

Abstract
Dilated cardiomyopathy is characterized by decreased contractile function and loss of myofibril organization. Previously unexplored structural and molecular events that precede and initiate dilation can now be studied in tropomodulin-overexpressing transgenic (TOT) mice exhibiting progressive dilated cardiomyopathy. Onset of dilation did not correspond to a change in transgene expression levels, which were more than threefold above normal at birth and remained elevated throughout postnatal life. Similarly, mitogen-activated protein kinase activation (p38, ERK1/ERK2, JNK1/JNK2) was not associated with dilation. In contrast, calcineurin was activated before dilation, presumably due to doubling of intracellular diastolic calcium levels in TOT cardiomyocytes. Amplitude of systolic calcium transients was greatly increased as well, demonstrating the novel and unique calcium handling profile of TOT cardiomyocytes. Loss of myofibril organization was not apparent by confocal microscopy until over 1 week after birth, although neonatal sarcomeric abnormalities were revealed by ultrastructural analysis. Rapid postnatal increases in heart:body weight ratio at 1.5 weeks were followed by two waves of mortality between 2 and 3 weeks after birth coincident with maturational stress. Ultimately, TOT pathogenesis is a compensatory response to altered sarcomeric structure driven by calcineurin activation within days after birth, making TOTs an excellent paradigm for studying the role of calcium overload in dilated cardiomyopathy.
Animal paradigms of dilated cardiomyopathy are useful model systems for understanding the progression of disease, identifying important molecular changes occurring during development, and testing treatment modalities. The utility of transgenic mice as a testing arena for hypothetical causes and treatments of human cardiovascular disease depends, in part, on characterizing molecular and cellular changes in the transition from normal to failing myocardium. Understanding the progression of dilated cardiomyopathy will be critical in the design of treatment modalities to intervene at the molecular level in the initial stages of pathogenesis. 1,2 Early animal studies of dilated cardiomyopathy used exogenous treatments such as infection, toxins, or drugs to cause disease. 3 The advent of cardiomyopathic transgenic mouse models now allows researchers to alter the genetic program of heart development in a consistent fashion. Maintaining mice as an inbred homozygous colony eliminates complex genetic background variables, allowing the study to focus on effects of transgene expression without variable penetrance between individuals. Aside from minimal variation between subject animals, a transgenic paradigm with predictable onset, defined time window of pathogenesis, and unambiguous knowledge of transgene function and effect(s) facilitates the study of cardiomyopathic disease.
Cardiomyopathic transgenic mouse models often lack characterization of molecular and cellular changes occurring throughout various stages of pathogenesis, particularly the early phase. These investigations will be critical for understanding disease progression and determining similarities between mouse paradigms and inherited cardiomyopathic conditions in man. In human populations, phenotypic variation in progression and severity of disease complicates interpretation of the underlying etiology. 4 Despite this limitation, population-based longitudinal analyses are necessary to achieve an accurate description of the progression, pathology, and variability of the cardiomyopathy as exemplified by studies such as Framingham. 5,6 Transgenic mice can be created and propagated using genetically identical individuals, circumventing concerns related to genetic background heterogeneity that are inescapable in human populations. The combination of genetic homogeneity and comparable environment of the population greatly simplifies interpretation of results. Thus, studying the epidemiology of tropomodulin (Tmod)-overexpressing transgenic (TOT) mice provides valuable insight into the nature of their cardiomyopathy. Because TOTs predictably develop dilated cardiomyopathy within 2 weeks after birth, 7 they provide an attractive model system to study the molecular and structural changes before and during the progressive phase of disease. Furthermore, characteristics of TOT cardiomyopathy such as loss of myofibril organization and diminished contractility are hallmarks of progressive and chronic dilated cardiomyopathy in humans. 8-10
TOT utility as a paradigm of human cardiomyopathy depends on defining relationships between essential disease characteristics in man and mouse. The goal of this report was to detail the fundamental properties of TOT pathogenesis to establish the paradigm. TOT cardiomyopathy was thought to arise from Tmod-mediated myofibril degeneration, 11 but the dilated phenotype apparently stems from resultant calcineurin activation driving an impaired hypertrophic response. 12 The utility of TOTs in testing pharmaceutical intervention was recently demonstrated by treatment of TOTs with cyclosporin, a calcineurin inhibitor, to inhibit development of the dilated phenotype. 12 Calcineurin activation is important in experimental hypertrophy 13 and human cardiomyopathy, 14 presumably due to chronic elevation of intracellular calcium. Results presented here indicate that calcium signaling is likely to be a primary effector in the initiation of TOT pathogenesis, leading to molecular and histological changes before development of the dilated cardiomyopathic phenotype within 1 week after birth. Concurrently and shortly thereafter, environmental factors such as maturational stress and litter size influence TOT survival, reminiscent of reported associations between environment and outcome in human cardiomyopathic conditions. 6,15 Molecular and structural analyses identified mechanisms causing cardiac dilation, and population analyses demonstrated associations between survival, postnatal development, and the environment. Collectively, these findings establish fundamental parallels between the TOT paradigm and human disease, with implications not only for the development and interventional treatment of TOT cardiomyopathy, but also for dilated cardiomyopathy in general.
Materials and Methods
TOT Breeding
TOTs were established as previously described. 7 The TOT population was derived from a single FVB/N line inbred to transgene homozygosity for increased production, creation of uniformly affected litters, and facilitation of population-based analyses. Mating of the founder line 65 transgenic female and nontransgenic male produced heterozygous first generation (F1) offspring. These F1 siblings were then mated to produce second generation (F2) mice with litter ratios of 1 normal: 2 heterozygotes: 1 homozygote. The heterozygotes showed elevated Tmod expression without juvenile morbidity or mortality (data not shown) and are the subject of ongoing study. F3 homozygotes developed acute dilated cardiomyopathy with variation in mortality despite consistent onset and progression of the disease.
Immunoblot Analyses
Hearts were collected at the indicated time points and washed in phosphate buffered saline, frozen in liquid nitrogen, crushed with a mortar and pestle, and reconstituted in buffer A (20 mmol/L sodium phosphate, pH 7.0, 150 mmol/L NaCl, 2 mmol/L MgCl2, 0.1% NP-40, 10% glycerol, 10 nmol/L cypermethrin, 10 nmol/L okadaic acid, 100 μmol/L phenylasine oxide, 10 mmol/L NaF, 10 mmol/L sodium pyrophosphate, 1 mmol/L dithiothreitol, 100 μmol/L sodium orthovanadate, 10 μg/μl pepstatin, 10 μg/μl leupeptin, 10 μg/μl aprotinin, 10 μg/μl tosyl-l-lysine chloromethyl ketone, 10 μg/μl N-tosyl-l-phenyl-alanine chloromethyl ketone). Approximately 50 μg of protein powder was added to 200 μl of buffer A and light sonication was performed to lyse remaining cells. For calmodulin immunoprecipitations, 300 μg of protein extract in buffer A was mixed with 5 μg of calmodulin antibody (Zymed, South San Francisco, CA) and 30 μl of protein A/G agarose (Santa Cruz Biotechnology, Santa Cruz, CA) and incubated at 4°C with gentle rocking for 2 hours. Samples were then gently centrifuged at 800 × g for 3 minutes in a microfuge at 4°C, at which time the supernatant was discarded and the agarose pellet was resuspended in 100 μl of buffer A and spun again. This wash was repeated two additional times. After the last spin the agarose pellet was resuspended in 20 μl of sodium dodecyl sulfate (SDS) sample loading buffer and boiled. These samples were then loaded on a polyacrylamide gel for subsequent Western blot analysis. For tropomodulin and signal transduction immunoblots, protein concentration of heart lysates was determined using the Bio-Rad DC Protein assay (#500–0116, Bio-Rad, Melville, NY). 100 μg/lane of protein sample was mixed with an equal volume of sample buffer, boiled for 5 minutes, cooled to room temperature, and loaded onto 12.5% SDS-polyacrylamide gels for electrophoresis. Separated proteins were transferred in Tris-glycine immunoblot buffer at 70V and 4°C overnight to PVDF membrane (#RPN 2020F, Hybond-P PVDF, Amersham, Arlington Heights, IL). The next day, membranes were rinsed in water and stained with 0.2% Ponceau S (#BP103-10, Fisher, Pittsburgh, PA) solution containing 3% TCA (#T-4885, Sigma, St. Louis, MO) to confirm uniform protein transfer and comparable loadings between samples. The blot was rinsed again in water, washed in TBST buffer (50 mmol/L Tris-HCl pH 7.6, 150 mmol/L NaCl, and 0.1% Tween 20), and blocked in TBST containing 5% Amersham blocking buffer (w/v). Antibodies used for the primary incubation included: anti-tropomodulin (affinity-purified polyclonal rabbit antibody produced against bacterially expressed recombinant tropomodulin), anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH; #RDI-TRK5G4–6C5, Research Diagnostic Industries, Flanders, NJ), anti-phosphotyrosine (#P11120, Transduction Labs, Lexington, KY), anti-ERK 1 & 2, anti-phospho-p38, anti-phospho-mitogen-activated protein kinase (MAPK); (#9101S, #9213, #9211, #9211S, and #9101S, New England Biolabs, Beverly, MA) and anti phospho-JNK (#SC-6254, Santa Cruz). Membranes were incubated with primary antibodies either at 4°C overnight on a rocking platform shaker or for 2 hours at room temperature on an orbital platform shaker. After labeling, membranes were washed 3 times in TBST for 10 minutes per wash and then incubated with appropriate species-specific alkaline phosphatase-conjugated secondary antibodies for enhanced chemifluorescence as directed by the manufacturer (ECF kit, Amersham). Chemifluorescent images were scanned using a Storm 860 (Molecular Dynamics, Sunnyvale, CA).
Intracellular Calcium Analyses
Cardiomyocytes were isolated and calcium measurements were performed as previously described. 16 Briefly, isolated hearts were perfused with low Ca2+ Joklik medium containing collagenase type I and type II enzyme. Measurement of intracellular free Ca2+ transients were obtained by loading isolated cells with Fura-2 a.m. dye at 37°C for 30 minutes. After loading, cells were washed and resuspended in oxygenated physiological buffer and imaged using a photo scan dual-beam spectro-photometer coupled to a microscope equipped with optics for ultraviolet viewing. Intracellular free Ca2+ was monitored as the ratio of 340:380 nm fluorescence of Fura-2, and data are reported as the 340:380 nm ratio. Two control and two TOT mice were used at 13 weeks of age, based on prior experiments which established the appropriate age for calcium measurements. Results with younger control animals show variable measurements, complicating interpretation of transgenic data (data not shown). Eight cardiomyocytes were measured from each animal.
Microscopic Analyses
Light and electron microscopy were performed as previously described. 7,17 For the confocal time course analysis, hearts were removed from mice at 1, 5, 8, 11, 14, and 17 days after birth, rinsed in PBS, and fixed in 2% paraformaldehyde, 10 mmol/L HEPES, pH 7.0, overnight at 4°C on a rocking platform shaker. The next day, hearts were transferred to PBS containing 30% sucrose and left overnight to equilibrate at 4°C on a rocking platform shaker. Cryoblocks were made by embedding hearts in Tissue Freezing Medium (#H-TFM, Triangle Biomedical Sciences, Durham, NC) and freezing in melting isopentane which was frozen over liquid nitrogen. Sections 7 to 10 μm thick were collected onto glass slides (SuperFrost Plus, Fisher), air-dried for at least 1 hour, and used immediately or stored at 4°C. Immunofluorescence labeling of heart sections was performed with antibodies to Tmod (affinity purified rabbit anti-Tmod prepared against bacterially expressed protein) and sarcomeric α-actinin (#A-7811, Sigma). Slides were rehydrated in PBS for 10 minutes, washed in PBS containing 0.1 mol/L glycine for 5 minutes, rinsed briefly in PBS, and then permeabilized in PBS containing 15% methanol for 15 minutes. After another brief rinse in PBS, sections were further permeabilized for 20 minutes in PBS containing 0.5% Triton X-100, washed twice in 3 minute PBS rinses, and blocked for 1 hour in PBS containing 10% horse serum. Primary antibodies diluted in PBS containing 10% horse serum were applied overnight at 4°C. The next day, slides were washed 3 times in PBS for 10 minutes per wash, labeled at room temperature in the dark with secondary antibodies diluted 1:100 in PBS (#711-095-152: anti-rabbit FITC-conjugate; #115-075-146: anti-mouse Texas red conjugate, both from Jackson Immunoresearch, West Grove, PA). After secondary labeling, slides were washed 3 times in PBS for 5 minutes per wash and mounted for viewing in Vectashield medium (Vector Labs, Burlingame, CA). Confocal images were acquired using a Molecular Dynamics CLSM 2010 as previously described. 7 Morphometric scans were taken using a 2× objective and quantitated using Imagespace software (Molecular Dynamics).
TOT Tracking and Statistical Analysis
TOT population data were gathered and analyzed using Progeny v2.0 (Genetic Data Systems, Mishawaka, IN). Progeny combines the use of pedigree tree drawing with data entry and database relational analysis. Statistical analyses of litter survival were performed using SAS (SAS Institute, Cary, NC). Data were checked for completeness, and range and crosschecks were performed to find possible entry or transcription errors. Spearman correlation coefficients were calculated between days of survival and heart-to-body weight ratio and litter size. Subsequent analyses included analysis of covariance, where survival was the dependent variable and litter and litter size were the independent variables. This analysis was performed to account for the assumed similarity of mice from the same litter.
Results
Tmod Levels Are Elevated at Birth and Maintain Increased Expression
Immunoblot analyses were performed to determine the accumulation of Tmod protein during postnatal development (Figure 1) ▶ . Protein lysates were separated by SDS-PAGE (Figure 1A) ▶ , transferred to nitrocellulose, and the region of the blot corresponding to the Mr of Tmod was excised from the blot for immunolabeling (Figure 1B) ▶ . Tmod expression in TOTs was estimated relative to control samples (Figure 1B ▶ , nontransgenic lanes) and quantitative analysis was standardized against GAPDH (data not shown). Tmod level in control nontransgenic animals remained constant throughout the time course investigated, so one representative time point was shown for each experiment. Two separate experiments (Figure 1A ▶ , left- and right-hand gels) showed Tmod expression was elevated over threefold at birth (Figure 1 ▶ , TOT, 0 days) and showed a trend toward increasing expression up to almost sixfold over the next 2.5 weeks (Figure 1 ▶ , TOT, 17 days). Comparable increases in expression were found using the second control sample (Figure 1 ▶ , nontransgenic, 2 days) compared with samples taken from TOTs ranging in age (Figure 1 ▶ , TOT, 2–8 days).
Figure 1.

Tropomodulin expression levels in TOTs from birth to 17 days. Coomassie SDS-PAGE and the immunoblot show increased level of Tmod protein in TOT hearts. Equivalent amounts of cardiac protein lysates were loaded and subjected to overnight electrophoresis followed by either staining with Coomassie brilliant blue dye (A) or transfer to nitrocellulose. The immunoblot region corresponding to the Mr of Tmod (∼43 kd) was excised and labeled with anti-Tmod antibody (B). Bound antibody was detected by enhanced chemifluorescence and signal was quantitated by fluoroimager analysis (see Methods). Samples loaded in each lane are indicated across the top of the gel. Two nontransgenic heart samples of different ages were used as references in separate experiments. In comparisons to these two nontransgenic samples, samples from TOTs show increases ranging from 3.5-fold at birth to almost sixfold at postnatal day 17. Mobility (Mr × 10−3) of molecular weight standards is shown at the right of the gel.
Altered Signal Transduction in TOTs before Onset of Structural Pathology
Activation of the calcium-regulated phosphatase calcineurin has been associated with dilated cardiomyopathy in TOT hearts. 12 To quantify activated calcineurin during postnatal development, immunoprecipitation was performed with calmodulin-specific antibody followed by a calcineurin Western blot (Figure 2A) ▶ . Because activated calcineurin is tightly associated with calmodulin, this precipitation assay indicates the fraction of calcineurin in the activated state. The absolute level of calcineurin and calmodulin proteins in cardiac protein extracts was assessed by Western blot. Absolute levels of calcineurin were increased at every time point assayed, while calmodulin levels were invariant. Activated calcineurin was increased 1.9-fold at day 3, 7.7-fold at day 5, 4.0-fold at day 7, and 2.4-fold at day 24. Absolute calcineurin protein levels were also up-regulated 1.6-fold at day 3, 2.4-fold at day 5, 2.6-fold at day 7, and 2.0-fold at day 24. The increase in calcineurin protein and activated state from days 3 through 7 before onset of dilation suggests a close association with the initiation of TOT cardiomyopathy.
Figure 2.
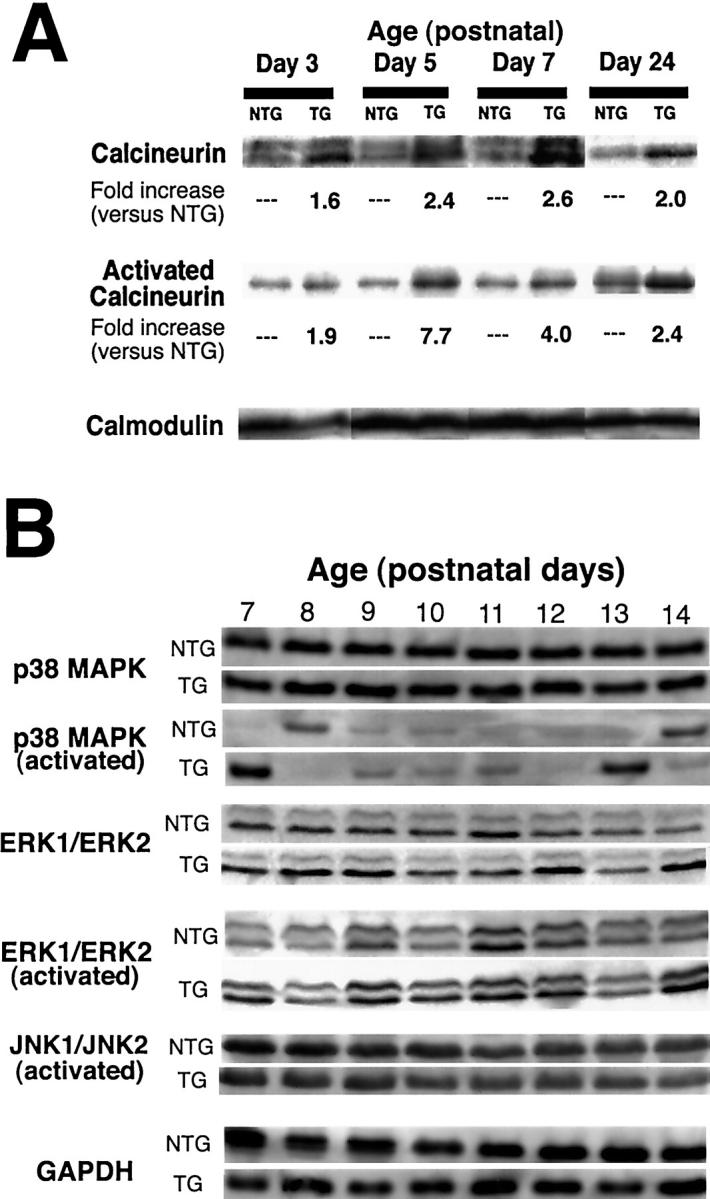
Signal transduction immunoblots of TOTs. Immunoblot analysis of heart lysates prepared at various days after birth as indicated showing calcineurin (A) and MAPK family members (B). TOT calcineurin (A, top row) and activated calcineurin (A, middle row) levels are increased in TOTs relative to nontransgenics throughout early development. Invariant calmodulin levels show equivalence of immunoprecipitation loading between samples (A, bottom row). Calcineurin appearance as a single (days 3, 5, and 7) or double (day 24) band depends on electrophoretic conditions and should not be interpreted as a change in protein expression or modification. Within the MAPK family (B), total p38 levels are constant throughout postnatal days 7 to 14 in both nontransgenics and TOTs (B, top rows, p38). Increases in p38 activity shown are typical of biphasic activation spikes occurring between postnatal days 7 to 10 and again on or after postnatal day 13 (B, bottom rows, p38). Transient p38 activation also showed variable intensity in both nontransgenic and TOT samples (data not shown). Neither ERK1/ERK2 or JNK1/JNK2 showed changes in activation throughout the time course of postnatal days 7 to 14 (B, ERK and JNK). GAPDH blot shows equivalence of loading between sample lanes (B, GAPDH).
Induction of hypertrophy is associated with activation of the MAPK signal transduction pathway. 18 Immunoblot analyses were performed to determine the activation state of MAPK family members p38, ERK1/ERK2, and JNK from 7 to 14 days after birth (Figure 2B) ▶ . Total cellular p38 MAPK level was comparable between TOTs and nontransgenic controls and transient activation of p38 was present (Figure 2B ▶ , upper rows). Repeated studies of p38 activation during the postnatal period showed these transient activity spikes vary in both intensity and timing, occurring between postnatal days 7 and 10 and again on or after postnatal day 13 (data not shown). Thus, p38 MAPK may participate in normal postnatal cardiac hypertrophy, but the transient nature of activation and intensity in the TOT samples suggests it is not a primary effector of Tmod-induced cardiomyopathy. Analysis using antibodies to additional hypertrophic pathway mediators ERK 1/ERK2 as well as JNK showed no significant difference in levels of activated protein between controls and transgenics (Figure 2B ▶ , middle rows). Control probing of the blot strips with antibody to GAPDH shows equivalence of sample loadings between days and between control and TOT samples (Figure 2B ▶ , bottom rows).
Increased Basal Level and Amplitude of Calcium Transients in TOT Cardiomyocytes
Activation of calcineurin suggested increased basal calcium level in TOT cardiomyocytes. Intracellular calcium as determined by Fura-2 ratios in cardiomyocytes from TOTs was significantly increased 89% (P < 0.0001) in basal (diastolic) level compared to nontransgenic controls (Figure 3, A and B) ▶ . The magnitude of this increase is brought into context by noting basal calcium levels in TOT cells are approximately equivalent to peak systolic calcium levels (baseline + amplitude) in normal mice (1.7 versus 1.6, respectively). Furthermore, the amplitude of the TOT transient calcium transient is 128% greater than calcium transients in nontransgenic cells (Figure 3C ▶ , 1.6 versus 0.7, respectively; P < 0.0001), raising peak systolic calcium level in TOT cells twofold above nontransgenic levels (3.6 versus 1.6, respectively). The time to reach 80% reduction from peak calcium level (T80) was significantly longer by 17% (P = 0.043) in TOT cardiomyocytes, presumably due to the high systolic calcium load (Figure 3D) ▶ .
Figure 3.

Altered calcium handling in TOT cardiomyocytes. Calcium transients in isolated ventricular myocytes measured by Fura-2 ratios shown as time plot (A) and pooled baseline (B) and amplitude (C) values. B: Basal calcium level in normal mice (0.978 ± 0.148) is 57% lower than in TOTs (1.745 ± 0.259). C: Peak amplitude in controls (0.836 ± 0.309) is 43% lower than in TOTs (1.936 ± 0.774). D: Time to reduce systolic calcium level by 80% (T80) was prolonged by 17% in TOTs compared to normal mice. Values indicated by asterisks were significantly different from normal control values by Student’s t-test (B and C, P < 0.0001; D, P = 0.043).
Subtle Sarcomeric Dysgenesis Evident at Birth Becomes Overt 1 Week Later
The earliest observed clinical changes in TOTs symptomatic of cardiomyopathic changes occur at least 10 days after birth. 7 Immunofluorescence analysis was performed to examine Tmod distribution throughout the heart as well as myofibril organization during the first 2.5 weeks after birth. Age-matched hearts from control nontransgenics and TOTs were sectioned and labeled with antibodies to Tmod and α-actinin (Figure 4) ▶ . TOT hearts showed intense Tmod reactivity compared to nontransgenic controls throughout the 6 time points sampled, ranging from 1 to 17 days after birth (Figure 4 ▶ , upper rows). Overall, both control and TOT samples labeled uniformly throughout the myocardium. Myofibril organization was subjectively evaluated by using α-actinin to label Z-disks followed by observation of striation alignment and periodicity (Figure 4 ▶ , lower rows). Sarcomeric periodicity in neonatal nontransgenics (Figure 4 ▶ , normal fibrils, 1 and 4 days) was relatively poor, but showed progressive improvement and clear striations at later time points (Figure 4 ▶ , 8, 11, 14, and 17 days). In comparison, myofibrils in neonatal TOTs and nontransgenics appeared similar in degree of organization (Figure 4 ▶ , 1 and 4 days). By 8 days after birth, subtle indications of sarcomeric dysgenesis such as poor alignment and wavy appearance were noted in the TOT myofibrils (Figure 4 ▶ , Tg fibrils, 8 days). Progressive deterioration of sarcomeric structure was evident in TOT sections throughout the remainder of the time course (Figure 4 ▶ , Tg fibrils, 11, 14, and 17 days).
Figure 4.

Immunofluorescence analysis of TOTs from 1 to 17 days after birth. Hearts were removed from TOTs at various days after birth and labeled with antibody to tropomodulin (Tmod, upper rows) or actinin (lower rows). Nontransgenic hearts show modest but consistent anti-tropomodulin immunoreactivity throughout the time course (top row). In comparison, TOT hearts show significantly greater tropomodulin staining as early as 1 day after birth (second row). Anti-α-actinin labeling shows development of myofibril organization throughout the first week after birth, with clear striations evident by postnatal day 8 (lower two rows). At postnatal day 8, myofibrils in TOT hearts show subtle signs of deterioration with smearing of α-actinin staining and loss of alignment. Progressive myofibril degeneration in TOT hearts continues through postnatal day 17.
Sarcomere organization was examined by electron microscopy at early time points (Figure 5) ▶ where, by confocal microscopy, myofibril integrity appeared comparable between control nontransgenic and TOT myocardial samples. Control myofibrils at birth showed slight curvature of Z-disk structure and myofibril alignment (Figure 5, A and B) ▶ , presumably due to the nascent assembly and increased workload demands on contractile structure in the neonate. Degenerative changes in TOT sarcomere organization were apparent at birth relative to control nontransgenic samples. Although Z-disks were present throughout the TOT heart sections, structures were often poorly spaced (Figure 5C) ▶ or unaligned (Figure 5D) ▶ . Control myofibril organization improved through postnatal days 3 (Figure 5E) ▶ and 7 (Figure 5H) ▶ of the experiment, unlike TOT myofibril organization, which showed widening areas of Z-disk curvature and myofibril misalignment (Figure 5, F, G, and I) ▶ . The retention of Z-disk banding, albeit compromised, probably accounts for why myofibril organization appeared intact in confocal observations of early time points (Figure 4) ▶ . The higher resolution of electron microscopy allowed for visualization of sarcomeric abnormalities that were not apparent at the limit of confocal analyses.
Figure 5.
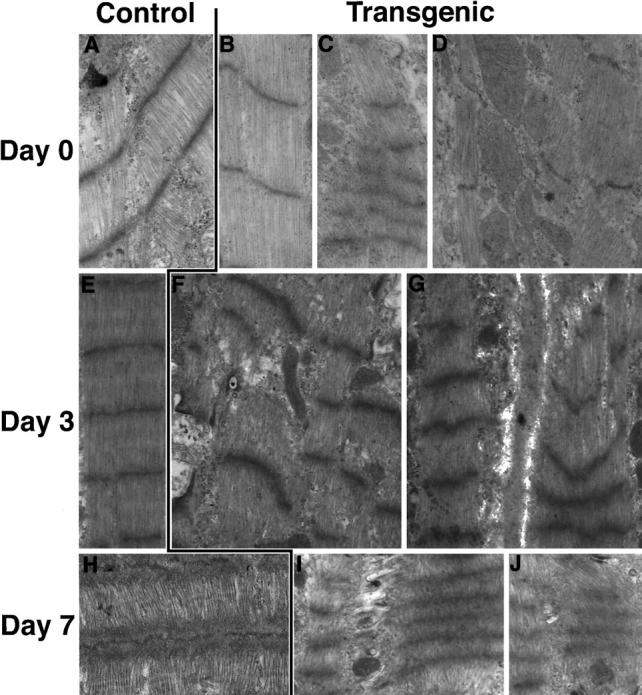
Electron microscopy of control nontransgenics versus TOTs. Representative views of left ventricular sections from mice at day of birth (top row) and postnatal days 3 (middle row) or 7 (bottom row). At birth, control samples (A and B) showed many organized sarcomeres with well spaced but slightly curved Z-disks. In comparison, TOT samples showed many areas where Z-disks were irregularly spaced (C) or poorly aligned (D). At postnatal day 3, sarcomeres of control hearts were well organized and regularly spaced (E), whereas TOT sarcomeres showed curved and compressed Z-disk structures with variable spacing of Z-disk alignment between neighboring myofibrils (F and G). At postnatal day 7, ordered myofibril organization of control samples (H) was clearly distinct from TOT sarcomeres, which showed increasing compression of Z-disk structure and concomitant loss of myofibril alignment (I). Original magnifications, ×20,000 (A, C, E-I) and ×30,000 (B, D).
Trends in TOT Mortality and Cardiac Pathogenesis
Actuarial data were collected from a population of 278 TOTs that died of cardiomyopathic disease to determine trends in survival and correlate the course of the disease with postnatal development. TOT mortality was concentrated primarily within 6 weeks after birth (161 dead, 58% of the population), although some TOTs can live over 1 year (Figure 6A) ▶ . Examination of early mortality from birth to 50 days of age showed two distinct peaks coincident with stressful transitions in maturation of the young mouse (Figure 6B) ▶ . The first wave of increased mortality around 15 to 16 days after birth (Figure 6B ▶ , arrow 1) correlated with increased activity as eyes opened and the mice explored their environment. The second peak of increased mortality at 21 days after birth coincided with weaning age (Figure 6B ▶ , arrow 2). Mortality occurring between 30 and 50 days (Figure 6B ▶ , arrow 3) was associated with the development of anasarca, which was evident in 50% of the mice that died during this time (n = 11 of 22). Symptomatically, these edematous and proptotic mice (Figure 7) ▶ resemble clinical anasarca related to secondary complications of poor heart function reported in human cardiomyopathy, 19 and mice exhibiting this condition never survive. Collectively, one-half of the TOT population died between 14 and 22 days after birth with losses at days 14 and 21 accounting for 11 and 14% of all deaths, respectively. Cumulative TOT population data indicated that survival showed a significant association (P < 0.0001) with litter size (Figure 8) ▶ . Specifically, smaller litters showed the highest mortality rate and average age at death increased in correlation with a larger numbers of siblings. This result is consistent with the interpretation that gestational and/or postnatal environmental factors play a significant role in the lethality of TOT cardiomyopathy.
Figure 6.

Population analyses of TOTs. Graphs showing TOT mortality plot of all spontaneous deaths (A; n = 278), the subset of deaths at less than 50 days of age (B; n = 161), all heart:body weight ratios (C), and the subset of heart:body weight ratios at less than 50 days of age (D). A: Mortality of TOTs shows a predominant cluster of mortality occurring within 1 month after birth. Bracketed area along x axis shows region of plot expanded on in B. B: Mortality within 50 days after birth. Two distinct peaks are evident at postnatal days 15 and 16 (arrow 1) and at day 22 (arrow 2). A third cluster of deaths contains TOTs that developed anasarca (at arrow 3 within bracket). C: Heart:body weight ratio of TOTs shows a trend toward increasing ratio with age. Bracketed area along x axis shows region of plot expanded on in D. D: Heart:body weight ratios within 50 days after birth. Ratios for selected nontransgenic controls of various ages ranging from 1 to 3 weeks consistently range from 5.0–5.2 (dotted bars). In comparison, ratios in TOT hearts were comparable to control values through postnatal day 8 and then rose approximately threefold within a 48-hour period (at arrow within bracket) to reach a plateau, which was maintained.
Figure 7.
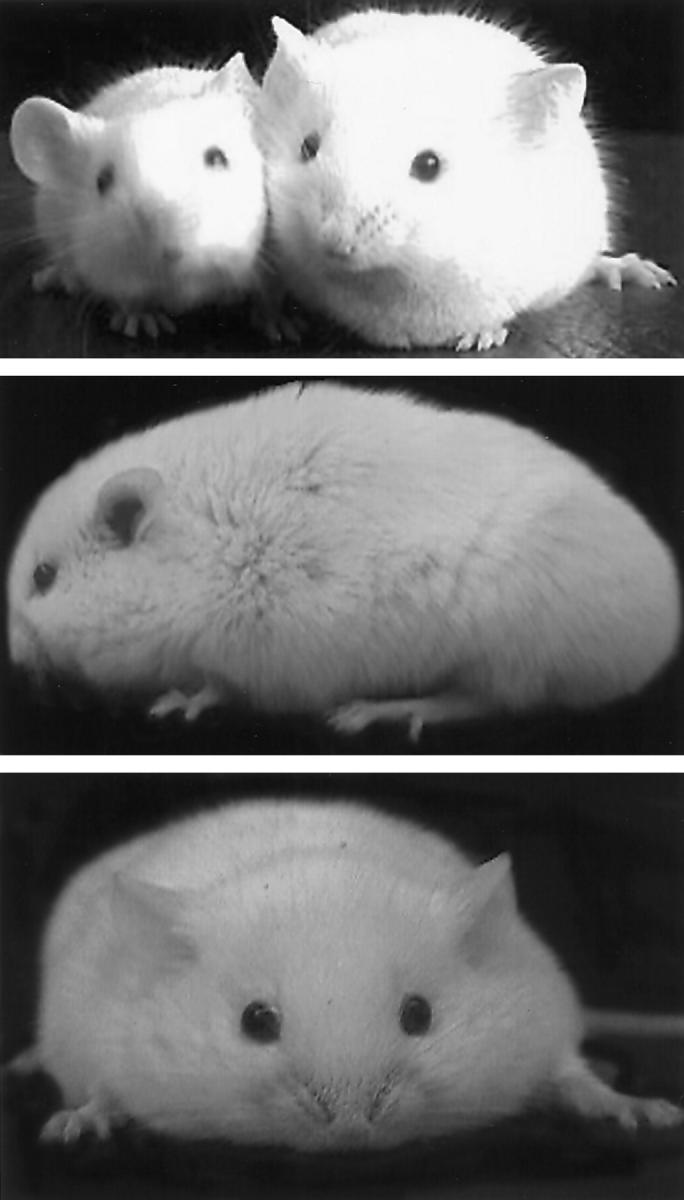
Older TOTs exhibit anasarca. Top: Age-matched nontransgenic and TOT mouse (on right) showing external signs of edematous swelling. Middle and bottom: Side and front views of TOT with anasarca shows swollen body. Body weights of the anasarca TOTs were approximately 40% higher than those of nontransgenics (n = 11 for each group), and mice developing anasarca usually died within 3 to 4 days after onset.
Figure 8.

Effect of litter size on TOT survival. The line was fitted using a smoothed cubic spline function. A significant association was found between TOT survival and litter size, by Spearman correlation analysis (r = 0.23, P < 0.0001). Analysis of covariance confirmed this result after accounting for litter effect (two or more mice in the analysis coming from the same litter; P < 0.01).
Heart:body weight ratios for TOTs ranging in age from 4 to 390 days (Figure 6C) ▶ increased quickly after birth and remained high throughout the lifetime of the mouse. A significant difference between TOT and control nontransgenic ratios was apparent by 9 days after birth (Figure 6D ▶ , at arrow showing bracketed region). However, increased TOT heart:body weight ratio plateaued rapidly within 48 hours after birth and did not change significantly throughout the study. In general, disproportionate increases in TOT heart weight occur around 1.5 weeks after birth, with most of the cardiac enlargement occurring in a very short time of 2 to 3 days. The average heart:body weight ratio for TOTs was 15.0 ± 4.0 mg/g compared to an average of 5.3 ± 0.2 mg/g for nontransgenic controls.
Chamber Dilation without Increasing Wall Thickness in TOT Hearts
TOT hearts were measured to determine ventricular wall and septal thickness as well as left ventricular chamber cross-sectional area. Three time points of 8, 11, and 14 days were chosen to observe changes before and after development of increased heart:body weight ratio (Figure 6) ▶ . Ventricular wall (Figure 9, A and C) ▶ and septal thickness (Figure 9B) ▶ remained comparable without significant differences (P > 0.1) between controls and TOTs throughout the time points investigated, consistent with the lack of hypertrophic response previously reported. 6 Left ventricular chamber cross-sectional area was consistently greater in TOTs than in nontransgenic controls (Figure 9D) ▶ . The trend toward increasing left ventricular chamber size by day 11 (0.3 > P > 0.2) became significant by day 14 (P < 0.03).
Figure 9.

Chamber volume and wall thickness of TOT hearts. Measurements in μm for wall thickness of right ventricle (A), septum (B), and left ventricle (C). Measurements were taken at three time points for control (open bars, n = 4) and TOTs (shaded bars, n = 5). No significant differences between the two groups were evident (P > 0.2). D: Cross sectional area measurements (in mm2) for the left ventricles of control (open bars, n = 4) and TOTs (shaded bars, n = 5) at indicated time points. Area measurements were consistently higher in TOTs, becoming significant by day 14 (* P < 0.03).
Discussion
A major limiting factor in human studies of dilated cardiomyopathy is the detection and diagnosis of early pathological changes. Because ventricular chamber enlargement is the defining morphological characteristic of dilated cardiomyopathy, initial assessment of TOTs was targeted at identifying the earliest detectable signs of ventricular pathology. Gross pathological features such as dilation of chambers and increased heart:body weight ratio were not evident until over 1 week after birth (Figures 6 and 9) ▶ ▶ . However, marked elevation of Tmod expression at birth (Figure 1) ▶ suggested initial stages of TOT cardiomyopathy were underway by late gestation or early postnatal development. Early postnatal irregularities in sarcomeric periodicity evident by ultrastructural analysis (Figure 5) ▶ progressed to widespread loss of Z-disk structural integrity and myofibril organization, apparent by confocal microscopy (Figure 4) ▶ several days later. Ultrastructural analysis before development of marked cardiomyopathic changes showed the initial stages of pathogenesis before onset of secondary compensatory effects, which complicate the interpretation of transgene mechanism. Intracellular structural anomalies reveal the immediate and direct effects of transgene expression, specifically that neonatal expression of Tmod weakens sarcomere structure and compromises myofibril alignment, eventually leading to gross pathology over 1 week later.
Based on knowledge of Tmod function in regulation of actin filament length from in vitro 11,20,21 and in vivo 7 studies, it is reasonable to postulate that myofibrillogenesis is impaired in the TOTs. Tmod binding to tropomyosin on the slow-growing (pointed) end of an actin filament inhibits polymerization. 19 Normally present in low levels relative to actin, excess Tmod level dissociates tropomyosin from actin filaments. 22 Because tropomyosin stabilizes actin filaments, 23 Tmod-mediated uncoating renders actin filaments susceptible to depolymerization and breakdown. This scenario is consistent with the in vitro effect of Tmod overexpression in cardiomyocytes where decreased I-band width, sarcomere shortening, and myofibril degeneration were observed. 11 Similar in vivo effects were noted after Tmod overexpression in the heart including loss of myofibril organization and thin filament disarray. 7 Resultant degeneration of myofibril structure leads to impaired contractility typically associated with development of dilated cardiomyopathy. Underlying pathogenic causes of impaired contractility can be attributed to loss of myofibril integrity as in TOT, 7 defective force transmission from the myofibril to myocardium as in muscle LIM protein (MLP)-deficient mice, 24 or transcriptional down-regulation of unidentified target genes essential for cardiomyocyte function and/or viability as in dominant-negative cAMP response element binding protein (CREB)-overexpressing mice. 25 All three lines share a similar phenotype, but the underlying causes are different: the first from overexpression of a sarcomeric component (Tmod), the second from deletion of a cytoskeletal protein component (MLP), and the third from overexpression of a mutated transcription factor (CREB). A condition similar to anasarca in TOTs, referred to as massive generalized edema, has been reported to occur in CREB-induced cardiomyopathy. 25 Molecular mechanisms causing pathological changes in these transgenic lines are unrelated, but they are likely to impact on interdependent processes. Loss of myofibrils probably contributes to degeneration of the myocardial cytoskeleton and vice versa. Interdependence of myofibril formation and attachment via the cytoskeleton is observed in cardiomyocyte cultures where contractility stimulates myofibril organization 26 and decreased tension results in myofibril breakdown and reorganization. 27 Shared phenotypic characteristics between the TOT, CREB, and MLP mouse models will help to advance understanding of the pathogenesis of dilated cardiomyopathy. However, in comparison to CREB and MLP, a combination of biochemical, in vitro, and in vivo analyses have extensively defined the functional activity of Tmod. The apparently direct causal relationship between Tmod overexpression, myofibril degeneration, and loss of contractile function makes this an attractive model system to study the process of dilated cardiomyopathy development.
TOT cardiomyopathy was initially described as a “failed hypertrophic response.” 7 However, TOT hearts show a significant increase in heart:body weight ratio (Figure 6) ▶ , which could be interpreted as hypertrophic growth. Hypertrophic and dilated changes can occur concurrently in cardiomyopathy, but TOT hearts show little evidence of a hypertrophic phenotype. Five independent observations point to a lack of hypertrophy in TOT hearts: i) no change in expression of some genes associated with hypertrophy such as skeletal actin, 7 ii) activation of calcineurin signaling (a powerful mediator of hypertrophy 13 ) fails to induce typical hypertrophic changes, iii) ventricular wall thickening typically associated with hypertrophy does not occur in TOT hearts (Figure 9) ▶ , iv) TOT cardiomyocytes are generally elongated without increased width, and cardiomyocytes with increased width typical of hypertrophic enlargement are never observed (Sussman, unpublished observation), and v) TOT hearts progress directly to failure without ever undergoing a transitory enhancement of contractile function, as is sometimes observed with hypertrophic mouse models of cardiomyopathy. The potential contribution of fluid retention in initial stages of dilation was examined in postnatal day 12 TOTs, but dry weight measurements indicate that contribution of fluid to heart:body weight is comparable between postnatal day 12 TOTs and nontransgenic controls (data not shown). Thus, we postulate that changes in TOTs arise from cardiomyocyte elongation and possibly cell slippage, both processes associated with increasing ventricular chamber volume characteristic of dilation.
Because TOT cardiac morphology can be rescued by inhibition of calcineurin without improving contractility or altering tropomodulin level, 12 TOT cardiac dilation is caused by secondary compensatory mechanism(s) rather than a primary effect of tropomodulin expression. However, the previous study did not establish a temporal relationship between calcineurin activation and the onset of the cardiomyopathic phenotype. In this report we demonstrate that calcineurin protein levels and activation state are significantly increased as early as 3 days after birth (Figure 2) ▶ . Increased expression and activation of calcineurin was maintained throughout the progressive development phase of TOT cardiomyopathy, implicating calcineurin as the regulator of a secondary reactive response that leads to the dilated phenotype. This idea is supported by the demonstration of substantially elevated diastolic and peak systolic calcium levels in TOT cardiomyocytes (Figure 3) ▶ . Increased calcium levels coupled with calcineurin activation in TOT cardiomyocytes suggests the following mechanism of cardiomyopathic pathogenesis: altered sarcomeric architecture caused by tropomodulin overexpression leads to compensatory increases in intracellular calcium to enhance contractility, and chronic elevation of calcium levels activate the calcium-sensitive phosphatase calcineurin. This hypothesis is consistent with our previous report that calcineurin inhibitory drugs (cyclosporin and FK506) prevent the initiation and progression of cardiac dilation, which is characteristic of TOT hearts. 12 Collectively, these findings correlate with other models of heart disease where alterations of sarcomeric structure/function are associated with altered calcium dynamics. 28-30 However, the enhanced basal and systolic calcium level coupled to the heart failure phenotype of TOTs is without precedent. Although calcium level determinations were performed on 13-week-old TOTs, it is reasonable to expect a similar rise in calcium levels within cardiomyocytes of younger TOTs. Calcium-dependent calcineurin activation in neonatal mice (Figure 2) ▶ serves as circumstantial evidence that TOT cardiomyocytes possess elevated calcium levels within the first week after birth. The 13-week time point was chosen for calcium ratio imaging to maintain consistency with previously published studies 16 and provide a high yield of viable cells. Technical considerations of the collagenase perfusion prevent the use of hearts from neonatal animals but, by confocal microscopy, the extent of myofibril degeneration observed between individual cardiomyocytes of young (2–3 weeks) and old (24–30 weeks) TOT mice are indistinguishable (data not shown). Thus, at least from a structural level, there is no reason to anticipate that cardiomyocytes from older animals would differ significantly from those of younger mice. Pilot experiments using cardiomyocytes from 4-week-old nontransgenic controls and TOTs showed substantial heterogeneity of calcium handling between cells within each preparation, perhaps as a consequence of ongoing postnatal developmental growth. As techniques to assess intracellular calcium levels improve, it will be interesting to examine intracellular calcium levels in relation to neonatal growth and TOT pathogenesis. At present, the circumstantial evidence of calcineurin activation (Figure 2) ▶ together with calcium measurements from isolated cardiomyocytes is consistent with a syndrome of myofibril dysgenesis leading to compensatory calcium increases. This combination of conditions in TOTs offers a unique paradigm to assess the mechanism underlying altered calcium handling in dilated cardiomyopathy.
The notion that calcineurin acts as a primary regulatory pathway for initiation of compensatory cardiomyopathic changes in TOTs is further supported by the analysis of MAPK signaling factors. The three main branches of the MAPK signaling cascade (p38, ERK1/2, and JNK1/2) were analyzed during the initiation and progression of TOT cardiac dilation and no significant association between any MAPK signaling factors and disease was found. p38 activation apparently participates in normal postnatal cardiac growth as seen in nontransgenic controls (Figure 2B) ▶ , possibly as a mediator of cardiac hypertrophy. 18 Variation in the timing and intensity of p38 activation is presumably due to differences in maturational rate of the mice and development of the myocardium. Overall, MAPK signaling factors are not associated with the reactive dilated response that is characteristic of TOT hearts.
Participation of calcineurin signaling in TOT pathogenesis together with increased calcium levels in TOT cardiomyocytes (Figure 3) ▶ indicate a primary role for calcium signaling pathways in the etiology of TOT disease. Presumably, rising calcium level results from decreased calcium uptake by the sarcoplasmic reticulum during relaxation, as observed in the failing heart. 33 Abnormal regulation of calcium currents in cardiac hypertrophy and failure leads to prolongation of the action potential, altering both contractile and relaxation properties. 34 As a consequence, altered expression of many calcium transport and regulatory proteins are observed in human and experimental mouse cardiomyopathies. For example, sarco/endoplasmic reticulum Ca2+-ATPase mRNA expression was significantly depressed in samples from 3-week-old TOTs. 7 Altering calcium level is one mechanism to regulate inotropy, so it is reasonable to speculate that calcium levels initially rise as a compensatory effort to increase contractility in the TOT heart. Activation of calcineurin within 5 days after birth suggests that elevation of calcium levels occurs early in TOT pathogenesis before marked changes are apparent in cardiac morphology. Subsequent deterioration of myofibril organization and altered expression of calcium handling proteins would provoke further elevation of intracellular calcium. Interestingly, pathologically high calcium levels may be necessary for TOT survival: breeding TOTs into a phospholamban-null background, which should enhance calcium reuptake into the sarcoplasmic reticulum, dramatically increased TOT mortality (Delling and Molkentin, unpublished observation). Rising calcium within TOT cardiomyocytes is likely to have consequences for multiple calcium-dependent signal transduction pathways in addition to calcineurin, including selected protein kinase C isoforms 35 and tyrosine kinases such as pyk2. 36 Preliminary results show pyk2 activation in TOT hearts, which probably exacerbates loss of force transmission from cardiomyocytes to the myocardium by affecting focal adhesion contact organization (data not shown).
The contributory effect of normal cardiac development to postnatal cardiomyopathy should be considered when describing phenotypes, especially in view of the significant number of mouse models exhibiting pathology within the first month after birth. 7,13,31,32 Rapidly changing demands on the heart at birth 37,38 coupled with postnatal cardiac hyperplasia and hypertrophy 39,40 require cardiac adaptation involving processes similar to those observed in conjunction with cardiomyopathy. Typically, increased heart weight and cell volume, altered myofibril organization and contractility, and chamber dilatation are considered characteristic cardiomyopathic changes when occurring late in life. The resilient postnatal heart can accommodate these normal developmental stresses. However, the TOT heart with impaired myofibrillogenesis is presumably unable to meet demands of normal postnatal hypertrophy, leading to a cyclic amplification of hypertrophic signaling, which is maladaptive and leads to decompensation. The quiescent period of comparable heart:body weight ratios between nontransgenic and TOTs through 9 days after birth was followed by a rapid onset of increased heart weight occurring within 48 hours, after which the elevated heart weight:body weight ratio was maintained at a consistently high level. The swift cardiac adaptive response and prolonged plateau of enlargement indicates that the TOT heart rapidly compensated for loss of contractility up to an inherent limit in the progression of cardiac dilation.
Initially, extended longitudinal studies of TOTs were hampered by high mortality rates within 1 month after birth. 7 However, a small percentage of severely affected mice survived. The TOT population was increased by breeding surviving mice, resulting in uniformly affected homozygous litters. With the breeding program established, the longitudinal study of TOT pathogenesis was started. Using the TOT relational database for analysis of phenotypic trends, developmental aspects of TOT cardiomyopathy became apparent, and early mortality at 2 weeks after birth narrowed the window for onset of TOT pathogenesis to neonatal life. Variable mortality between genetically identical siblings suggested environmental factors influenced TOT survival beyond the critical 1-month period. The hypothesis of postnatal litter size influencing survival in TOTs can be tested by removing some siblings at birth to decrease the number of offspring. The association between TOT survival and larger litter size (Figure 8) ▶ may stem from developmental retardation and slower maturation of mice reared in large litters compared to small litters, 41,42 allowing postnatal cardiac hypertrophy to progress more gradually. This survival analysis is, to our knowledge, the first demonstration of an environmental factor impacting on survival in a mouse cardiomyopathic model.
Hallmarks of dilated cardiomyopathy such as poor systolic performance and increased ventricular chamber cavity 8-10 are seen in several cardiomyopathic transgenic lines 7,24,25 but the unique combination of features in TOTs offers a useful paradigm for understanding dilated cardiomyopathy. First, TOTs suffer from a thin filament-based sarcomeric disease, a myofibril component now receiving more attention due the discovery of actin-based familial cardiomyopathy. 43 Second, TOTs exhibit dilation in the absence of the significant hypertrophic response and fibrosis often seen in transgenic cardiomyopathies. 13,31 Third, the myofibril disarray and lack of ventricular wall thickening found in TOTs is reminiscent of histological reports describing some human dilated cardiomyopathies. 8-10,44-46 Fourth, circumstantial evidence from genetic studies suggests Tmod as a candidate gene for human cardiomyopathy. 47 Fifth, extensive molecular and biochemical characterizations defining the interaction between Tmod, tropomyosin, and actin provide a clear mechanism of action for Tmod overexpression in vivo. Last, but not least, TOT cardiomyopathy involves presence of the transgene together with compensatory failed hypertrophy, a combinatorial mechanism leading to disease which has not been addressed in previous studies and may be relevant to human cardiomyopathic disease. To elucidate how hypertrophy contributes to pathogenesis and identify molecular mechanism(s) involved in the TOT paradigm, cross-breeding of TOTs to other transgenic lines with altered postnatal cardiac development is underway.
Acknowledgments
We would like to acknowledge support from the Marion-Merrill Dow fund for establishing cardiomyopathic transgenic mice, Angela Walker for generating the population graphs, and the contribution of Jon Neuman in the creation of our TOTs.
Footnotes
Address reprint requests to Mark A. Sussman, The Children’s Hospital and Research Foundation, Division of Molecular Cardiovascular Biology, Room 3033, 3333 Burnet Avenue, Cincinnati, Ohio 45229. E-mail: sussman@heart.chmcc.org.
Supported by a grant-in-aid from the American Heart Association Ohio Affiliate, a Scientist Development Grant (9630047N) and a grant-in-aid (9750638N) from the American Heart Association National, and a National Institutes of Health R29 award (HL58224–01).
References
- 1.Patterson JH, Adams KF, Jr: Pathophysiology of heart failure: changing perceptions. Pharmacotherapy 1996, 16:27S-37S [PubMed] [Google Scholar]
- 2.Garg R, Packer M, Pitt B, Yusuf S: Heart failure in the 1990s, evolution of a major public health problem in cardiovascular medicine. JACC Supp A 1993, 22:3A-5A [DOI] [PubMed] [Google Scholar]
- 3.Abelmann WH: The dilated cardiomyopathies: experimental aspects. Cardiol Clinics 1988, 6:219-231 [PubMed] [Google Scholar]
- 4.Manolio TA, Baughman KL, Rodeheffer R, Pearson TA, Bristow JD, Michels VV, Abelmann WH, Harlan WR: Prevalence and etiology of idiopathic dilated cardiomyopathy: summary of a National Heart, Lung, and Blood Institute workshop. Am J Cardiol 1992, 69:1458-1466 [DOI] [PubMed] [Google Scholar]
- 5.Ho KK, Pinsky JL, Kannel WB, Levy D: The epidemiology of heart failure: the Framingham study. J Am Coll Cardiol Supp A 1993, 22:6A-13A [DOI] [PubMed] [Google Scholar]
- 6.Kannel WB: Some lessons in cardiovascular epidemiology from Framingham. Am J Cardiol 1976, 37:269-282 [DOI] [PubMed] [Google Scholar]
- 7.Sussman MA, Welch S, Cambon N, Klevitsky R, Hewett T, Price RL, Witt SA, Kimball TR: Myofibril degeneration caused by tropomodulin overexpression in juvenile mice leads to dilated cardiomyopathy. J Clin Invest 1998, 101:51-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schaper J, Froede TA, Hein St, Buck A, Hashizume H, Speiser B, Friedl A, Bleese N: Impairment of the myocardial ultrastructure and changes of the cytoskeleton in dilated cardiomyopathy. Circulation 1991, 83:504-514 [DOI] [PubMed] [Google Scholar]
- 9.Izumi T, Hattori A, Higuma N, Tamura K: Cardiac myofibril disorietation and Z-band abnormalities in idiopathic dilated cardiomyopathy: an electron microscope study. Arch Histol Jpn 1978, 41:293-308 [DOI] [PubMed] [Google Scholar]
- 10.Jindal N, Talwar KK, Chopra P: Ultrastructural and histological study of endomyocardial biopsies from patients of dilated cardiomyopathy: a comparative evaluation and their clinical correlation. Indian Heart J 1994, 46:329-334 [PubMed] [Google Scholar]
- 11.Sussman MA, Baque S, Uhm C-S, Daniels MP, Price RL, Simpson D, Terracio L, Kedes L: Altered expression of tropomodulin in cardiomyocytes disrupts the sarcomeric structure of myofibrils. Circ Res 1998, 82:94-105 [DOI] [PubMed] [Google Scholar]
- 12.Sussman MA, Lim HW, Gude N, Taigen T, Olson EN, Robbins J, Colbert MC, Gualberto A, Wieczorek DF, Molkentin JD: Prevention of cardiac hypertrophy in mice by calcineurin inhibition. Science 1998, 281:1690-1693 [DOI] [PubMed] [Google Scholar]
- 13.Molkentin JD, Lu JR, Antos CL, Marham B, Richardson J, Robbins J, Grant SR, Olson E: A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 1998, 93:215-228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lim HW, Molkentin JD: Calcineurin and human heart failure. Nat Med 1999, 5:246-247 [DOI] [PubMed] [Google Scholar]
- 15.Kloner RA, Leor J, Poole WK, Perritt R: Population-based analysis of the effect of the Northridge earthquake on cardiac death in Los Angeles County, California. J Am Coll Cardiol 1997, 30:1174-1180 [DOI] [PubMed] [Google Scholar]
- 16.Kadambi VJ, Ponniah S, Harrer JM, Hoit BD, Dorn II GW, Walsh RA, Kranias EG: Cardiac-specific overexpression of phospholamban alters kinetics and resultant cardiomyocyte mechanics in transgenic mice. J Clin Invest 1996, 97:533–539 [DOI] [PMC free article] [PubMed]
- 17.Price RL, Chintanowonges C, Shirashi I, Bork TK, Terracio L: Local and regional variations in myofibrillar patterns in looping rat hearts. Anat Rec 1996, 245:83-93 [DOI] [PubMed] [Google Scholar]
- 18.Zechner D, Thuerauf DJ, Hanford DS, McDonough PM, Glembotski CC: A role for the p38 mitogen-activated protein kinase pathway in myocardial cell growth, sarcomeric organization, and cardiac-specific gene expression. J Cell Biol 1997, 139:115-127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Katz AM: Evolving concepts of heart failure: cooling furnace, malfunctioning pump, enlarging muscle: Part I. J Card Fail 1997, 3:319-334 [DOI] [PubMed] [Google Scholar]
- 20.Weber AM, Pennise CR, Babcock GG, Fowler VM: Tropomodulin caps the pointed ends of actin filaments. J Cell Biol 1994, 127:1627-1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fowler VM, Sussman MA, Miller PG, Flucher BE, Daniels MP: Tropomodulin is associated with the free (pointed) ends of the thin filaments in rat skeletal muscle. J Cell Biol 1993, 120:411-420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fowler VM: Tropomodulin: A cytoskeletal protein that binds to the end of erythrocyte tropomyosin and inhibits tropomyosin binding to actin. J Cell Biol 1990, 111:471-482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Broschat K: Tropomyosin prevents depolymerization of actin filaments from the pointed end. J Biol Chem 1990, 265:21323-21329 [PubMed] [Google Scholar]
- 24.Arber S, Hunter JJ, Ross J, Hongo M, Sansig G, Borg J, Perriard J-C, Chien KR, Caroni P: MLP-deficient mice exhibit a disruption of cardiac cytoarchitectural organization, dilated cardiomyopathy, and heart failure. Cell 1997, 88:393-403 [DOI] [PubMed] [Google Scholar]
- 25.Fentzke RC, Kocarz CE, Lang RM, Lin H, Leiden JM: Dilated cardiomyopathy in transgenic mice expressing a dominant-negative CREB transcription factor in the heart. J Clin Invest 1998, 101:2415-2426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharp WW, Terracio L, Borg TK, Samarel AM: Contractile activity modulates actin synthesis and turnover in cultured neonatal rat heart cells. Circ Res 1993, 73:172-183 [DOI] [PubMed] [Google Scholar]
- 27.Simpson DG, Sharp WW, Borg TK, Price RL, Terracio L, Samarel AM: Mechanical regulation of cardiac myocyte protein turnover and myofibrillar structure. Am J Physiol 1996, 270:C1075-1087 [DOI] [PubMed] [Google Scholar]
- 28.Delbridge LM, Satoh H, Yuan W, Bassani JW, Qi M, Ginsburg KS, Samarel AM, Bers DM: Cardiac myocyte volume, Ca2+ fluxes, and sarcoplasmic reticulum loading in pressure-overload hypertrophy. Am J Physiol 1997, 272:H2425-2435 [DOI] [PubMed] [Google Scholar]
- 29.Palmiter KA, Kitada Y, Muthuchamy M, Wieczorek DF, Solaro RJ: Exchange of β- for α-tropomyosin in hearts of transgenic mice indices changes in thin filament response to Ca2+, strong cross-bridge binding, and protein phosphorylation. J Biol Chem 1996, 271:11611-11614 [DOI] [PubMed] [Google Scholar]
- 30.Spindler M, Saupe KW, Christe ME, Sweeney HL, Seidman CE, Seidman JG, Ingwall JS: Diastolic dysfunction and altered energetics in the alphaMHC403/+ mouse model of familial hypertrophic cardiomyopathy. J Clin Invest 1998, 101:1775-1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muthuchamy M, Grupp IL, Grupp G, O’Toole BA, Kier AB, Boivin GP, Neumann J, Wieczorek DF: Molecular and physiological effects of overexpressing striated muscle β-tropomyosin in the adult murine heart. J Biol Chem 1995, 270:30593-30603 [DOI] [PubMed] [Google Scholar]
- 32.Colbert MC, Hall DG, Kimball TR, Witt SA, Lorenz JN, Kirby ML, Hewett TE, Klevitsky R, Robbins J: Cardiac compartment-specific overexpression of a modified retinoic acid receptor produces dilated cardiomyopathy and congestive heart failure in transgenic mice. J Clin Invest 1997, 100:1958-1968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mittmann C, Eschenhagen T, Scholz H: Cellular and molecular aspects of contractile dysfunction in heart failure. Cardiovasc Res 1998, 39:267-275 [DOI] [PubMed] [Google Scholar]
- 34.Richard S, Leclercq F, Lemaire S, Piot C, Nargeot J: Ca2+ currents in compensated hypertrophy, and heart failure. Cardiovasc Res 1998, 37:300-311 [DOI] [PubMed] [Google Scholar]
- 35.Bowling N, Walsh RA, Song G, Estridge T, Sandusky GE, Fouts RL, Mintze K, Pickard T, Roden R, Bristow MR, Sabbah HN, Mizrahi JL, Gromo G, King GL, Vlahos CJ: Increased protein kinase C activity and expression of Ca2+-sensitive isoforms in the failing human heart. Circulation 1999, 99:384-391 [DOI] [PubMed] [Google Scholar]
- 36.Li X, Dy RC, Cance WG, Graves LM, Earp HS: Interactions between two cytoskeleton-associated tyrosine kinases: calcium dependent tyrosine kinase and focal adhesion kinase. J Biol Chem 1999, 274:8917-8924 [DOI] [PubMed] [Google Scholar]
- 37.Friedman AH, Fahey JT: The transition from fetal to neonatal circulation: normal responses and implications for infants with heart disease. Sem Perinatol 1993, 17:106-121 [PubMed] [Google Scholar]
- 38.Teitel DF: Circulatory adjustments to postnatal life. Sem Perinatol 1988, 12:96-103 [PubMed] [Google Scholar]
- 39.Rakusan K, Cicutti N, Flanagan MF: Changes in the microvascular network during cardiac growth, development, and aging. Cell Mol Biol Res 1994, 40:117-122 [PubMed] [Google Scholar]
- 40.Hudlicka O: Mechanical factors involved in the growth of the heart and blood vessels. Cell Mol Biol Res 1994, 40:143-152 [PubMed] [Google Scholar]
- 41.Nagy ZM, Porada KJ, Anderson JA: Undernutrition by rearing in large litters delays the development of reflexive, locomotor, and memory processes in mice. J Comp Physiol Psychol 1977, 91:682-696 [DOI] [PubMed] [Google Scholar]
- 42.Ryan V, Wehmer F: Effect of postnatal litter size upon adult aggression in the laboratory mouse. Dev Psychobiol 1975, 8:363-370 [DOI] [PubMed] [Google Scholar]
- 43.Olson TM, Michels VV, Thibodeau SN, Tai Y-S, Keating MT: Actin mutations in dilated cardiomyopathy, a heritable form of heart failure. Science 1998, 280:750-752 [DOI] [PubMed] [Google Scholar]
- 44.Hein S, Schaper J: Pathogenesis of dilated cardiomyopathy and heart failure: insights from cell morphology and biology. Curr Opin Cardiol 1996, 11:293-301 [DOI] [PubMed] [Google Scholar]
- 45.Ferrans VJ: Pathologic anatomy of the dilated cardiomyopathies. Am J Cardiol 1989, 64:9C-11C [DOI] [PubMed] [Google Scholar]
- 46.Beltrami CA, Finato N, Rocco M, Feruglio GA, Puricelli C, Cigola E, Sonnenblick EH, Olivetti G, Anversa P: The cellular basis of dilated cardiomyopathy in humans. J Mol Cell Cardiol 1991, 27:291-305 [DOI] [PubMed] [Google Scholar]
- 47.Krajinovic M, Pinamonti B, Sinagra G, Vatta M, Severini GM, Milasin J, Falaschi A, Camerini F, Giacca M, Mestroni L, : the Heart Muscle Disease Study Group: Linkage of familial dilated cardiomyopathy to chromosome 9. Am J Hum Genet 1995, 57:846-852 [PMC free article] [PubMed] [Google Scholar]