

Abstract
Light chain deposition disease (LCDD) and light chain amyloidosis (AL) are disorders of monoclonal immunoglobulin deposition in which normally soluble serum precursors form insoluble deposits in tissues. A common feature in both is the clonal proliferation of B-cells that produce pathogenic light chains. However, the deposits in LCDD differ from those in AL in that they are ultrastructurally granular rather than fibrillar and do not bind Congo red or colocalize with amyloid P component or apolipoprotein E. The reason(s) for their differences are unknown but are likely multifactorial and related to their protein conformation and their interaction with other molecules and tissue factors in the microenvironment. Knowledge of the primary structure of the light chains in LCDD is very limited. In the present study two new κ1 light chains from patients with LCDD are described and compared to seven other reported κ-LCDD proteins. The N-terminal amino acid sequences of light chain GLA extracted from the renal biopsy and light chain CHO from myocardial tissue were each identical to the respective light chains isolated from the urines and to the V-region amino acid sequences translated from the cloned cDNAs obtained from bone marrow cells. The germline V-region sequences, determined from the genomic DNA in both and in MCM, a previously reported κ1 LCDD light chain, were identical and related to the L12a germline gene. The expressed light chains in all three exhibit amino acid substitutions that arise from somatic mutation and result in increased hydrophobicity with the potential for protein destabilization and disordered conformation.
The monoclonal immunoglobulin deposition diseases, which include nonamyloid light chain deposition disease (LCDD) and light chain amyloidosis (AL), have in common monoclonal immunoglobulin synthesis by an expanded clone of B cells leading to the deposition of insoluble light chains in systemic organs with displacement and eventual destruction of parenchymal cells and organ dysfunction. 1 However, LCDD and AL differ in several ways. In the case of AL the deposits are congophilic, have a fibrillar ultrastructure, are more frequently derived from λ than κ light chain, have a patchy distribution within and among systemic organs, and colocalize with amyloid P component, apolipoprotein-E (apo-E), and glycosaminoglycans (GAGs). 2,3 In contrast, the deposits in LCDD are noncongophilic, have a granular rather than fibrillar ultrastructure, and are more frequently κ than λ. In cases completely examined at autopsy, deposits are uniformly distributed in all the basement membranes of systemic organs 4,5 and do not associate with amyloid P component or apo-E. 2 These differences are of fundamental importance in determining the mechanisms of protein deposition in tissues and the process of fibrillogenesis possibly relevant to other common types of amyloidosis, such as Alzheimer’s disease. However, the biophysical basis for their differences is poorly understood.
Knowledge of the primary structure of the protein deposits in LCDD is very limited; in only nine cases of LCDD has the complete light chain variable region (V-region) sequence been published. 6-13 Thus, a comparison of the nonfibrillar and fibrillar forms of deposits in LCDD and AL that could identify differences in their primary structures and might relate to their dissimilar properties is hampered by the apparent scarcity of cases of LCDD to study and the unavailability of large amounts of tissues from postmortem examinations for biochemical analysis. This limitation is now partially overcome by microextraction methods to isolate and obtain the amino-terminal sequence of light chain deposits from milligram amounts of diagnostic biopsy tissues, 14,15 as well as the application of molecular techniques to obtain the light chain V-region amino acid sequence translated from cloned cDNA of bone marrow cells. These methods, applied to more readily available biopsy tissues, allow the opportunity to build a primary structure data base for comparison with nonpathogenic Bence-Jones light chains as well as those in AL disease, with the goal of elucidating the mechanism(s) of tissue deposition and fibrillogenesis.
In a previous case of LCDD, we reported the biochemical data of a V-region κ1 (V-κ1) nonamyloidotic immunoglobulin light chain, MCM, obtained by extraction of deposits from myocardial tissue in which five unique amino acid substitutions were identified. 11 We now report two new κ1 LCDD proteins: GLA and CHO. In both we determined the N-terminal amino acid sequences of the light chains isolated from the tissues and urines, the complete V-region light chain amino acid sequences deduced from the cloned cDNAs, and the nucleotide germline sequences from genomic DNA. We also determined the germline sequences from genomic DNA of MCM. We conclude that the amino acid substitutions identified in GLA and CHO, as well as in MCM, are due to somatic mutations and contribute to protein instability, aggregation, and the deposition of the light chains in the tissues.
Materials and Methods
Patients
The diagnosis of LCDD was made in two patients who presented with renal disease and whose renal biopsy tissues showed monotypic κ light chain deposits.
A 61-year-old caucasian female (GLA) presented with increasing renal functional impairment. Physical examination revealed no edema or other abnormalities. The serum creatinine rose from 0.8 to 2.6 mg/dl over an 8-month period and to 3.7 mg/dl at the ninth month, the time of admission and renal biopsy. The urine showed microhematuria and a urine protein excretion of 0.16–0.37 g/24 hours. The serum cholesterol level was 270 mg/dl, C3/C4 was 129/40 mg/dl (normal), and hematocrit was 32%. A diagnosis of κ LCDD was made by renal biopsy. Immunoelectrophoresis revealed no monoclonal protein in the serum and a mixture of albumin and monoclonal Bence-Jones κ light chain, estimated at 30 mg/dl, in 100-fold concentrated urine. A bone marrow biopsy showed features of a B-lymphocyte neoplasm. Despite chemotherapy with Melphalan and Prednisone, irreversible renal failure developed, requiring maintenance hemodialysis.
The second patient, a 64-year-old Asian female (CHO), was admitted to the hospital for complaints of fatigue, anorexia, back pain, and acute renal failure that developed after abdominal computed tomography (CT) with radio contrast. She was known to have normal renal function, with a serum creatinine of 0.9 mg/dl, 2 years before. After CT studies the creatinine was 5.7 mg/dl and rose to 9.1 mg/dl 4 days later at the time of admission. The past medical history was unremarkable except for hepatitis B surface antigenemia for many years. Her blood pressure was normal and there was no edema. The significant laboratory findings included urine protein excretion, 1.0 g/24 hour; microhematuria; blood urea nitrogen, 100 mg/dl; serum Ca, 10.4 mg/dl; Phos, 6.7 mg/dl. A diagnosis of κ LCDD was made by renal biopsy. The initial serum and urine immunoelectrophoresis showed no monoclonal Ig, but subsequently Bence-Jones κ light chain was identified in concentrated urine, and lytic lesions were detected in the skull. Bone marrow examination revealed multiple myeloma. Renal failure worsened and expiration occurred 2 months after admission. A postmortem examination was performed.
Pathological and Immunohistological Studies
Paraffin sections of formalin-fixed tissues were stained with hematoxylin and eosin, periodic acid silver methenamine, and Congo red. Standard immunofluorescence microscopy examination of frozen sections was performed on renal biopsy tissues from both patients and on systemic tissues obtained at necropsy from CHO. Sections were incubated with a panel of fluorescein-labeled rabbit polyclonal anti-human Ig chain-specific antibodies (γ, μ, α, κ, and λ), C3, C1q, fibrin and unlabeled rabbit polyclonals anti-amyloid P component (Dako, Carpenteria, CA), and anti-apo-E (Chemicon, Temecula, CA), followed by fluorescein-conjugated swine anti-rabbit Ig (Dako). Immunoperoxidase examinations of formalin-fixed, paraffin-embedded bone marrow biopsy specimens from both patients incubated with polyclonal anti-κ and anti-λ antibodies (Dako) were performed as described. 16
Electron microscopic studies were carried out on ultrathin sections of glutaraldehyde-fixed epon-embedded renal biopsy tissues and on frozen cardiac tissue stored at −70°C.
Light Chain Isolation from Tissue and Urine Specimens
The GLA residual 1-mm 3 frozen renal biopsy tissue was washed three times in 500 μl of 50 mmol/L phosphate/150 mmol/L NaCl (PBS) (pH 7.2) and centrifuged at 2500 rpm. The tissue pellet was placed in dissociating buffer (50 mmol/L Tris, pH 6.8, 2% sodium dodecyl sulfate, 5% glycerol, 0.1% bromophenol blue), incubated at (80°C) for 2 hours with continuous agitation and centrifuged at 2500 rpm for 5 minutes. The supernatant was boiled for 5 minutes after the addition of dithiothreitol (DTT) to a final concentration of 100 mmol/L.
Light chain deposits were extracted from CHO myocardial tissue stored at −70°C as previously described. 11 Briefly, tissue (5–8 g) was repeatedly homogenized in 10 mmol/L phosphate/2.7 mmol/L KCl/137 mmol/L NaCl, pH 7.4, with a cocktail of protease inhibitors (Complete, plus 1 μmol/L pepstatin and 1 μmol/L leupeptin, all from Boehringer Mannheim, Indianapolis, IN), and 1 mmol/L EDTA, followed by centrifugation. The pellet was extracted with 6 mol/L guanidine-HCl in 50 mmol/L Tris-HCl, pH 10.2, containing 170 mmol/L DTT under constant stirring for 48 hours at room temperature. The extracted material was centrifuged at 100,000 × g for 1 hour at 4°C, and the crude extract was dialyzed at 4°C in membrane tubing with a cutoff of 2 kd against distilled water and lyophilized.
The GLA and CHO 24-hour urine specimens were dialyzed against PBS in tubing with a molecular mass cutoff of 2 kd and concentrated 100-fold. A sample diluted 1:3 in PBS was filtered through a 0.45-μm disk (Millipore, Bedford, MA) and run three times through a 1-ml KappaLock sepharose column (Zymed Lab, San Francisco, CA) according to the manufacturer’s instructions. The column was washed with 10 column volumes of PBS before the bound κ chains were eluted with 2 column volumes of 500 mmol/L propionic acid (Fisher Scientific, Pittsburgh, PA). All of the eluates were lyophilized.
Gel Electrophoresis and Western Blotting Analysis
The protein extracts from the renal biopsy and myocardial tissues and the purified Bence-Jones protein from the urines were analyzed by fractionation in 12.5% and/or 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and electrotransferred onto polyvinyldifluoride membranes in 3-cyclo-hexylamino-1-propanesulfonic acid (Sigma Chemical Co., St. Louis, MO) buffer, pH 11, containing 10% methanol. The membranes were washed in distilled deionized water. Strips removed for immunoblotting were blocked for 1 hour in PBS–0.05% Tween 20 containing 5% nonfat dry milk and 1% bovine serum albumin, incubated with polyclonal rabbit anti-human κ antibody 1:500 (Dako) for 11/2 hours at room temperature, washed three times in PBS–0.1% Tween 20, followed by incubation with horseradish peroxidase-conjugated donkey anti-rabbit Ig (1:2500; Amersham, Arlington Heights, IL) for 2 hours. The reaction was developed with 4-chloro-1-naphthol peroxidase substrate system (Kirkegaard and Perry Laboratories, Gaithersburg, MD) or by fluorograms prepared with an ECL Western blotting kit (Amersham) according to the manufacturer’s specifications. For sequence purposes, membranes were stained with 0.1% Coomassie blue R 250 in 40% methanol–1% acetic acid (high-performance liquid chromatography (HPLC) grade), destained with 50% methanol (HPLC grade), and extensively washed in deionized water, and the bands were excised and sequenced. Automated Edman degradation analysis was carried out on a 477A microsequencer, and the resulting phenylthiohydantoin derivatives were identified using the on-line 120 A PTH analyzer (Applied Biosystems, Foster City, CA).
cDNA Cloning
The amino-terminal sequence of the light chain deposits extracted from the renal biopsy and the myocardial tissue, as well as the amino acid sequences obtained from the light chains isolated from the urines of both subjects, indicated that both light chains belonged to the V-κ1 subgroup. 17 Accordingly, oligonucleotide forward and reverse primers were synthesized as described. 9 Total RNA isolated from the GLA frozen residual diagnostic bone marrow aspirate and CHO frozen vertebral bone marrow fragments obtained at autopsy were used in reverse transcriptase-polymerase chain reaction (RT-PCR) experiments to isolate and clone the cDNAs. Briefly, bone marrow cells were washed with 10 mmol/L phosphate-buffered saline, pH 7.4, and 150 mmol/L NaCl, and total RNA was isolated by the guanidine isothiocyanate method, using Trizol LS (Gibco BRL, Gaithersburg, MD). Reverse transcription of RNA was performed with a first-strand cDNA synthesis kit (Boehringer Mannheim), using the avian myeloblastosis virus reverse transcriptase with the downstream primer. PCR amplification of the first-strand cDNA produced by reverse transcription was performed by introducing the upstream amplimer in 1× PCR buffer. Each PCR cycle, consisting of a denaturation step (94°C/1 minute), an annealing step (42°C/1 minute), and an elongation step (72°C/2 minute), was repeated 30 times. For control purposes RNA samples were pretreated with RNase A for 30 min at 37°C and subjected to RT-PCR. In these controls as well as in RT-PCR of RNA extracted from other diagnostic bone marrows without plasma cell dyscrasia, no specific amplification was observed. PCR products were fractionated on 5% polyacrylamide gels and visualized under UV light. The resulting PCR products were subcloned into the pCR2.1 vector (TA cloning kit; Invitrogen, Carlsbad, CA), and the isolated recombinant plasmids were sequenced by the dideoxy chain termination method in both directions.
Germline V-Region Sequences
Genomic DNA was isolated from lymphocytes in fresh peripheral blood (GLA) and from stored frozen cardiac tissues (CHO, MCM) obtained at necropsy. Specific PCRs were set up using forward and reverse primers: 5′-GTCTTCCYAYAATATGATC-3′ and 5′-AGGACCACT-CTCAGCTGATA-3′, respectively, for gene L12. Five hundred nanograms of each DNA were added to 25 pg of each primer, 200 μmol/L dNTP, and 1.5 units of Taq DNA polymerase in a final volume of 50 μl of appropriate buffer (Pharmacia, Uppsala, Sweden), denatured at 94°C for 5 minutes, and amplified through 30 cycles, including 30-second steps at 94°C, 55°C, and 72°C, followed by elongation at 72°C for 10 minutes. To rule out possible errors of Taq polymerase, at least two independent PCR reactions were performed on DNA extracted from each specimen. Products were ligated to pCR2.1 vector, and a minimum of 10 clones from each case were sequenced in both directions.
Structural Analysis
The spatial locations of the amino acid substitutions of the GLA and CHO light chains were computationally mutated on the backbone structure of REI, a κ1-soluble nonpathogenic Bence-Jones light chain of known three-dimensional structure, 18,19 with the computer program Insight II (Biosym, San Diego, CA).
Isoelectric Points of κ-Light Chains
The theoretic isoelectric points of the V-region Ig light chains were determined as previously described. 20 In this calculation cysteine residues involved in disulfide bond function were assumed to be nonionizable.
Results
Pathological and Immunohistological Studies
In the GLA renal biopsy tissue there were 29 glomeruli, four of which were obliterated by sclerosis. All of the remaining glomeruli exhibited normal cellularity, and there were no mesangial nodules. A few had thickened wrinkled basement membranes but most had minimal abnormalities. There was widespread interstitial edema and fibrosis, focal lymphocytic infiltration, and focal tubular atrophy. No prominent casts with multinucleated giant cells were seen. Congo red stain for amyloid was negative. Immunofluorescence microscopy demonstrated bright staining of all glomerular, tubular, and vascular basement membranes for κ (Figure 1A) ▶ , but not λ light chains; γ, μ, or α heavy chains; C3; C1q; fibrin; amyloid P component; or apoE (not shown). Clustered granular deposits in the glomerular and tubular basement membranes (Figure 1B) ▶ , typical of LCDD, were found ultrastructurally. A bone marrow aspirate and biopsy exhibited increased lymphocytes, dispersed as well as in small aggregates, some with plasmacytoid differentiation; cell marker analysis revealed predominantly B cells expressing surface immunoglobulin of the κ isotype with a κ:λ ratio of 30. A diagnosis of lymphoplasmacytoid lymphoma was rendered.
Figure 1.

A and B: GLA renal biopsy tissue. A: Immunofluorescence micrograph demonstrates immunostaining for κ light chain in glomerular and tubular basement membranes. B: Electron micrograph of a tubule shows clustered granular electron-dense deposits (arrows) in basement membrane. C and D: CHO myocardial tissue. C: Immunofluorescence micrograph reveals perimyocytic deposits staining for κ. D: No staining for λ light chain. Magnification: A, ×200; B, ×10,000; C and D, ×300.
In the CHO renal biopsy specimen all of the nine glomeruli present appeared normal; there was moderate interstitial edema and lymphocytic cell infiltration and many tubular protein casts, some surrounded by multinucleated giant cells, as seen in Bence-Jones cast nephropathy. Immunofluorescence microscopy revealed strong staining of glomerular, tubular, and vascular basement membranes for κ but not λ light chain nor γ, μ, or α heavy chains; C3; C1q; fibrin; amyloid P component; or apoE. Clustered, granular electron-dense deposits in glomerular and tubular basement membranes were ultrastructurally typical of LCDD. The bone marrow biopsy displayed nodules and sheets of κ-bearing plasma cells and only a few scattered λ-staining cells, diagnostic of myeloma. At autopsy the remarkable findings were a firm enlarged 400-g heart with left ventricular hypertrophy and a liver with nodular cirrhosis. There were no vertebral lytic lesions. Light microscopic examination confirmed the renal and bone marrow biopsy findings. The myocardium exhibited mild interstitial widening by loose eosinophilic material, but no evidence of inflammation or muscle damage. Congo red stains of all systemic organs showed no amyloid deposits. Immunofluorescence microscopy demonstrated deposits of κ (Figure 1C) ▶ , but not λ light chain (Figure 1D) ▶ , around cardiac myocytes, in fibrous septae and vascular walls, and diffusely in the basement membranes of all systemic organs.
Western Blotting, Amino Acid Sequencing, and cDNA Cloning
The electrotransferred material extracted from the tissues and the urines of both subjects is shown in Figure 2 ▶ . The GLA extract from the kidney biopsy tissue (lane 1) yielded intact protein and fragments reactive with anti-κ (lane 2). The sequences obtained from all bands were homologous to the intact N-terminus of the κ1 light chain subgroup (Figure 3A) ▶ ; the 13-kd protein yielded the sequence from residues 1–8 (16 kd, 1–15; 17 and 18 kd, 1–10; and the 28-kd protein the sequence from positions 1–6). The urine eluate from the KappaLock column was mainly comprised of a single protein of 28 kd (Figure 2 ▶ , lane 3) that was reactive with anti-κ (Figure 2 ▶ , lane 4) and revealed the sequence from positions 1 to 35.
Figure 2.

Fifteen percent SDS-PAGE and Western blot analysis of GLA and CHO κ light chains. Extracted light chains from GLA kidney biopsy tissue (lane 1) and CHO cardiac tissue (lane 5) were stained with Coomassie blue. Immunoreactivity was demonstrated for the extracted proteins GLA (lane 2) and CHO (lane 6) with anti-κ. Urine eluates are for GLA (lane 3) and CHO (lane 7) after KappaLock purification, stained with Coomassie blue. Western blot analysis was made of GLA (lane 4) and CHO (lane 8) urine eluates immunostained with anti-κ.
Figure 3.

cDNA sequence of the V-region of the monoclonal κ1 light chains GLA (A) and CHO (B) cloned from bone marrow plasma cells. Numbering is according to Kabat et al. 17
The transferred guanidine extract CHO from cardiac tissue (Figure 2 ▶ , lane 5), also consisting of intact protein and fragments, demonstrated immunoreactivity with anti-κ (Figure 2 ▶ , lane 6); the sequence, homologous with the N-terminus of the κ1 light chain subgroup (Figure 3B) ▶ , was obtained from bands at 6.5 kd yielding residues 1–10 (8 kd, 1–16; and 28 kd, 1–39). A 7-kd protein gave the sequence from residues 51–70. Light chain from the urine eluted from the KappaLock column, composed of a 28-kd protein (Figure 2 ▶ , lane 7), was immunoreactive with anti-κ (Figure 2 ▶ , lane 8) and rendered the sequence from residues 1–37.
Several independent cDNA κ clones of GLA, isolated from reverse-transcribed RNA obtained from bone marrow cells, were sequenced and found to be identical. The same was true for cDNA κ clones of CHO. The deduced amino acid sequence (Figure 3, A and B) ▶ was in full agreement with the N-terminal amino acid sequence retrieved from the tissues and the urines. When the cDNA sequences were compared with the V-κ1 germline sequences, the best homology was found with the L12a gene. 21
Germline V-Region Sequences
The cloned and sequenced nucleotides of the full-length germline V-κ genes from genomic DNA, obtained from GLA and CHO, as well as MCM, were all identical to the L12a germline subset of the κ1 light chain. In Figure 4 ▶ , a comparison of the germline sequence and the translated sequences from the cDNAs of GLA and CHO, together with the previously extracted and sequenced tissue deposits of MCM, 11 demonstrates that the amino acid substitutions are the result of somatic mutation in the proliferating clones instead of inherited susceptible variant genes. As shown, there are a total of nine mutations in GLA, seven in CHO and 13 in MCM. The mutations occur throughout the domain: in GLA two are in FR1, one is in CDR1, three are in FR2, and one each in FR3 and CDR3; in CHO one is in FR1, one in CDR2, two in FR3, and three are in CDR3; in MCM the mutations are three in CDR1, two in FR2, one in CDR2, three in FR3, and four in CDR3. The spatial locations of the substitutions in GLA and CHO are depicted in Figure 5, A and B ▶ .
Figure 4.

Comparison of the amino acid sequence encoded by the V-κ1 L12a subgroup germline gene with GLA, CHO and MCM 11 sequences.
Figure 5.
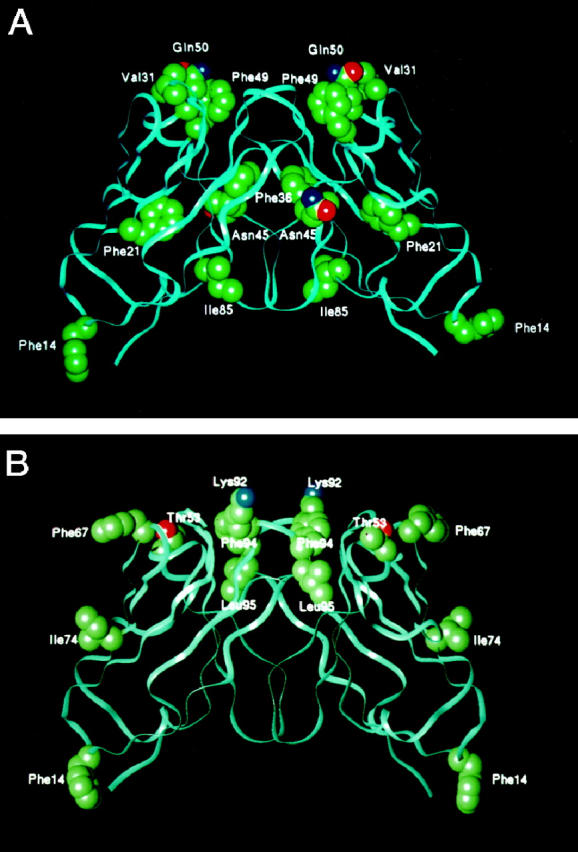
Variable-region light chain dimers of GLA (A) and CHO (B), showing the spatial location of amino acid substitutions. The backbone structure of κ1 light chain REI (Brookhaven Protein Databank entry REI) was computationally mutated to introduce the residues at the positions shown. Side-chain atoms represented in solid spheres are carbon (green), oxygen (red), and nitrogen (blue).
An analysis of GLA, CHO, and MCM light chains reveals a number of significant somatic mutations that have potential physicochemical and pathogenetic consequences. In both GLA and CHO the Ser→Phe substitution at position 14 breaks the hydrogen bonding pattern in the beta turn of FR1 and results in exposure of the hydrophobic Phe to the solvent. The mutation Ile→Phe at position 21 in GLA interferes with packing by replacing a moderate-sized hydrophobic side chain with a much larger hydrophobic Phe. At position 66 in MCM a highly conserved small Gly is replaced by the bulky side chain of Glu that may disrupt packing and the turn in FR3. The Ser→Phe mutation at position 67 in CHO interferes with the turn in the FR3 by inserting a hydrophobic Phe into the solvent. In MCM, a Ser→Lys at position 77 may destabilize the turn in FR3; the replacement of Asp with Ile at residue 82 in MCM is severely destabilizing by removing a highly conserved salt bridge with Arg61 and replacement of a strongly hydrophilic residue with a hydrophobic side chain. The replacement of Thr→Ile at position 85 in GLA results in a hydrophobic side chain exposed to solvent. At position 95, the Ser→Leu in CHO and Ser→Pro in GLA and MCM increase hydrophobicity and potentially destabilize the turn in CDR3. Taken together, GLA, CHO, and MCM all exhibit destabilizing mutations that increase hydrophobicity.
In Figure 6 ▶ , constructed models depicting the hydrophobic residues in solvent-exposed CDR-presenting surfaces of the V-region dimers of GLA, CHO, and MCM are compared to that of REI, a nonpathogenic κ1 light chain; 18,19 all three of the LCDD proteins show an increased proportion of hydrophobic side chains exposed to the solvent that could contribute to protein destabilization. Trp at position 32 in the CDR1 is a genetically encoded feature of κ1a (gene, L12a) light chain, and it is not found in κ1b (gene O18-O8) protein REI.
Figure 6.

The hydrophobic residue content of the CDR-presenting surfaces of V-region light chain dimers of proteins MCM, GLA, and CHO are compared to that of REI. Atoms of hydrophobic amino acids are represented by solid green spheres. The models were constructed by using the program Insight II (Molecular Simulations) to replace amino acids in the crystallographic structure of REI (pdb 1 rei) with the variations found in the amino acid sequences of the three V-region light chains associated with LCDD. All show an increased proportion of hydrophobic side chains accessible to solution. Arrows indicate Trp32, a genetically encoded feature of κ1a (gene: L12a) proteins not found in the κ1b (gene: O18–O8) protein REI. The fully exposed nature of the indole side chain of Trp32, as modeled, has been confirmed by the crystallographically determined structure of the human anti-gp41 Fab, 3D6 (pdb1dfb), which incorporates a κ1a light chain. 39 The view is down the twofold axis of the V-domain dimer.
Isoelectric Points
The calculated pI values were 6.42 (5.6–7.5) and 9.22 for GLA and CHO, respectively, and 8.21 for MCM. 11
Discussion
In the present report the structure of two new κ1-LCDD proteins, GLA and CHO, obtained by N-terminal sequence analysis of light chain deposits extracted from the tissues, light chains isolated from the urines, and the V-region amino acid sequences deduced from the cloned and sequenced cDNAs, and MCM, 11 are compared to the germline sequence determined from genomic DNA in each. The findings indicate that the amino acid substitutions in GLA, CHO, and MCM are the result of somatic mutations in the proliferating clones rather than from inherent variant alleles.
Although there are well over 150 published cases of nonamyloidotic LCDD, the primary structure of the involved light chains has been determined in only 11 cases. Nine, including GLA and CHO (Table 1) ▶ , are κ light chain, and two are λ light chain. 12 One (MCM) was obtained by sequence analysis of light chain deposits extracted from tissue, 11 two (SCI, BURN) by sequence analysis of the monoclonal light chain from the urine, 7,13 and the others (ISE, REV, BLU, FRA, BOU, and RAC) were deduced from cloned cDNAs. 6,8-10,12
Table 1.
Amino Acid Substitutions in V-Region κ-Light Chains in LCDD
| Genes: | L12a | L2–L16 | B3 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Protein: | CHO | GLA | MCM | ISE | REV | SCI | BLU | BURN | FRA | ||
| FR-1 1-23 | 03 | V>I* | 1 | ||||||||
| 04 | M>L | 1 | |||||||||
| 09 | A>G | 1 | |||||||||
| 14 | S>F | S>F | 2 | ||||||||
| 21 | I>F | 1 | |||||||||
| CDR-I 24-34 | 27 | Q>L | Q>L | 2 | |||||||
| 27a | S>G | 1 | |||||||||
| 27b | V>L | 1 | |||||||||
| 27c | L>F | 1 | |||||||||
| 27d | Y>F | 1 | |||||||||
| 27e | S>N | 1 | |||||||||
| 27f | S>P | S>T | 2 | ||||||||
| 28 | N>R | 1 | |||||||||
| 30 | S>G | S>N | 2 | ||||||||
| 31 | S>V | S>N | S>I | S>T | S>Y | N>I | 6 | ||||
| 32 | Y>F | Y>C | 2 | ||||||||
| 34 | A>N | 1 | |||||||||
| FR-II 35-49 | 36 | Y>F | 1 | ||||||||
| 38 | Q>E | 1 | |||||||||
| 42 | K>Q | 1 | |||||||||
| 43 | P>T | 1 | |||||||||
| 45 | K>N | K>N | K>Q | 3 | |||||||
| 49 | Y>F | 1 | |||||||||
| CDR-II 50-56 | 50 | K>Q | G>S | 2 | |||||||
| 51 | A>T | 1 | |||||||||
| 52 | S>T | 1 | |||||||||
| 53 | S>T | S>T | S>N | T>I | T>I | T>S | 6 | ||||
| 55 | E>Q | E>G | 2 | ||||||||
| FR-III 57-88 | 59 | P>S | 1 | ||||||||
| 64 | G>A | 1 | |||||||||
| 66 | G>E | 1 | |||||||||
| 67 | S>F | 1 | |||||||||
| 68 | G>E | 1 | |||||||||
| 70 | D>N | 1 | |||||||||
| 72 | T>S | 1 | |||||||||
| 74 | T>I | T>N | 2 | ||||||||
| 76 | S>T | S>T | 2 | ||||||||
| 77 | S>K | S>G | S>I | S>R | S>R | 5 | |||||
| 81 | E>G | 1 | |||||||||
| 82 | D>I | 1 | |||||||||
| 83 | F>L | 1 | |||||||||
| 85 | T>I | V>L | V>T | 3 | |||||||
| 87 | Y>F | 1 | |||||||||
| CDR-III 89-97 | 89 | Q>H | 1 | ||||||||
| 90 | Q>H | 1 | |||||||||
| 91 | Y>I | 1 | |||||||||
| 92 | N>K | N>F | N>D | N>G | N>E | Y>S | 6 | ||||
| 93 | N>D | N>D | S>T | S>T | 4 | ||||||
| 94 | Y>F | Y>L | T>A | 3 | |||||||
| 95 | S>L | (P)† | (P) | (P) | P>L | 2 |
* Germline > variation.
† (Proline) encoded by a germline allele.
All of the nine κ-LCDD proteins mentioned thus far (Table 1) ▶ are V-region light chains that belong to three different light chain subgroups. Four of them (CHO, GLA, MCM, ISE) belong to the κ1 subgroup, three (BLU, BURN, FRA) to κIV, and two (REV, SCI) belong to κIII. The two λ-LCDD proteins (BOU, RAC) belong to the λII subgroup. Among the nine κ-LCDD proteins summarized in Table 1 ▶ , all have amino acid substitutions that occur in all domains, but more frequently in the CDRs than in the FRs (51 versus 37), possibly reflecting immunoglobulin antibody specificity that is antigen driven. Collectively there are six in the FR1, 21 in the CDR1, eight in the FR2, 12 in the CDR2, 23 in the FR3, and 18 in the CDR3.
All κ-LCDD proteins exhibit one and as many as three mutations that can be expected to be structurally and functionally significant in affecting protein destabilization; in each at least one mutation is in a turn/loop exposed to solvent. All κ LCDD proteins characterized to date have come from the three major germline genes that encode a Trp residue at solvent accessible positions, ie, L12a, Trp32; L2–L16, Trp94; B3, Trp50. Trp, together with the other hydrophobic residues at the surface sufficient to exclude the light chain from the solvent, could cause aggregation and precipitation in tissues. Hydrophobic residues (Figure 6) ▶ are increased in relation to REI, and such substitutions in the FR regions (Table 1) ▶ could distort the β structure, producing amorphous rather than fibrillar deposits.
The diffuse organ distribution of nonamyloidotic deposits in MCM and CHO and in other cases of LCDD 4,5 is in contrast to the more patchy distribution of deposits in AL disease. The uniform light chain deposits in systemic basement membranes of MCM suggested a unique affinity for some component of the basement membrane, possibly due to an immunological or electrostatic interaction. The calculated pI values of the V-region amino acid sequences in MCM (8.21), as well as pI values calculated from published sequences of ISE and BLU (8.20, 8.19, respectively), were high. 11 This observation raised the possibility that the affinity might be due to charge interactions between cationic light chains and anionic GAGs in basement membranes, 22 a mechanism of immune complex deposition demonstrated in vivo in a murine model. 23 The calculated isoelectric point of the complete V-region in CHO is also high (9.22). In GLA the pI is lower (5.5–7.5). The high pIs of the light chains in LCDD are in contrast to the acidic pIs of amyloidogenic Bence-Jones proteins (mean, 4.8 ± 1.1), 24 suggesting a different mechanism for tissue deposition. Furthermore, if one compares the pI values determined by the N-terminal 50 amino acids encoded by the κ germline genes, one finds acidic pI values for κ1b (6.4) and κ2 peptides (3.6–4.5). In contrast, all of the other genes encode basic 50-mers, with 10.4, 9.9, and 9.8 deduced for κ1a, κ3a, and κ4, respectively, the three gene families for which LCDD representatives have been identified to date.
Why some Bence-Jones proteins are soluble and nonpathogenic, whereas others form either fibrillar or nonfibrillar granular aggregates in AL or LCDD, or even coexisting fibrillar and nonfibrillar deposits, 25 remains elusive. In AL, although the primary protein structure likely plays an important role in fibril formation, amyloidogenic motifs have not been identified in the primary sequence data of more than 60 light chains extracted from amyloid tissue deposits. Moreover, the identity of the complete primary structure of both the deposited amyloid fibril protein and the soluble precursor Bence-Jones protein DIA 26 and the amino-terminal identity of extracted light chains from coexisting fibrillar deposits of AL and granular deposits of LCDD 25 argue in favor of extrinsic factors and other molecules that might play a part in the processing of susceptible light chains. For example, the detection of amyloid P component and apo-E in amyloid, but not in nonamyloid light chain deposits, 2 suggests a function, as yet unclear, for these molecules. On the other hand, spontaneous in vitro fibril formation of amyloid β peptides 27 and other proteins not known to cause amyloidosis 28,29 relates to a physical-chemical environment favoring intermolecular interactions.
It is now apparent that a number of different diseases, including AL and LCDD, involve aberrant protein folding. 30,31 Based upon the accumulated data, LCDD and AL are considered to be conformational disorders. The evidence suggests that somatic mutational effects may be responsible for destabilization of the normal soluble globular light chain structure. 32 The increased hydrophobicity of GLA, CHO, and MCM relative to REI shown in Figure 6 ▶ and similar observations reported by others 33,34 could promote the formation of aggregation-prone, partially unfolded intermediates, thereby reducing solubility and favoring deposition in tissues. In vitro studies have shown that the Bence-Jones light chains in LCDD and AL were more unstable than a nonpathogenic Bence-Jones protein, and AL light chain was more unstable than the LCDD light chain. 35 In the context of current concepts of protein folding, 36,37 the interesting question posed is the relationship between LCDD and AL, and whether LCDD represents a partially unfolded or misfolded intermediate species stabilized in a transition state that favors off-pathway aggregation and precipitation in tissues, or alternatively is a preamyloid form of disease with restriction of maturation on pathway to fibril formation. The latter possibility is supported by both in vivo and in vitro ultrastructural observations showing a transition between granular and fibrillar structures in tissue deposits in a patient with coexistent LCDD and AL, 38 and the progressive formation in solution of Congophilic fibrils within granular aggregates. 29 As new data develop on protein folding and protein-protein interactions, it is likely that new insights into the different disease expressions exhibited in AL and LCDD and, importantly, the mechanisms of fibrillogenesis will be achieved.
Footnotes
Address reprint requests to Dr. Gloria Gallo, New York University Medical Center, 560 First Avenue, New York, NY 10016. E-mail: gloria.gallo@med.nyu.edu.
Supported in part by a grant from the National Institutes of Health (AR 02594, Merit), by the U.S. Department of Energy, Office of Health and Environmental Research (contract W-31-109-ENG), and by the U.S. Public Health Service (grant DKY 3757).
R. V. and F. G. contributed equally to this work.
P. A.’s current address: Hôpital Necker, 161 rue de Sèvres, 75743 Paris CEDEX 15, France.
References
- 1.Gallo G, Picken M, Buxbaum J, Frangione B: The spectrum of monoclonal immunoglobulin deposition disease associated with immunocytic dyscrasias. Semin Hematol 1989, 3:234-245 [PubMed] [Google Scholar]
- 2.Gallo G, Wisniewski T, Choi-Miura NH, Ghiso J, Frangione B: Potential role of apolipoprotein E in fibrillogenesis. Am J Pathol 1994, 145:526-530 [PMC free article] [PubMed] [Google Scholar]
- 3.Lyon AW, Narindrasorasak S, Young ID, Anastassiades T, Couchman JR, McCarthy KJ, Kisilevsky R: Co-deposition of basement membrane components during the induction of murine splenic AA amyloid. Lab Invest 1991, 64:785-790 [PubMed] [Google Scholar]
- 4.Randall RE, Williamson WC, Jr, Mulinax F, Tung MY, Still MJ: Manifestations of systemic light chain deposition. Am J Med 1976, 60:293-299 [DOI] [PubMed] [Google Scholar]
- 5.Ganeval D, Mignon F, Preud’homme JL, Noel L-H, Morel-Maroger L, Droz D, Brouet JC, Mery J, Grunfeld J-P: Visceral deposition of monoclonal light chains and immunoglobulins: a study of renal and immunopathologic abnormalities. Adv Nephrol 1982, 11:25-63 [Google Scholar]
- 6.Cogne M, Preud’homme JL, Bauwens M, Touchard G, Aucouturier P: Structure of a monoclonal κ chain of the V-κIV subgroup in the kidney and plasma cells in light chain deposition disease. J Clin Invest 1991, 87:2186-2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bellotti V, Stoppini M, Merlini G, Zapponi MC, Meloni ML, Banfi G, Ferri G: Amino acid sequence of κ Sci, the Bence Jones protein isolated from a patient with light chain deposition disease. Biochim Biophys Acta 1991, 1097:177-182 [DOI] [PubMed] [Google Scholar]
- 8.Khamlichi AA, Aucouturier P, Silvain C, Bauwens M, Touchard G, Preud’homme J-L, Nau F, Cogne M: Primary structure of a monoclonal κ chain in myeloma with light chain deposition disease. Clin Exp Immunol 1992, 87:122-126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rocca A, Khamlichi AA, Aucouturier P, Noel LH, Denoroy JL, Preud’homme JL, Cogne M: Primary structure of a variable region of the V-κ1 subgroup (ISE) in light chain deposition disease. Clin Exp Immunol 1993, 91:506-509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Decourt C, Cogne M, Rocca A: Structural peculiarities of a truncated VκIII immunoglobulin light chain in myeloma with light chain deposition disease. Clin Exp Immunol 1996, 106:357-361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gallo G, Goni F, Boctor F, Vidal R, Kumar A, Stevens FJ, Frangione B, Ghiso J: Light chain cardiomyopathy: structural analysis of the light chain tissue deposits. Am J Pathol 1996, 148:1397-1406 [PMC free article] [PubMed] [Google Scholar]
- 12.Decourt C, Touchard G, Preud’homme J-L, Vidal R, Beaufils H, Diemert M-C, Cogne M: Complete primary sequences of two λ immunoglobulin light chains in myelomas with nonamyloid (Randall-type) light chain deposition disease. Am J Pathol 1998, 153:313-318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stevens FJ, Weiss DT, Solomon A: Structural bases of light chain-related pathology. Zanetti M Capra JD eds. The Antibodies, 1999, vol 5.:pp 175-208 Harwood Academic Publishers, Amsterdam [Google Scholar]
- 14.Kaplan B, German G, Pras M: Isolation and characterization of amyloid proteins from milligram amounts of amyloid-containing tissue. J Liq Chromatogr 1993, 16:249-268 [Google Scholar]
- 15.Kaplan B, Yakar S, Kumar A, Pras M, Gallo G: Immunochemical characterization of amyloid in diagnostic biopsy tissues. Amyloid Int J Exp Clin Invest 1997, 4:80-86 [Google Scholar]
- 16.Gallo G, Feiner H, Chuba J, Beneck D, Marion P, Cohen DH: Characterization of tissue amyloid by immunofluorescence microscopy. Clin Immunol Immunopathol 1986, 39:479-490 [DOI] [PubMed] [Google Scholar]
- 17.Kabat EA, Wu TT, Perry HM, Gottesman KS, Foeller C: Human κ Light Chain Subgroup I. Sequences of Proteins of Immunological Interest. 1991, :pp 103-112 National Institutes of Health, Bethesda, MD, [Google Scholar]
- 18.Epp O, Lattman EF, Schiffer M, Huber R, Palm W: The molecular structure of a dimer composed of the variable portions of the Bence-Jones protein REI refined at 2.0-A resolution. Biochemistry 1975, 14:4943-4952 [DOI] [PubMed] [Google Scholar]
- 19.Choathia C, Lesk AM: Canonical structure for the hypervariable regions of the immunoglobulins. J Mol Biol 1987, 196:901-917 [DOI] [PubMed] [Google Scholar]
- 20.Skoog B, Wichman A: Calculation of the isoelectric points of polypeptides from the amino-acid composition. Trends Anal Chem 1986, :82-83 [Google Scholar]
- 21.Klein R, Jaenichen R, Zachau HG: Expressed human immunoglobulin κ genes and their hypermutation. Eur J Immunol 1993, 23:3248-3271 [DOI] [PubMed] [Google Scholar]
- 22.Kanwar YS, Farquhar MG: Presence of heparan sulfate in glomerular basement membrane. Proc Natl Acad Sci USA 1979, 76:1303-1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gallo GR, Caulin-Glaser T, Lamm ME: Charge of circulating immune complexes as a factor in glomerular basement membrane localization in mice. J Clin Invest 1981, 67:1305-1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bellotti V, Merlini G, Bucciarelli E, Perfetti V, Quaglini S, Ascari E: Relevance of class, molecular weight and isoelectric point in predicting human light chain amyloidogenicity. Br J Haematol 1990, 74:65-69 [DOI] [PubMed] [Google Scholar]
- 25.Kaplan B, Vidal R, Kumar A, Ghiso J, Frangione B, Gallo G: Amino-terminal identity of co-existent amyloid and non-amyloid immunoglobulin κ light chain deposits. A human disease to study alterations of protein conformation. Clin Exp Immunol 1997, 110:472-478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klafki H-W, Kratzin HD, Pick AI, Eckart K, Karas M, Hilschmann N: Complete amino acid sequence determinations demonstrate identity of the urinary Bence Jones protein (BJP-DIA) and the amyloid fibril protein (AL-DIA) in a case of AL-amyloidosis. Biochemistry 1992, 31:3265-3272 [DOI] [PubMed] [Google Scholar]
- 27.Soto C, Frangione B: Two conformational states of amyloid β-peptide: implication for the pathogenesis of Alzheimer’s disease. Neurosci Lett 1995, 186:115-118 [DOI] [PubMed] [Google Scholar]
- 28.Jimenez JL, Guijarro JI, Orlova E, Zurdo J, Dobson CM, Sunde M, Saibil HR: Cryo-electron microscopy structure of an SH3 amyloid fibril and model of the molecular packing. EMBO J 1999, 18:815-821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chiti F, Webster P, Taddei N, Clark A, Stefani M, Ramponi G, Dobson CM: Designing conditions for in vitro formation of amyloid protofilaments and fibrils. Proc Natl Acad Sci USA 1999, 96:3590-3594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soto C, Ghiso J, Frangione B: Alzheimer’s amyloid β aggregation is modulated by the interaction of multiple factors. Alzheimer’s Res 1997, 3:215-222 [Google Scholar]
- 31.Carrell RW, Lomas DA: Conformational disease. Lancet 1997, 350:134-138 [DOI] [PubMed] [Google Scholar]
- 32.Hurle MR, Helms LR, Li L, Chan W, Wetzel R: A role for destabilizing amino acid replacements in light chain amyloidosis. Proc Natl Acad Sci USA 1994, 91:5446-5450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deret S, Chomilier J, Huang DB, Preud’homme J-L, Stevens FJ, Aucouturier P: Molecular modeling of immunoglobulin light chains implicates hydrophobic residues in non-amyloid light chain deposition disease. Protein Eng 1997, 10:1191-1197 [DOI] [PubMed] [Google Scholar]
- 34.Raffen R, Dieckman LJ, Szpunar M, Wuschl C, Pokkuluri PR, Dave P, Stevens PW, Cai X, Schiffer M, Steven F: Physicochemical consequences of amino acid variations that contribute to fibril formation by immunoglobulin light chains. Protein Sci 1999, 8:509-517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bellotti V, Stoppini M, Mangione PP, Fornasieri A, Min L, Merlini G, Ferri G: Structural and functional characterization of three human immunoglobulin kappa light chains with different pathologic implications. Biochim Biophys Acta 1996, 1317:161-167 [DOI] [PubMed] [Google Scholar]
- 36.Capaldi AP, Ferguson SJ, Radford S: The Greek key protein apo-pseudoazurin folds through an obligate on-pathway intermediate. J Mol Biol 1999, 286:1621-1632 [DOI] [PubMed] [Google Scholar]
- 37.Radford SE, Dobson CM: From computer simulations to human disease: emerging themes in protein folding. Cell 1999, 97:291-298 [DOI] [PubMed] [Google Scholar]
- 38.Stokes MB, Jagirdar J, Burchstin O, Kornacki S, Kumar A, Gallo G: Nodular pulmonary immunoglobulin light chain deposits with coexistent amyloid and nonamyloid features in an HIV-infected patient. Mod Pathol 1997, 10:1059-1065 [PubMed] [Google Scholar]
- 39.He XM, Ruker F, Casale E, Carter DC: Structure of a human monoclonal antibody Fab fragment against gp41 of human immunodeficiency virus type I. Proc Natl Acad Sci USA 1992, 89:7154-7158 [DOI] [PMC free article] [PubMed] [Google Scholar]