengtsson BA: Giant cell arteritis. Rheum Dis Clin North Am 1995, 21:1013-1026 [PubMed] [Google Scholar]
- 3.Kaiser M, Weyand CM, Bjornsson J, Goronzy JJ: Platelet-derived growth factor, intimal hyperplasia, and ischemic complications in giant cell arteritis. Arthritis Rheum 1998, 41:623-633 [DOI] [PubMed] [Google Scholar]
- 4.Evans JM, O’Fallon WM, Hunder GG: Increased incidence of aortic aneurysm and dissection in giant cell (temporal) arteritis: a population-based study. Ann Intern Med 1995, 122:502-507 [DOI] [PubMed] [Google Scholar]
- 5.Weyand CM, Goronzy JJ: Giant cell arteritis as an antigen-driven disease. Rheum Dis Clin North Am 1995, 21:1027-1039 [PubMed] [Google Scholar]
- 6.Brack A, Geisler A, Martinez-Taboada VM, Younge BR, Goronzy JJ, Weyand CM: Giant cell vasculitis is a T cell-dependent disease. Mol Med 1997, 3:530-543 [PMC free article] [PubMed] [Google Scholar]
- 7.Weyand CM, Schonberger J, Oppitz U, Hunder NN, Hicok KC, Goronzy JJ: Distinct vascular lesions in giant cell arteritis share identical T cell clonotypes. J Exp Med 1994, 179:951-960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wagner AD, Bjornsson J, Bartley GB, Goronzy JJ, Weyand CM: Interferon-gamma-producing T cells in giant cell vasculitis represent a minority of tissue-infiltrating cells and are located distant from the site of pathology. Am J Pathol 1996, 148:1925-1933 [PMC free article] [PubMed] [Google Scholar]
- 9.Wolinsky H, Glagov S: Nature of species differences in the medial distribution of aortic vasa vasorum in mammals. Circ Res 1967, 20:409-421 [DOI] [PubMed] [Google Scholar]
- 10.Risau W: Mechanisms of angiogenesis. Nature 1997, 386:671-674 [DOI] [PubMed] [Google Scholar]
- 11.Zetter BR: Angiogenesis and tumor metastasis. Annu Rev Med 1998, 49:407-424 [DOI] [PubMed] [Google Scholar]
- 12.Miller JW: Vascular endothelial growth factor and ocular neovascularization. Am J Pathol 1997, 151:13-23 [PMC free article] [PubMed] [Google Scholar]
- 13.Folkman J: Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med 1995, 1:27-31 [DOI] [PubMed] [Google Scholar]
- 14.Bussolino F, Mantovani A, Persico G: Molecular mechanisms of blood vessel formation. Trends Biochem Sci 1997, 22:251-256 [DOI] [PubMed] [Google Scholar]
- 15.Ferrara N, Davis-Smyth T: The biology of vascular endothelial growth factor. Endocr Rev 1997, 18:4-25 [DOI] [PubMed] [Google Scholar]
- 16.Basilico C, Moscatelli D: The FGF family of growth factors and oncogenes. Adv Cancer Res 1992, 59:115-165 [DOI] [PubMed] [Google Scholar]
- 17.Bikfalvi A, Klein S, Pintucci G, Rifkin DB: Biological roles of fibroblast growth factor-2. Endocr Rev 1997, 18:26-45 [DOI] [PubMed] [Google Scholar]
- 18.Hunder GG, Bloch DA, Michel BA, Stevens MB, Arend WP, Calabrese LH, Edworthy SM, Fauci AS, Leavitt RY, Lie JT: The American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum 1990, 33:1122-1128 [DOI] [PubMed] [Google Scholar]
- 19.Brack A, Rittner HL, Younge BR, Kaltschmidt C, Weyand CM, Goronzy JJ: Glucocorticoid-mediated repression of cytokine gene transcription in human arteritis-SCID chimeras. J Clin Invest 1997, 99:2842-2850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baldursson O, Steinsson K, Bjornsson J, Lie JT: Giant cell arteritis in Iceland: an epidemiologic and histopathologic analysis. Arthritis Rheum 1994, 37:1007-1012 [DOI] [PubMed] [Google Scholar]
- 21.Weyand CM, Tetzlaff N, Bjornsson J, Brack A, Younge B, Goronzy JJ: Disease patterns and tissue cytokine profiles in giant cell arteritis. Arthritis Rheum 1997, 40:19-26 [DOI] [PubMed] [Google Scholar]
- 22.Couffinhal T, Kearney M, Witzenbichler B, Chen D, Murohara T, Losordo DW, Symes J, Isner JM: Vascular endothelial growth factor/vascular permeability factor (VEGF/VPF) in normal and atherosclerotic human arteries. Am J Pathol 1997, 150:1673-1685 [PMC free article] [PubMed] [Google Scholar]
- 23.Kwon HM, Sangiorgi G, Ritman EL, McKenna C, Holmes DRJ, Schwartz RS, Lerman A: Enhanced coronary vasa vasorum neovascularization in experimental hypercholesterolemia. J Clin Invest 1998, 101:1551-1556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumamoto M, Nakashima Y, Sueishi K: Intimal neovascularization in human coronary atherosclerosis: its origin and pathophysiological significance. Hum Pathol 1995, 26:450-456 [DOI] [PubMed] [Google Scholar]
- 25.Williams JK, Armstrong ML, Heistad DD: Vasa vasorum in atherosclerotic coronary arteries: responses to vasoactive stimuli and regression of atherosclerosis. Circ Res 1988, 62:515-523 [DOI] [PubMed] [Google Scholar]
- 26.Barger AC, Beeuwkes R, Lainey LL, Silverman KJ: Hypothesis: vasa vasorum and neovascularization of human coronary arteries: a possible role in the pathophysiology of atherosclerosis. N Engl J Med 1984, 310:175-177 [DOI] [PubMed] [Google Scholar]
- 27.Barker SG, Talbert A, Cottam S, Baskerville PA, Martin JF: Arterial intimal hyperplasia after occlusion of the adventitial vasa vasorum in the pig. Arterioscler Thromb 1993, 13:70-77 [DOI] [PubMed] [Google Scholar]
- 28.Martin JF, Booth RF, Moncada S: Arterial wall hypoxia following hyperfusion through the vasa vasorum is an initial lesion in atherosclerosis. Eur J Clin Invest 1990, 20:588-592 [DOI] [PubMed] [Google Scholar]
- 29.Cuevas P, Gonzalez AM, Carceller F, Baird A: Vascular response to basic fibroblast growth factor when infused onto the normal adventitia or into the injured media of the rat carotid artery. Circ Res 1991, 69:360-369 [DOI] [PubMed] [Google Scholar]
- 30.Weyand CM, Hicok KC, Hunder GG, Goronzy JJ: Tissue cytokine patterns in patients with polymyalgia rheumatica and giant cell arteritis. Ann Intern Med 1994, 121:484-491 [DOI] [PubMed] [Google Scholar]
- 31.Fais S, Burgio VL, Silvestri M, Capobianchi MR, Pacchiarotti A, Pallone F: Multinucleated giant cells generation induced by interferon-gamma: changes in the expression and distribution of the intercellular adhesion molecule-1 during macrophages fusion and multinucleated giant cell formation. Lab Invest 1994, 71:737-744 [PubMed] [Google Scholar]
- 32.Most J, Neumayer HP, Dierich MP: Cytokine-induced generation of multinucleated giant cells in vitro requires interferon-gamma and expression of LFA-1. Eur J Immunol 1990, 20:1661-1667 [DOI] [PubMed] [Google Scholar]
- 33.Weyand CM, Wagner AD, Bjornsson J, Goronzy JJ: Correlation of the topographical arrangement and the functional pattern of tissue-infiltrating macrophages in giant cell arteritis. J Clin Invest 1996, 98:1642-1649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsurumi Y, Murohara T, Krasinski K, Chen D, Witzenbichler B, Kearney M, Couffinhal T, Isner JM: Reciprocal relation between VEGF and NO in the regulation of endothelial integrity. Nat Med 1997, 3:879-886 [DOI] [PubMed] [Google Scholar]
- 35.Nikkari ST, Hoyhtya M, Isola J, Nikkari T: Macrophages contain 92-kd gelatinase (MMP-9) at the site of degenerated internal elastic lamina in temporal arteritis. Am J Pathol 1996, 149:1427-1433 [PMC free article] [PubMed] [Google Scholar]
- 36.Lie JT: Histopathologic specificity of systemic vasculitis. Rheum Dis Clin North Am 1995, 21:883-909 [PubMed] [Google Scholar]
- 37.Rittner HL, Kaiser M, Brack A, Szuleda LI, Goronzy JJ, Weyand CM: Tissue destructive macrophages in giant cell arteritis. Circ Res (in press) [DOI] [PubMed]