

Abstract
We have characterized amyloid β peptide (Aβ) concentration, Aβ deposition, paired helical filament formation, cerebrovascular amyloid angiopathy, apolipoprotein E (ApoE) allotype, and synaptophysin concentration in entorhinal cortex and superior frontal gyrus of normal elderly control (ND) patients, Alzheimer’s disease (AD) patients, and high pathology control (HPC) patients who meet pathological criteria for AD but show no synapse loss or overt antemortem symptoms of dementia. The measures of Aβ deposition, Aβ-immunoreactive plaques with and without cores, thioflavin histofluorescent plaques, and concentrations of insoluble Aβ, failed to distinguish HPC from AD patients and were poor correlates of synaptic change. By contrast, concentrations of soluble Aβ clearly distinguished HPC from AD patients and were a strong inverse correlate of synapse loss. Further investigation revealed that Aβ40, whether in soluble or insoluble form, was a particularly useful measure for classifying ND, HPC, and AD patients compared with Aβ42. Aβ40 is known to be elevated in cerebrovascular amyloid deposits, and Aβ40 (but not Aβ42) levels, cerebrovascular amyloid angiopathy, and ApoE4 allele frequency were all highly correlated with each other. Although paired helical filaments in the form of neurofibrillary tangles or a penumbra of neurites surrounding amyloid cores also distinguished HPC from AD patients, they were less robust predictors of synapse change compared with soluble Aβ, particularly soluble Aβ40. Previous experiments attempting to relate Aβ deposition to the neurodegeneration that underlies AD dementia may have failed because they assayed the classical, visible forms of the molecule, insoluble neuropil plaques, rather than the soluble, unseen forms of the molecule.
Several previous studies 1-5 have reported observing patients who had no overt symptoms of dementia antemortem but, at autopsy, were found to have profuse plaques and tangles sufficient to otherwise qualify for the diagnosis of Alzheimer’s disease (AD). Based on these characteristics, we have termed such patients high pathology controls (HPCs) as compared with the usual nondemented elderly controls (ND) who come to autopsy without history of dementia or significant AD pathological findings. 5
HPC patients may be of great importance in unraveling the relative contributions and sequencing of different postmortem pathological features in AD neurodegeneration and dementia. In some cases, for example, they appear to be very early AD patients just at the margins of expression of overt clinical disease. 2 We have also found that HPC patients show little or no evidence of neocortical or limbic synapse loss, 5 underscoring the critical relationship of neuritic degeneration to dementia. Similarly, by seeking other AD pathophysiological processes that are not extant or not yet fully extant in HPC patients, insight into their significance for the clinical expression of AD may be gained.
Finally, the inclusion of HPC patients in AD studies may offer advantages for correlational statistics because these patients often provide an intermediate subset between ND and AD groups. In scatter plots depicting plaque counts versus dementia scores, for example, the data for ND and AD patients tend to cluster at extremes (by definition, ND patients have few plaques and low dementia scores and AD patients have many plaques and high dementia scores). 6 Under these circumstances, correlational statistics essentially draw a straight line connecting the two clusters of data as if there were a continuum of points between them, leading to the potentially spurious inference that the more plaques patients have the more demented they will become. 7 In fact, if one looks at the data for either group alone, the correlation may not be significant at all. For this reason, the ability of HPC patients to provide a more continuous spectrum of data between ND and AD patients lends increased substance to correlational inferences about those groups. Similarly, the demonstration that a significant correlation across several experimental groups continues to hold when only one group or another is examined also strengthens any within-subjects inferences that might be drawn from the data.
In the present research, we quantified amyloid β peptide (Aβ) concentrations, plaque type, apolipoprotein E (ApoE) allele frequency, cerebrovascular amyloid angiopathy, and paired helical filament formation in entorhinal cortex and superior frontal gyrus of ND, HPC, and AD patients. In contrast to previous qualitative observations wherein an absence of cored Aβ deposits was suggested to discriminate HPC from AD patients, 3,8 we found the type and number of Aβ deposits to be similar in these groups. Rather, of the many variables studied, soluble Aβ concentrations best distinguished HPC from AD patients. Soluble Aβ40, in particular, was a very robust predictor of synapse loss, giving significant inverse correlations over all patients and within the AD group alone.
Materials and Methods
Patient Samples
From among 124 routine brain autopsies of ND patients without prior medical history of dementia, eight were obtained that appeared to exhibit sufficient neocortex Aβ plaques and entorhinal cortex neurofibrillary tangles to otherwise qualify for the diagnosis of AD. 9 These HPC patients were contrasted with eight randomly selected ND patients who had limited AD pathology and no prior medical history of dementia and eight randomly selected AD patients who had previously received a clinical diagnosis of probable AD that was confirmed neuropathologically at autopsy. Post hoc evaluation of the samples by a neuropathologist (T. Beach) using CERAD pathological criteria 9 and Braak staging 10 confirmed the initial assignment of patients to groups with the possible exception of one ND patient who might well have qualified for the HPC group and one HPC patient who could equally have been assigned to the ND group. Since 1) the evaluations were post hoc, 2) the primary intent of including an HPC group was to provide a more continuous range of pathology than afforded by ND and AD patients only, and 3) the statistical assessments were not materially affected by reassigning the patients, the original assignments were maintained. The ND, HPC, and AD patients evaluated here represent a second sample from our autopsy population and differ from those we previously tested for inflammatory correlates of dementia. 5 Absence of material symptoms of dementia in HPC and ND patients was confirmed by reference to medical records and interviews with relatives, attending physicians, and nurses. Nonetheless, in the absence of more definitive premortem cognitive status data, the present research focuses on a correlate of dementia that is quantifiable postmortem, synapse loss, 11,12 and not on dementia itself.
Brain Samples and Processing
Brains were removed at autopsy, weighed, and immersed in ice-cold 0.1 mol/L phosphate buffer (pH 7.4). They were then sectioned coronally at 1-cm intervals, and blocks of the entorhinal cortex (including the transentorhinal area) at the level of the anterior hippocampus and superior frontal gyrus at the level of the genu of the corpus callosum were dissected. These samples were post-fixed for 24 to 36 hours in ice-cold 4% buffered paraformaldehyde (pH 7.4), cut at 40 μm on a freezing microtome or paraffin embedded and cut at 6 μm on a rotary microtome, and subjected to histochemical and immunohistochemical procedures. Ventricular cerebrospinal fluid (CSF) samples and contralateral entorhinal cortex and superior frontal gyrus samples were snap frozen and stored at −80°C until assay by ELISA and other methods.
Soluble and Insoluble Aβ40 and Aβ42 Europium Immunoassay (EuIA)
As previously described, 13 soluble and insoluble Aβ40 and Aβ42 were measured using EuIA methods, with rabbit polyclonal antisera R163 and R165 (P. Metha, New York Institute for Mental Disabilities, Staten Island, NY) as capture antibodies for Aβ40 and Aβ42, respectively, and Europium-labeled mouse monoclonal antibody 4G8 (Senetec & Mease, Maryland, MO) as the detection antibody. Data for total soluble Aβ (Aβ40 plus Aβ42), total insoluble Aβ (Aβ40 plus Aβ42), and Aβ40 and Aβ42 alone were evaluated. We note that, while correctly distinguishing the Aβ40 from the Aβ42 carboxy terminus of the peptides, our EuIA assay does not discriminate full-length Aβ from Aβ fragments. Such a discrimination could be an important target for future research, as Aβ17–42, for example, has been reported to be the major constituent of the diffuse plaque. 14 The soluble and insoluble Aβ fractions were prepared by finely mincing 200 mg of unfixed entorhinal cortex or superior frontal gyrus from each patient, followed by homogenization in a glass Ten Broeck tissue grinder in the presence of 4 ml of 20 mmol/L Tris/HCl buffer (pH 7.4) containing 3 mmol/L EDTA, 500 μg/L leupeptin, 700 μg/L pepstatin, 350 mg/L phenylmethylsulfonyl fluoride, 100 mg/L 1,10-phenanthroline, and 100 mg/L benzamidine. The brain homogenates were spun at 220,000 × g in polyallomer tubes in a Sorval AH-650 rotor for 1 hour at 4°C. For the estimation of soluble Aβ, an aliquot of 100 μl of supernatant was assayed by EuIA, as described above. For insoluble Aβ quantitation, the pellet was dissolved in 5 ml of 98% glass-distilled formic acid and centrifuged at 220,000 × g for 30 minutes at 4°C. An aliquot of 50 μl was diluted 10X with 0.25 mol/L Tris/HCl (pH 7.4) plus 30% acetonitrile, then pH adjusted to 7.4 with 10 N NaOH using a microelectrode. After further dilution (5X) with 20 mmol/L Tris/HCl buffer plus 0.05% Tween-20, an aliquot of 100 μl was submitted for EuIA. Because the factors that determine Aβ solubility in tissue are incompletely understood, the techniques used here necessarily establish a working definition of soluble versus insoluble Aβ, as in previous studies. 15 That is, those molecules that remain in the aqueous supernatant after centrifugation at 220,000 × g for 1 hour are considered as soluble Aβ, and those Aβ aggregates that sediment under these conditions are considered as insoluble Aβ. 15 By ultrafiltration and molecular sieving the soluble fraction appears to contain Aβ oligomers that fall within the following size ranges: >100 kd, 30 to 100 kd, 10 to 30 kd, and <10 kd, corresponding to assemblies of >22 molecules, 7 to 22 molecules, 3 to 7 molecules, and monomers/dimers. 15
CSF Aβ42
CSF samples were submitted blind to Athena Diagnostics, (Worcester, MA), and levels of Aβ42 were quantified as previously described. 16
Immunohistochemistry and Quantification of Plaque Type, Cerebrovascular Amyloid Angiopathy, and Paired Helical Filament Formation
Routine thioflavin S histofluorescence staining to reveal Aβ deposits and neurofibrillary pathology were performed on the entorhinal cortex and superior frontal gyrus sections. Immunohistochemistry of Aβ used mouse monoclonal antibodies 6E10 and 4G8 from Senetek & Mease at 1:500 and 1:1000 dilutions, respectively. No material differences were observed in staining by these antibodies, and the mean is presented here. Aβ deposits were distinguished as having a discernible Aβ-immunoreactive dense core surrounded by Aβ-immunoreactive neurites or as having diffuse Aβ immunoreactivity only. 17 Paired helical filament immunohistochemistry used anti-PHF1 antibody (gift of S. Greenberg) at a 1:500 dilution to reveal penumbras of presumably dystrophic neurites surrounding amyloid cores. 18,19 All immunohistochemistry was performed free-floating at the same time and in the same wells for ND, HPC, and AD patients. As in our previous research, 5 counts of the various elements at ×100 final magnification were recorded by an observer blind to disease state. For each section, two columns of five nonoverlapping fields stretching from the pial surface to the beginning of the white matter were assayed. The columns were located approximately one-third of the way in from the beginning and end of each structure. Cerebrovascular amyloid angiopathy was quantified by a blind observer on a scale of 0 to 3, with 0 representing no visible vascular-associated Aβ deposits and 3 representing profuse deposition. Representative examples of thioflavin histofluorescent plaques and tangles, Aβ-immunoreactive plaques with and without cores, paired helical filament occurrence around plaques, and cerebrovascular amyloid angiopathy are given in Figure 1 ▶ .
Figure 1.
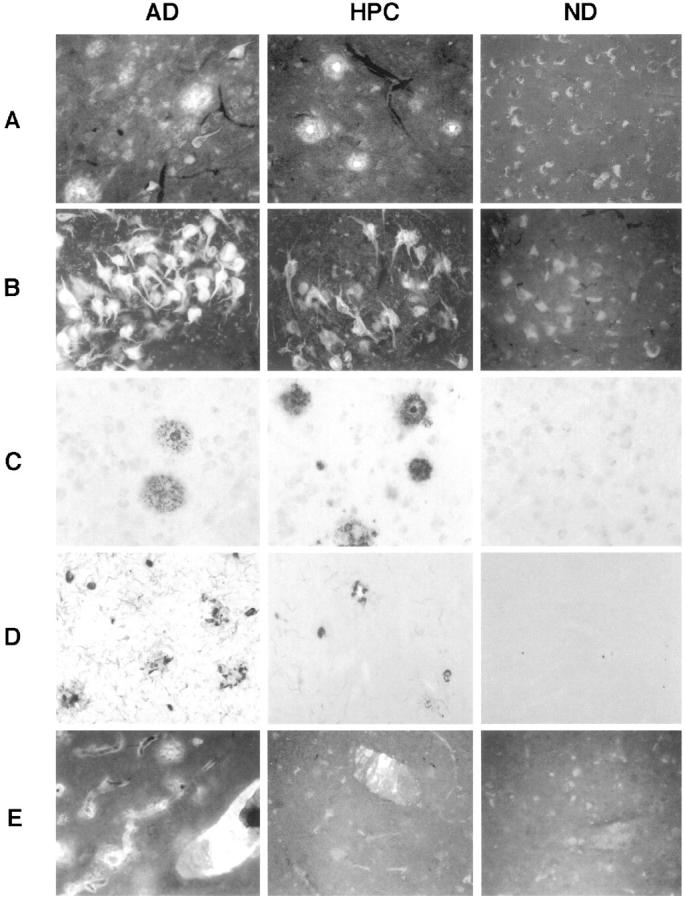
Representative examples in AD patients (left panels), HPC patients (middle panels), and ND patients (right panels) of thioflavin histofluorescent plaques (A) and tangles (B), Aβ immunoreactive plaques with and without cores (C), paired helical filament occurrence around plaques (D), and cerebrovascular amyloid angiopathy (E).
Synaptic Density Estimates
Protein extracts from entorhinal cortex and superior frontal gyrus homogenates of ND, HPC, and AD patients were subjected to standard synaptophysin Western blot analysis and densitometry as previously described. 5
Apolipoprotein E Allele Frequency
Genomic DNA was extracted 20 from approximately 50-mg cerebellar samples from each patient. The purified DNA was then subjected to PCR amplification using allele-specific oligonucleotide probes that span positions 112 and 158 of the ApoE locus. Amplified ApoE PCR products were digested with restriction enzyme HhaI and electrophoresed for classification on polyacrylamide gels. 21
Statistics
Parametric outcome measures that could be assayed in both entorhinal cortex and superior frontal gyrus were first evaluated by two-way repeated measures analysis of variance (ANOVA), with disease state (AD, HPC, or ND) as the first factor and brain region (entorhinal cortex or superior frontal gyrus) as the second, repeated-measures factor. For variables with overall significance, comparisons were then made between HPC and ND patients only and between HPC and AD patients only using the same two-way repeated-measures ANOVA approach. Nonparametric outcome measures (cerebrovascular amyloid angiopathy, ApoE allele frequency) were evaluated by the Kruskal-Wallis statistic (comparisons across all three groups) and the Mann-Whitney U statistic (comparisons between two groups). Correlations of a priori interest were performed using Pearson’s R statistic. To ensure that significant correlations did not result simply from an anchoring of the data at one extreme by ND patients, Pearson’s tests were re-run using HPC and AD patients only. The CSF Aβ42 data point for one patient was excluded from analysis because it exhibited a mean value more than seven standard deviations from the other patients and more than twice that of the next highest patient. This exclusion altered the significance test by ANOVA from P < 0.10 to P < 0.01. We note that previous studies have found significant differences in CSF Aβ42 when AD patients were compared with ND patients (cf Ref. 16 ).
Results
Patient Entry Characteristics
Summary data for the experiments are given in Table 1 ▶ , including patient entry statistics. Patients in the AD, HPC, and ND groups were well matched, with statistically similar values for age, gender, postmortem interval, and length of sample storage. For ND patients, the median Braak stage was II, the median CERAD plaque score was 0, and the median CERAD neuropathology diagnosis was “not AD”. For HPC patients, the median Braak stage was IV, the median CERAD plaque score was B, and the median CERAD neuropathology diagnosis was “possible AD”. For AD patients, the median Braak stage was VI, the median CERAD plaque score was C, and the median CERAD neuropathology diagnosis was “definite AD”.
Table 1.
Patient Entry and Experimental Data (¯X ± SEM)
| Variable | Patient condition | ||
|---|---|---|---|
| ND | HPC | AD | |
| Age (years) | 80 ± 2 | 82 ± 3 | 81 ± 2 |
| Sex (M/F) | 6/2 | 6/2 | 6/2 |
| CSF Aβ42 (pg/ml) | 232.4 ± 25.0 | 224.8 ± 18.8 | 162.9 ± 2.9 |
| Autolysis (hours) | 3.1 ± 0.4 | 3.7 ± 0.9 | 3.5 ± 0.2 |
| Brain weight (g) | 1271.9 ± 50.2 | 1155.0 ± 39.9 | 1112.5 ± 65.0 |
| ApoE allele distribution | |||
| E2/E2 | 00% | 13% | 00% |
| E2/E3 | 25% | 00% | 00% |
| E3/E3 | 75% | 50% | 25% |
| E2/E4 | 00% | 00% | 00% |
| E3/E4 | 00% | 38% | 38% |
| E4/E4 | 00% | 00% | 38% |
| Entorhinal cortex | |||
| Synaptophysin (OD) | 8.8 ± 0.6 | 8.1 ± 0.6 | 5.9 ± 0.3 |
| Insoluble Aβ40 (μg/g) | 0.8 ± 0.3 | 1.4 ± 0.3 | 53.7 ± 25.9 |
| Insoluble Aβ42 (μg/g) | 8.3 ± 4.7 | 64.2 ± 10.8 | 117.3 ± 18.1 |
| Soluble Aβ40 (pg/g) | 1.9 ± 0.6 | 4.5 ± 0.9 | 66.5 ± 18.7 |
| Soluble AB42 (pg/g) | 0.0 ± 0.0 | 6.2 ± 3.0 | 15.5 ± 5.9 |
| Thioflavin plaques (count/mm2) | 0.4 ± 0.3 | 25.8 ± 4.5 | 46.6 ± 13.1 |
| Aβ plaques, no core (count/mm2) | 0.8 ± 0.8 | 13.9 ± 2.5 | 17.9 ± 2.1 |
| Aβ plaques, core (count/mm2) | 0.0 ± 0.0 | 0.5 ± 0.1 | 0.7 ± 0.2 |
| PHF1 plaques (count/mm2) | 1.1 ± 0.6 | 1.1 ± 0.4 | 4.5 ± 0.9 |
| Amyloid angiopathy (0–3) | 0.0 ± 0.0 | 0.1 ± 0.1 | 1.5 ± 0.5 |
| Thioflavin tangles (count/mm2) | 0.8 ± 0.3 | 12.3 ± 2.4 | 48.1 ± 11.2 |
| Superior frontal gyrus | |||
| Synaptophysin (OD) | 10.2 ± 0.4 | 9.5 ± 0.4 | 7.0 ± 0.5 |
| Insoluble Aβ40 (μg/g) | 1.1 ± 0.3 | 7.4 ± 4.1 | 105.4 ± 40.2 |
| Insoluble Aβ42 (μg/g) | 11.9 ± 5.6 | 108.8 ± 9.1 | 142.1 ± 15.6 |
| Soluble Aβ40 (pg/g) | 2.5 ± 1.5 | 14.0 ± 6.2 | 103.8 ± 18.4 |
| Soluble AB42 (pg/g) | 0.0 ± 0.0 | 4.0 ± 2.7 | 6.7 ± 3.7 |
| Thioflavin plaques (count/mm2) | 7.3 ± 4.1 | 77.3 ± 12.3 | 76.6 ± 11.2 |
| Aβ plaques, no core (count/mm2) | 0.0 ± 0.0 | 25.4 ± 4.6 | 31.2 ± 3.3 |
| Aβ plaques, core (count/mm2) | 0.1 ± 0.1 | 2.2 ± 0.5 | 1.5 ± 0.5 |
| PHF1 plaques (count/mm2) | 0.3 ± 0.3 | 0.3 ± 0.2 | 3.8 ± 1.2 |
| Amyloid angiopathy (0–3) | 0.0 ± 0.0 | 0.8 ± 0.3 | 1.9 ± 0.5 |
| Thioflavin tangles (count/mm2) | 0.0 ± 0.0 | 0.1 ± 0.1 | 8.1 ± 3.8 |
Synaptic Density
As in our previous study, 5 HPC patients had significantly higher (>36%) synaptophysin immunoreactivity on Western blot analysis compared with AD patients (F1,14 = 25.8, P < 0.0003), whereas they did not differ from ND patients on this measure (F1,14 = 1.9, P > 0.50, 7% difference; Table 1 ▶ ).
Aβ Deposition
The HPC group exhibited significantly greater numbers of thioflavin histofluorescent plaques (>1200%; F1,14 = 35.2, P < 0.0001), plaques without cores (>4800%; F1,14 = 38.3, P < 0.0001), or plaques with cores (>2700%; F1,14 = 26.6, P < 0.0002) compared with the ND group, whereas these measures failed to distinguish HPC from AD patients. Indeed, HPC patients had only 16% fewer thioflavin-positive plaques (F1,14 = 0.6, P > 0.50), 20% fewer plaques without cores (F1,14 = 1.4, P > 0.50), and 27% more plaques with cores (F1,14 = 0.6, P > 0.50). Across the brain regions evaluated, none of these measures was a significant within-subjects correlate of synapse loss when all of the groups or the HPC and AD groups together were considered.
Soluble and Insoluble Aβ40 and Aβ42 Concentrations (Figure 2) ▶
Figure 2.

Soluble and insoluble Aβ40 and Aβ42 concentrations in superior frontal gyrus and entorhinal cortex of ND, HPC, and AD patients.
Like the Aβ plaques that can be seen under the microscope, concentrations of insoluble Aβ (insoluble Aβ40 plus Aβ42) distinguished HPC from ND patients (F1,14 = 89.2, P < 0.0001), but failed to discriminate HPC from AD patients (F1,14 = 2.8, P > 0.30). Although in some instances there was a substantial difference among group means, insoluble Aβ levels within groups were often highly variable. Mean insoluble Aβ40 in AD entorhinal cortex, for example, was 53.7 ± 25.9 μg/g, some 38-fold higher than the mean for HPC entorhinal cortex. However, one-half of the AD patients actually had concentrations less than 4 μg/g, substantially overlapping the values for HPC patients and leading to a nonsignificant comparison.
In contrast to the results for insoluble Aβ, HPC patients had sevenfold higher soluble Aβ concentrations than ND patients (F1,14 = 10.0, P < 0.008) and sevenfold lower soluble Aβ concentrations than AD patients (F1,14 = 24.2, P < 0.003). Soluble Aβ levels were a significant, inverse, within-subjects correlate of synapse loss regardless of whether all groups (r = −0.58, P < 0.0001) or the HPC and AD groups (r = −0.53, P < 0.002) were considered (Figure 3, A–C) ▶ . The association of soluble Aβ40 with synapse changes was especially striking and could be observed in AD patients alone even in a single brain region (Figure 3D) ▶ . Soluble Aβ42, measured in ventricular CSF, also distinguished HPC from AD (F1,14 = 9.3, P < 0.01) but not ND (F1,14 = 0.1, P > 0.5) patients.
Figure 3.

Synaptic density estimates from synaptophysin Western blot analysis (synaptic density OD) as a function of entorhinal cortex and superior frontal gyrus soluble Aβ concentrations. Upper left panel: Sum of soluble Aβ40 plus Aβ42 versus synaptophysin optical density (OD) in all patients. Upper right panel: Sum of soluble Aβ40 plus Aβ42 in HPC and AD patients only. Correlations within individual structures or for soluble Aβ40 and Aβ42 alone were also highly significant and gave trends similar to those illustrated here. For example, the bottom panels show the data for soluble Aβ40 in superior frontal gyrus of all patients (left) or AD patients alone (right).
With respect to Aβ amino acid length, Aβ40, whether in the soluble or insoluble form, more consistently defined HPC and AD patients than Aβ42. For example, the comparison of HPC with AD patients barely reached significance for insoluble Aβ42 (F1,14 = 7.2, P < 0.02) and did not reach significance for soluble Aβ42 (F1,14 = 1.4, P > 0.50). By contrast, HPC and AD patients differed significantly, sometimes by as much as 50-fold, with respect to Aβ40 (for the soluble form, F1,14 = 18.0, P < 0.0009; for the insoluble form, F1,14 = 5.3, P < 0.04).
Paired Helical Filament Formation
Paired helical filament formation in the form of thioflavin histofluorescent neurofibrillary tangles was significantly greater in HPC compared with ND patients (>1400%; F1,14 = 22.8, P < 0.0003), whereas it was significantly lower (<80%) in HPC compared with AD patients (F1,14 = 8.7, P < 0.02). PHF1 immunoreactivity of neurites surrounding amyloid cores was also significantly lower (>80%) in HPC compared with AD patients (F1,14 = 11.9, P < 0.004), but was statistically similar (0% difference) in HPC and ND patients (F1,14 = 0.0, P > 0.50). Both measures were significant, inverse, within-subjects correlates of synaptic loss (for tangles, Rall groups = −0.45, P < 0.002, and RHPC+AD = −0.38, P < 0.04; for PHF1 immunoreactivity, Rall groups = −0.48, P < 0.006, and RHPC+AD = −0.44, P < 0.02; Figure 4 ▶ ).
Figure 4.

Synaptic density estimates from synaptophysin Western blot analysis as a function of entorhinal cortex and superior frontal gyrus paired helical filament immunoreactive (PHF+) plaques/mm 2 (left panel) or thioflavin histofluorescent tangles/mm 2 (right panel). Data points for ND, HPC, and AD patients are included.
Cerebrovascular Amyloid Angiopathy
This variable successfully discriminated HPC from ND patients (U = 88, P < 0.02) and HPC from AD patients (U = 62, P < 0.007).
Apolipoprotein E Allele Frequency
Of the ND patients sampled, none were heterozygous or homozygous for the ApoE4 allele. Of the HPC patients sampled, three were heterozygous and none were homozygous for the E4 allele. Of the AD patients sampled, three were heterozygous and three were homozygous for the E4 allele. These distributions resulted in a significant difference in ApoE4 allele frequency among the groups (KW = 9.99, P < 0.01). We note that patients were selected for study without any prior knowledge of their ApoE types.
Although all of the measures of visible AD pathology (ie, thioflavin histofluorescent plaques, diffuse plaques, cored plaques, PHF1-immunoreactive plaques, and thioflavin histofluorescent tangles) exhibited significant effects of ApoE4 allele frequency, it is important to recognize that our study and others (cf Ref. 22 ) did not include balanced representation of ApoE4 allele frequency in all of the patient groups because the patients were randomly selected from the available autopsy pool. As a result, no patient in the ND group was either heterozygous or homozygous for the ApoE4 allele. Such a circumstance substantially violates ANOVA assumptions, and indeed, when only HPC and AD groups, both of which did contain ApoE4 patients, or AD patients alone were considered, the significant effects of ApoE4 allele frequency on plaque and tangle burden vanished. By contrast, across all subjects, HPC plus AD subjects, or AD patients alone, ApoE4 allele frequency had a significant effect on soluble Aβ40 concentrations (all subjects, F2,21 = 35.12, P < 0.0001; AD and HPC, F2,13 = 17.77, P < 0.002; AD only, F2,5 = 8.05, P < 0.03), insoluble Aβ40 concentrations (all subjects, F2,21 = 72.11, P < 0.0001; AD and HPC, F2,13 = 39.95, P < 0.0001; AD only, F2,5 = 12.00, P < 0.02), and soluble Aβ42 concentrations (all subjects, F2,21 = 16.71, P < 0.0001; AD and HPC, F2,13 = 7.98, P < 0.006; AD only, F2,5 = 27.45, P < 0.003). Insoluble Aβ42 concentrations were not significantly affected by ApoE4 allele frequency.
Although correlations of nonparametric variables with parametric variables can only be viewed as suggestive, clear within-subjects relationships of ApoE4 with increased levels of soluble Aβ40 (r = 0.85, P < 0.001) and insoluble Aβ40 (r = 0.53, P < 0.01) but not with levels of soluble or insoluble Aβ42 were observed. In turn, both increased ApoE4 allele frequency (r = 0.59, P < 0.01) and Aβ40 levels (soluble Aβ40, r = 0.67, P < 0.001; insoluble Aβ40, r = 0.53, P < 0.01) were highly associated with increased occurrence and severity of cerebrovascular amyloid angiopathy. ApoE4, Aβ40, and cerebrovascular amyloid angiopathy were equally or even more strongly associated when AD and HPC patients alone were considered. Conversely, in both all patients and AD plus HPC patients alone, the 42-amino-acid form of Aβ was not significantly correlated with ApoE frequency or cerebrovascular amyloid angiopathy.
Brain Regional Differences
Across patients, synaptophysin levels (F1,21 = 20.0, P < 0.006), tangle counts (F1,21 = 39.8, P < 0.0001), plaque counts (F1,21 = 32.1, P < 0.0005), soluble Aβ40 and Aβ42 (F1,21 = 5.2, P < 0.04), and insoluble Aβ40 and Aβ42 (F1,21 = 4.3, P < 0.05) all exhibited statistically significant differences when entorhinal cortex was compared with superior frontal gyrus, with the latter being higher on all measures except neurofibrillary tangles. Nonetheless, virtually all these measures in entorhinal cortex and superior frontal gyrus were significantly correlated across the two brain regions (synaptophysin, r = 0.45, P = 0.03; neurofibrillary tangles, r = 0.88, P < 0.001; thioflavin histofluorescent plaques, r = 0.70, P < 0.001; soluble Aβ40, r = 0.89, P < 0.001; soluble Aβ42, r = 0.64, P < 0.001; insoluble Aβ40, r = 0.85; P < 0.001; and insoluble Aβ42, r = 0.79, P < 0.001), suggesting that although the two brain regions have different predilections for AD changes, such changes occur in parallel in both structures. Similar results on brain regional changes in AD pathology were obtained in our previous study of ND, HPC, and AD patients. 5
Prediction of Synaptic Change
When all of the parametric variables were placed into a step-wise linear regression model with synapse change as the outcome measure, soluble Aβ40 and Aβ42 concentrations were found to account for 40% of the variance, with no other significant predictors. This remained true when AD patients only were considered (R2 = 0.38, P < 0.01). Of the two, soluble Aβ40 accounted for substantially more of the variance than soluble Aβ42. Forcing neurofibrillary tangles and PHF1-immunoreactive plaques into the model added 2% to the variance prediction. Forcing the remaining variables into the model added only an additional 2%.
Discussion
Comparisons of HPC with AD patients can help indicate pathophysiological processes that may be critical to the development of overt dementia. That is, one would expect a pathophysiologically relevant process to be present in AD patients but significantly reduced or absent in HPC or preclinical AD patients. Several of the variables assayed here meet this criterion, and several others do not.
Plaque type, whether distinguished by thioflavin histofluorescence or Aβ immunohistochemistry or by the presence or absence of a distinct core, did not discriminate HPC from AD patients, although it did distinguish HPC from ND patients. These findings argue against the assumption that HPC, early AD, or pathological aging patients fail to show symptoms of dementia or evidence of neurodegeneration, despite profuse Aβ deposition, because their plaques are almost wholly of the diffuse type. 3,8,23-25 HPC and AD patients shared statistically similar numbers of Aβ deposits without a discernible core. Likewise, the presence of plaques with a more compacted form, in particular, an aggregated Aβ core surrounded by a halo of Aβ-immunoreactive neurites, 17,23 had little predictive power. Indeed, if anything, HPC patients had more plaques with cores than AD patients (cf superior frontal gyrus, Table 1 ▶ ).
Like inflammation, which we examined in a previous study of HPC patients, 5 soluble Aβ (including the Aβ that has reached the CSF), cerebrovascular amyloid angiopathy, and paired helical filaments in the form of neurofibrillary tangles and neuropil threads surrounding amyloid cores did distinguish HPC from AD patients. Although soluble Aβ concentrations predicted synapse changes in the patients better than any of the other parametric variables studied, we would be remiss in failing to point out the striking absence of neurofibrillary tangle pathology in frontal neocortex of HPC patients. Only one of eight HPC patients exhibited any tangles in superior frontal gyrus, and these were sparse, whereas all eight HPC patients had many entorhinal cortex tangles. Nearly identical results were obtained in our previous research with six other HPC patients. 5
Of the different length Aβ fragments, Aβ40, whether in soluble or insoluble form, was a particularly strong correlate of synapse changes and readily discriminated HPC from AD patients. Aβ42 was much less robust in this regard and correlated poorly with synapse loss. Taken together with the data on soluble versus insoluble Aβ, our results therefore emphasize the potential importance of soluble Aβ40 and the relatively weak contribution of insoluble Aβ42 to AD pathophysiology. Although insoluble Aβ42 may be an essential element in neuropil Aβ deposition and a necessary participant in other neurodegenerative mechanisms, 13,23 neither it nor the other measures associated with it (eg, neuropil plaques) accounted for AD synapse loss or the absence of overt dementia in HPC patients. By contrast, our findings are consistent with previous results demonstrating an important role for the soluble forms of Aβ in AD pathology 15,26 and Aβ toxicity. 27,28 This may not be altogether surprising when one considers that soluble Aβ has the potential to affect neurons and neurites over a much wider area than insoluble Aβ, which is essentially pinned to fixed points in the neuropil in the form of plaques.
Both cerebrovascular amyloid angiopathy and ApoE4 allele frequency were highly correlated with Aβ40 levels and with each other, whereas they were not correlated with Aβ42 levels. The ANOVA results with ApoE also showed significant effects of ApoE4 on cerebrovascular amyloid and soluble amyloid, but not on diffuse plaques, neuritic plaques, or insoluble Aβ42, the primary constituent of the neuritic plaque. 29 These results confirm and extend those of Ishii et al, 30 who found ApoE4 allele frequency to correlate with Aβ40 but not Aβ42 concentrations, Greenberg et al, 31 who found ApoE4 allele frequency to correlate with earlier onset of hemorrhage in cerebrovascular amyloid angiopathy, and Alonzo and colleagues 32 and Roher and colleagues, 29 who found Aβ40 levels to be much higher in cerebrovascular amyloid deposits than neuropil amyloid deposits. Conversely, as expected for the major Aβ species in plaques, 29 Aβ42 levels were highly correlated with plaque density measurements, whereas Aβ40 levels were not. Collectively, then, our data do not confirm the suggestion that ApoE4 patients experience an increased neuritic amyloid plaque burden. 33 Rather, ApoE4 appears mainly to increase cerebrovascular amyloid burden, as previously reported, 33 as well as soluble amyloid burden.
In conclusion, many of the controversies about Aβ and its pathophysiological relevance may follow from the fact that the visible, insoluble form of the peptide has always been used in attempts to establish correlations with AD neurodegeneration and dementia. 2,4,6,7,12 Our data suggest that it is not the Aβ that we can see, the diffuse or classical plaque, but the soluble Aβ that we cannot see that best explains Aβ neurotoxicity.
Acknowledgments
We thank Heather Lampert and Rhonda Lee for assistance with the experiments and Dr. Michael Boss (Athena Diagnostics) for his help in the evaluation of CSF Aβ42 levels. This paper is dedicated to the memory of our colleague W. Harold Civin, M.D.
Footnotes
Address reprint requests to Dr. Joseph Rogers, Sun Health Research Institute, P.O. Box 1278, Sun City, AZ 85372. E-mail: jrogers@mail.sunhealth.org.
Supported by NIA AGO7367 (J. Rogers), NIA AG11925 (A. Roher), the Arizona Alzheimer’s Center (A. Roher, Y. Shen, and T. Beach), and the Alzheimer’s Association (J. Rogers and T. Beach).
References
- 1.Crystal H, Dickson D, Fuld P, Masur D, Scott R, Mehler M, Masdeu J, Kawas C, Aronson M, Wolfson L: Clinico-pathologic studies in dementia: nondemented subjects with pathologically confirmed Alzheimer’s disease. Neurology 1998, 38:1682-1687 [DOI] [PubMed] [Google Scholar]
- 2.Katzman R, Terry R, DeTeresa R, Brown T, Davis P, Fuld P, Renbing X, Peck A: Clinical, pathological, and neurochemical changes in dementia: a subgroup with preserved mental status and numerous neocortical plaques. Ann Neurol 1988, 23:138-144 [DOI] [PubMed] [Google Scholar]
- 3.Dickson DW, Crystal HA, Mattiace LA, Masur DM, Blau AD, Davies P, Yen SH, Aronson MK: Identification of normal and pathological aging in prospectively studied nondemented elderly humans. Neurobiol Aging 1991, 13:179-189 [DOI] [PubMed] [Google Scholar]
- 4.Arrigada PA, Marzloff K, Hyman BT: Distribution of Alzheimer-type pathologic changes in nondemented elderly individuals matches the pattern in Alzheimer’s disease. Neurology 1992, 42:1681-1688 [DOI] [PubMed] [Google Scholar]
- 5.Lue LF, Brachova L, Civin WH, Rogers J: Inflammation, Aβ deposition, and neurofibrillary tangle formation as correlates of Alzheimer’s disease neurodegeneration. J Neuropathol Exp Neurol 1996, 55:1083-1088 [PubMed] [Google Scholar]
- 6.Blessed G, Tomlinson BE, Roth M: The association between quantitative measures of dementia and of senile change in the cerebral gray matter of elderly subjects. Br J Psychiatry 1968, 114:797-811 [DOI] [PubMed] [Google Scholar]
- 7.Dekosky ST: Searching for the holy grail. What is the structural correlate of cognition? Neurobiol Aging 1995, 16:285-3047566338 [Google Scholar]
- 8.Dickson DW, Ivanushkin A, Heitner J, Yen SH, Davies P: Apolipoprotein E immunoreactivity is increased in amyloid deposits of Alzheimer’s disease, but not pathological aging or diffuse Lewy body disease. Iqbal K Mortimer JA Winblad B Wisniewski HM eds. Research Advances in Alzheimer’s Disease and Related Disordors. 1995, :pp 371-382 John Wiley & Sons, New York [Google Scholar]
- 9.Mirra SS, Hyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L, participating CERAD neuropathologists: The consortium to establish a registry for Alzheimer’s disease (CERAD). II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 1991, 41:479–481 [DOI] [PubMed]
- 10.Braak H, Braak F: Neuropathological staging of Alzheimer-related changes. Acta Neuropathol 1991, 82:239-259 [DOI] [PubMed] [Google Scholar]
- 11.Scheff SW, DeKosky ST, Price DA: Quantitative assessment of cortical synaptic density in Alzheimer’s disease. Neurobiol Aging 1990, 11:29-37 [DOI] [PubMed] [Google Scholar]
- 12.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R: Physical basis of cognitive alteration in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 1991, 30:572-580 [DOI] [PubMed] [Google Scholar]
- 13.Kuo YM, Emmerling MR, Bisgaier CL, Essenburg AD, Lampert HC, Roher AE: Elevated low density lipoprotein in Alzheimer’s disease correlates with brain Aβ 42 levels. Biochem Biophys Res Commun 1998, 252:711-715 [DOI] [PubMed] [Google Scholar]
- 14.Gowing E, Roher AE, Woods AS, Cotter RJ, Chaney MM, Little SP, Ball MJ: Chemical characterization of Aβ17–42 peptide, a component of the diffuse amyloid deposits of Alzheimer’s disease. J Biol Chem 1994, 269:10987-10990 [PubMed] [Google Scholar]
- 15.Kuo YM, Emmerling MR, Vigo-Pelfrey C, Kasunic TC, Kirkpatrick JB, Murdoch GH, Ball MJ, Roher AE: Water-soluble Aβ(N-40, N-42) oligomers in normal and Alzheimer disease brains. J Biol Chem 1996, 271:4077-4081 [DOI] [PubMed] [Google Scholar]
- 16.Galasko D, Chang L, Clark CM, Kaye J, Knopman D, Thomas R, Kholodenko D, Schenk D, Lieberburg I, Miller B, Green R, Basherad R, Kertiles L, Boss MA, Seubert P: High cerebrospinal fluid tau and low amyloid β42 levels in the clinical diagnosis of Alzheimer disease and relation to apolipoprotein E genotype. Arch Neurol 1998, 55:937-945 [DOI] [PubMed] [Google Scholar]
- 17.Wisniewski HM, Bancher C, Barcikowska M, Wen GY, Currie J: Spectrum of morphological appearance of amyloid deposits in Alzheimer’s disease. Acta Neuropathol 1989, 78:337-347 [DOI] [PubMed] [Google Scholar]
- 18.Otvos L, Feiner L, Lang E, Szendrei GI, Goedert M, Lee VM: Monoclonal antibody PHF-1 recognizes tau protein phosphorylated at serine residues 396 and 404. J Neurosci Res 1994, 39:669-673 [DOI] [PubMed] [Google Scholar]
- 19.Su JH, Cummings BJ, Cotman CW: Plaque biogenesis in brain aging and Alzheimer’s disease. I. Progressive changes in phosphorylation states of paired helical filaments and neurofilaments. Brain Res 1996, 739:79-87 [DOI] [PubMed] [Google Scholar]
- 20.Harrington CR, Louwagie J, Rossau R, Vanmechelen E, Perry RH, Perry EK, Xuereb JH, Roth M, Wischik CM: Influence of apolipoprotein E genotype on senile dementia of the Alzheimer and Lewy body types: significance for etiological theories of Alzheimer’s disease. Am J Pathol 1994, 145:1472-1484 [PMC free article] [PubMed] [Google Scholar]
- 21.Hixson JE, Vernier DT: Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J Lipid Res 1990, 31:545-548 [PubMed] [Google Scholar]
- 22.Sparks DL, Scheff SW, Liu H, Landers T, Danner F, Coyne CM, Hunsaker JC: Increased density of senile plaques (SP), but not neurofibrillary tangles (NFT), in non-demented individuals with the apolipoprotein E4 allele: comparison to confirmed Alzheimer’s disease patients. J Neurol Sci 1996, 138:97-104 [DOI] [PubMed] [Google Scholar]
- 23.Joachim CL, Selkoe DJ: The seminal role of β-amyloid in the pathogenesis of Alzheimer’s disease. Alz Dis Assoc Disord 1992, 6:7-34 [DOI] [PubMed] [Google Scholar]
- 24.Tagliavini F, Giaccone G, Frangione B, Bugianin O: Preamyloid deposits in the cerebral cortex of patients with Alzheimer’s disease and nondemented individuals. Neurosci Lett 1988, 93:191-196 [DOI] [PubMed] [Google Scholar]
- 25.Delaere P, Duyckaerts C, Masters C, Beyreuther K, Piette F, Hauw JJ: Large amounts of neocortical βA4 deposits without neuritic plaques nor tangles in a psychometrically assessed, non-demented person. Neurosci Lett 1990, 116:87-93 [DOI] [PubMed] [Google Scholar]
- 26.Teller JK, Russo C, DeBusk LM, Angelini G, Zaccheo D, Dagna-Bricarelli F, Scartezzini P, Bertolini S, Mann DM, Tabaton M, Gambetti P: Presence of soluble amyloid β-peptide precedes amyloid plaque formation in Down’s syndrome. Nature Med 1996, 2:93-95 [DOI] [PubMed] [Google Scholar]
- 27.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL: Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA 1998, 95:6448-6453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oda T, Wals P, Osterburg HH, Johnson SA, Pasinetti GM, Morgan TE, Rozovsky I, Stine WB, Snyder SW, Hozman TF, Finch CE: Clusterin (apoJ) alters the aggregation of amyloid β-peptide (Aβ1–42) and forms slowly sedimenting Aβ complexes that cause oxidative stress. Exp Neurol 1995, 136:22-31 [DOI] [PubMed] [Google Scholar]
- 29.Roher AE, Lowenson JD, Clarke S, Woods AS, Cotter RJ, Gowing E, Ball MJ: β-Amyloid-(42) is a major component of cerebrovascular amyloid deposits: implications for the pathology of Alzheimer’s disease. Proc Natl Acad Sci USA 1993, 90:10836-10840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishii K, Tamaoka A, Mizusawa H, Shoji S, Ohtake T, Frazer P, Takahashi H, Tsuji S, Gearing M, Mizutani T, Yamada S, Kato M, St George-Hyslop P, Mirra S, Mori H: Aβ40 but not Aβ42 levels in cortex correlate with apolipoprotein E ε4 allele dosage in sporadic Alzheimer’s disease. Brain Res 1997, 748:250-252 [DOI] [PubMed] [Google Scholar]
- 31.Greenberg SM, Briggs ME, Hyman BT, Kokoris GJ, Takis C, Kanter DS, Kase CS, Pessin MS: Apolipoprotein E ε4 is associated with the presence and earlier onset of hemorrhage in cerebral amyloid angiopathy. Stroke 1996, 27:1333-1337 [DOI] [PubMed] [Google Scholar]
- 32.Alonzo NC, Hyman BT, Rebeck GW, Greenberg SM: Progression of cerebral amyloid angiopathy: accumulation of amyloid-β40 in affected vessels. J Neuropathol Exp Neurol 1998, 57:353-359 [DOI] [PubMed] [Google Scholar]
- 33.Olichney JM, Hansen LA, Galasko D, Saitoh T, Hofstetter CR, Katzman R, Thal LJ: The apolipoprotein E ε4 allele is associated with increased neuritic plaques and cerebral amyloid angiopathy in Alzheimer’s disease and Lewy body variant. Neurology 1996, 47:190-196 [DOI] [PubMed] [Google Scholar]