

Abstract
Mutations in the human tau gene cause frontotemporal dementia and parkinsonism linked to chromosome 17. Some mutations, including mutations in intron 10, induce increased levels of the functionally normal four-repeat tau protein isoform, leading to neurodegeneration. We generated transgenic mice that overexpress the four-repeat human tau protein isoform specifically in neurons. The transgenic mice developed axonal degeneration in brain and spinal cord. In the model, axonal dilations with accumulation of neurofilaments, mitochondria, and vesicles were documented. The axonopathy and the accompanying dysfunctional sensorimotor capacities were transgene-dosage related. These findings proved that merely increasing the concentration of the four-repeat tau protein isoform is sufficient to injure neurons in the central nervous system, without formation of intraneuronal neurofibrillary tangles. Evidence for astrogliosis and ubiquitination of accumulated proteins in the dilated part of the axon supported this conclusion. This transgenic model, overexpressing the longest isoform of human tau protein, recapitulates features of known neurodegenerative diseases, including Alzheimer’s disease and other tauopathies. The model makes it possible to study the interaction with additional factors, to be incorporated genetically, or with other biological triggers that are implicated in neurodegeneration.
The tau protein belongs to a heterogeneous family of microtubule-associated proteins, predominantly expressed in neurons. 1 Alternative splicing of the human tau gene encodes at least six different mRNA species (6 kb) that yield six low-molecular-weight (LMW) tau protein isoforms ranging from 352 to 441 amino acids, which are differentially phosphorylated. 2,3 In addition, “big tau” protein is encoded by an 8-kb mRNA that contains an additional exon 4A (and/or exon 6), 4,5 resulting in isoforms ranging from 90–100 kd (MMW) to 110–120 kd (HMW). The LMW tau proteins are present in the central nervous system (CNS) and the peripheral nervous system (PNS), MMW tau isoforms are identified in the optic nerve, and HMW tau proteins are abundant in the PNS and spinal cord. 6 Since tau protein is required for neurite elaboration in different cell types, 7-10 a central role in neuronal process outgrowth and integrity is proposed. Moreover, selective inhibition of tau protein expression in primary cerebellar neurons prevents formation of neurite asymmetry and elaboration of an axon. 11 In tau protein-deficient mice a modest alteration of microtubular organization in small-caliber axons was observed. 12
Recently, missense and splice-site mutations were detected in the human tau gene in patients with inherited frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17). 13-19 Most of the exonic mutations located within the microtubule binding repeats rendered the tau protein less functional for microtubule binding and stabilization. 20,21 Thus the neurodegeneration appeared to be caused by reduced levels of functional tau protein. On the other hand, mutations in intron 10 and mutations N279K and S305N in exon 10 were shown to induce a preponderance of tau protein isoforms with four microtubule-binding repeats. 13,14,17,20,22 Likewise, the silent FTDP-17 mutation L284L, located in the exon 10 splicing silencer sequence, resulted in excess exon 10 inclusion. 23 These observations were interpreted to mean that increased levels of normal functional tau protein also could provoke neuronal dysfunction. Two previous studies showed a somatodendritic localization and hyperphosphorylation of the exogenous tau protein, similar to the pretangle changes that precede the neurofibrillary pathology in Alzheimer’s disease, in brain of mice transgenic for human tau protein. 24,25 However, neither neuronal dysfunction nor abnormal behavior has been demonstrated thus far in mice overexpressing any human tau isoform. 24,25
We have also tested the hypothesis that overproduction of tau protein with four microtubule-binding repeats can constitute a gain of a toxic function in vivo. We have generated transgenic mice that overexpress this tau protein isoform specifically in neurons. We report here that these transgenic mice develop an axonopathy that recapitulates some features of known neurodegenerative diseases, including Alzheimer’s disease and other tauopathies.
Materials and Methods
Production of Transgenic Mice
Human Tau40 cDNA, which was deduced from the cDNA clone tau40, 26 was ligated in the mouse thy-1 expression cassette. 27 A PvuI-NotI restriction fragment was microinjected, and transgenic founders were identified by Southern blotting of StuI-restricted mouse tail-biopt DNA. Routine genotyping of transgenic offspring, bred into the FVB/N genetic background, was performed on tail-biopt DNA by polymerase chain reaction (PCR).
Sensorimotor Tests
The behavioral experiments constitute a transversal study in which transgenic mice from different founder strains were tested at the age of 2–4 months. Five groups of mice were tested: 46 wild-type FVB mice, 31 htau40-1 heterozygous mice, 8 htau40-1 homozygous mice, 17 htau40-5 heterozygous mice, and 12 htau40-5 homozygous mice. The animals were subjected to three sensorimotor tasks designed to assess muscle strength, endurance, coordination, and equilibrium. 28
The forced swimming test was performed identically to a probe test in the Morris water maze test, as described before. 29 One-way analysis of variance (ANOVA) was used to test for differences in swimming distance between the five groups.
The second sensorimotor task measured the ability of the mouse to walk along a suspended narrow horizontal rod as an index of psychomotor integration and equilibrium. Each mouse was placed in the middle of a 50-cm-long aluminum rod of 14-mm diameter. The walking rod held an escape platform at each end and was positioned 50 cm above a flat surface. If the mouse succeeded in walking on the rod and/or reaching one of the escape platforms without falling off, it was scored as 1, and if the animal fell off, it was scored 0. The contingency χ 2 test was used for comparison between different groups. When χ 2 appeared to be significant, odds ratios and their two-sided 95% confidence intervals were calculated to determine the strength of the association.
Finally, the inverted wire mesh grid test referred to the ability to grasp an elevated horizontal wire grid and to remain suspended for 1 minute. The net was positioned 50 cm above a flat surface and measured 40 cm × 20 cm, with meshes of 0.5 cm × 0.5 cm. If the animal remained suspended for 1 minute, it was scored 1, and if the animal dropped off, it was scored 0. The contingency χ 2 test was used for comparison between different groups. When χ 2 appeared to be significant, odds ratios and their two-sided 95% confidence intervals were calculated to determine the strength of the association.
The statistical analyses were conducted with the SAS 6.12 computer package. All reported P values are two-sided and were considered statistically significant when P < 0.05 (the Bonferroni correction was applied for multiple testing).
Western Blotting
Brain and spinal cord tissue were homogenized in 2 ml and 350 μl, respectively, of buffer containing detergents, proteinase, and phosphatase inhibitors, ie, 0.1 mol/L 2-(N-morpholino)ethanesulfonic acid (pH 6.4), 0.5 mmol/L MgCl2, 1 mmol/L EDTA, 1 mmol/L EGTA, 1 mmol/L dithiothreitol, 5 μg/ml leupeptin, 5 μg/ml pepstatin, 200 μmol/L phenylmethylsulfonyl fluoride, 20 mmol/L NaF, 200 μmol/L sodium orthovanadate, 1 μmol/L okadaic acid, 5 μg/ml soybean trypsin inhibitor, 1% Triton X-100, 1% sodium desoxycholate, and 0.1% sodium dodecyl sulfate. Likewise, two sciatic nerves and 15 spinal ganglia of wild-type and htau40-1 heterozygous transgenic mice were dissected and homogenized in 100 μl and 50 μl homogenization buffer, respectively. After centrifugation (100,000 × g for 30 minutes at 4°C), the supernatant was denatured and reduced before separation on Tris-glycine-buffered polyacrylamide gels (8% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose filters as described. 29 Protein concentration of cleared homogenates was determined with the Bio-Rad Detergent Compatible protein assay (Bio-Rad Laboratories, Hercules, CA), and equal amounts were loaded.
Because the secondary goat anti-mouse antibody bound on Western blot to proteins of 55 and 67 kd, thereby interfering with the monoclonal anti-tau immunoresponses (especially of AT-8 and AT-180), supernatant of brain and spinal cord homogenates were incubated with immobilized protein-G (Pierce, Rockford, IL) at 4°C for 2.5 hours and purified from mouse IgG by centrifugation (8000 rpm, 5 minutes, 4°C). For the same reason, detection of human tau protein in spinal ganglia and sciatic nerve homogenates was performed with polyclonal antibody B19.
The phosphate-independent antibodies directed to tau protein were monoclonal antibodies HT-7 (Innogenetics, Ghent, Belgium) and Tau-5 (Pharmingen, San Diego, CA) and polyclonal antibody B19 (gift of J. P. Brion, Free University of Brussels, Brussels, Belgium). 30 Used monoclonal antibodies directed to phosphorylated tau protein epitopes are listed below.
To dephosphorylate the tau protein before densitometric quantification on Western blot, brain and spinal cord homogenates were diluted in a dephosphorylation buffer (Boehringer Mannheim) containing Escherichia coli alkaline phosphatase (type III; Sigma) at 50 units/ml and incubated for 2 hours at 25°C. Samples to be loaded on the gel were prepared as mentioned above.
Histochemistry, Immunohistochemistry, and Antibodies
For immunohistochemical detection of human tau protein in the htau40 transgenic mice, paraformaldehyde (4% in phosphate-buffered saline), fixed free-floating vibratome slices (40 μm) were incubated with different monoclonal and polyclonal antibodies. Brain and spinal cord sections were incubated with biotin-conjugated secondary antibody, submerged in Strept-ABComplex/horseradish peroxidase, and stained with 3,3′-diaminobenzidine tetrahydrochloride. The phosphate-independent antibodies directed to tau protein were HT-7, the epitope of which has been mapped on human tau between positions 159 and 163; Tau-5, directed to an epitope mapped in the middle of human and murine tau protein; and B19, reactive with the tau protein sequence 154–195. 30 Applied antibodies directed to phosphorylated tau protein were AT-8, AT-180, AT-270 (Innogenetics), and PHF-1 (gift of P. Davies, Albert Einstein College of Medicine, New York, NY). Their epitopes have been determined as phosphoSer199 and/or phosphoSer202, 31,32 phosphoThr231, 33 phosphoThr181, 33 and phosphoSer396 and/or phosphoSer404, 34 respectively. Antibodies Alz-50 35,36 and MC-1 37 (both gifts of P. Davies) are directed to paired helical filament tau protein. For detection of neurofilament subunit NF-H, antibodies SMI-31, SMI-32 (Affiniti, Nottingham, UK), and NF-200 (Sigma, St. Louis, MO) were applied. NF-M and NF-L were detected by antibodies NF-68 and NF-160 (Sigma), respectively. Antibodies NF-68, NF-160, and NF-200 are phosphate-independent; SMI-31 and SMI-32 are phosphate-dependent. Anti-glial fibrillary acidic protein (anti-GFAP) and ubiquitin were purchased (DAKO A/S, Glostrup, Denmark).
Sections of muscle were submitted to standard hematoxylin/eosin (H&E) staining. Tunel staining was applied to analyze the CNS for apoptotic neurons. Bielschowsky’s silver impregnation and thioflavine-S staining were used to determine the presence of tangle-like structures in neurons of the central nervous system of the transgenic mice. 29,38
Quantification of Axonopathy in the CNS
Four wild-type FVB mice, 3 htau40-1 and 3 htau40-5 homozygous mice, and 5 htau40-1 heterozygous mice were transcardially perfused with paraformaldehyde (4% in PBS). Brain and spinal cord were immersion-fixed overnight. Vibratome slices (40 μm) were generated from the right brain hemisphere. Eight cortical slices comprising the hippocampal structures were incubated with monoclonal antibody SMI-32. Immunoreactive axonal dilations with a diameter equal to or larger than the diameter of the nucleus of fifth-layer neocortical pyramidal neurons were counted. Likewise, vibratome sections were made of the mainly thoracal part of the spinal cord, and eight randomly taken thoracal sections were incubated with SMI-32. In parallel, the left hemisphere of each brain and the mainly thoracolumbal part of the spinal cord were embedded in paraffin. Microtome sections (6 μm) of brain and spinal cord were impregnated according to the method of Bielschowsky’s silver staining. The number of argyrophilic axonal dilations was counted in three series of three successive slices. Only cortical slices that comprised the hippocampus were considered in the quantitative analysis of the brain, and only axonal dilations equal to or exceeding the diameter of the nucleus of motor neurons were counted in the spinal cord sections. Three researchers independently performed counting, and the number of dilated axons was averaged for each section of spinal cord or hemisphere. Silver impregnation and SMI-32 immunostaining yielded similar results. The Kruskal-Wallis test was used to evaluate the statistical differences in the average number of axonal dilations between the different groups of transgenic mice.
Quantification of Neurons in the Ventral Horn of the Spinal Cord
Six wild-type FVB mice and six htau40-1 homozygous mice that were 3 months old were transcardially perfused with paraformaldehyde (4% in PBS). Spinal cords were immersion-fixed overnight and embedded in paraffin. Microtome sections (6 μm) of the thoracolumbal region were submitted to standard cresyl violet staining. Composite images from a 3CCD color video camera were collected and assembled with appropriate software (AIS/C 4.0; Imaging Research, St. Catharine’s, ON), and the number of neurons in the right ventral horn in three series of three successive sections was quantitated. Two researchers independently performed counting, and the number of neurons, averaged for each mouse, was statistically analyzed by the Kruskal-Wallis test.
Ultrastructural Analysis
For transmission electron microscopy (TEM), specific regions were excised from 40-μm-thick vibratome sections fixed in 4% paraformaldehyde and 0.1% glutaraldehyde in PBS. For immunogold labeling, samples incubated with AT-8 and Alz-50 after permeabilization with Triton X-100 were further incubated with a secondary antibody conjugated to 1.4-nm gold particles and enhanced with HQ-silver (Nanoprobes, Stony Brook, NY). All samples were postfixed with glutaraldehyde, embedded, and processed for electron microscopy. Samples were additionally postfixed with OsO4 before embedding.
Results
Generation of Human tau40 Transgenic Mice and Phenotyping
Transgenic mice that overexpress the four-repeat human tau protein isoform with two N-terminal inserts were generated using a recombinant DNA construct based on the mouse thy-1 gene (Figure 1a) ▶ . Three founder strains were selected, ie, htau40-1, htau40-2, and htau40-5, which transmitted the transgene in a stable Mendelian fashion. Transgenic mice from the three lines expressed the human tau protein exclusively in neurons. Western blotting of brain (Figure 1b) ▶ and spinal cord homogenates (Figure 1c) ▶ with the monoclonal antibody HT-7 demonstrated the highest expression of human tau protein in transgenic mice from strains htau40-1 and htau40-2. Western blotting with monoclonal antibody Tau-5 allowed us to determine the relative levels of human to mouse tau protein by densitometric scanning (Figure 1, b and c) ▶ . To obtain accurate quantification of total tau proteins, the tau isoforms were dephosphorylated by pretreatment of homogenates with alkaline phosphatase before application to the gel. The murine tau protein in brain and spinal cord homogenates in wild-type animals was used as an internal standard for normalization. In heterozygous mice of strain htau40-1, the level of human tau protein was about four-fold higher relative to endogenous mouse tau protein levels, whereas in strain htau40-5 human tau protein levels were 1.5 times those of murine tau protein. Human tau protein levels in spinal cord were also quantitated and estimated to be in the same range (Figure 1d) ▶ .
Figure 1.

Recombinant DNA construct used to generate htau40 transgenic mice and Western blot analysis of expression of human tau40 protein in brain, spinal cord, spinal ganglia, and sciatic nerve of selected transgenic mouse strains. a: Structure of the authentic mouse thy1 gene and the modified mini-gene construct yielding neuron-specific expression of the embedded cDNA. 43 Original exons are numbered and represented by black blocks. b: Western blotting of extracts of brain derived from wild-type and from transgenic htau40 mice with antibodies Tau-5 and HT-7. Approximately 10 μg of protein was loaded per lane. Samples were treated with alkaline phosphatase before application on the gel. c: Western blotting of extracts of spinal cord derived from wild-type and from transgenic htau40 mice with antibodies Tau-5 and HT-7. Approximately 10 μg of protein was loaded per lane. Samples were treated with alkaline phosphatase before loading on the gel. d: Numeric results from densitometric scanning and quantification of Western blots displayed in b and c. e: Western blotting of extracts of spinal ganglia (sp.g.) and sciatic nerves (sc.n.) derived from wild-type and htau40-1 transgenic mice with polyclonal antibody B19. Approximately 5 μg of protein was loaded per lane. WT, wild-type mice; H and HH, heterozygous and homozygous htau40 mice, respectively. The three different transgenic mouse strains are indicated as htau40-1, -2, and -5. All mice were about 3 months old (±1 week).
In addition, the presence of human tau protein was demonstrated in the PNS of htau40 transgenic mice, ie, in homogenates of sciatic nerves and of spinal ganglia (Figure 1e) ▶ . Moreover, axons originating from nerve cell bodies in the spinal ganglia were immunoreactive for monoclonal antibody HT-7 in the posterior column of the spinal cord (not shown).
Behavior and Motor Problems of htau40 Transgenic Mice
Homozygous htau40-1 and htau40-2 transgenic mice already displayed at weaning some neurological symptoms. When lifted by the tail, wild-type FVB mice exhibited an escape reflex by extending their legs, as opposed to homozygous transgenic mice, which flexed their hind limbs. Before the transgenic mice were subjected to the standard cognitive test, ie, the Morris water maze, 29 their motor abilities were examined in a forced swimming test. The swimming speed of htau40-1 homozygous mice was significantly lower than that of wild-type, heterozygous and homozygous htau40-5 mice. In 1 minute, homozygous htau40-1 mice traversed only about 70% of the distance covered by wild-type littermates (Figure 2b) ▶ . Subsequently, this motor disturbance, which prevented the mice from being tested in the Morris water maze, was studied in two additional sensorimotor tasks: rod walking and inverted grid hanging. This showed that the severity of the impairment was related to the level of expression of the transgene (Figure 2) ▶ . Compared to wild-type mice, fewer homozygous htau40-1, heterozygous htau40-1, and homozygous htau40-5 mice were able to walk on a rod. In comparison with wild-type mice, they were 90, 13, or 5 times more likely, respectively, to fall off the rod. Significantly more homozygous htau40-1 mice dropped off the rod, in comparison with homozygous and heterozygous mice of the strain with lowest expression, and more heterozygous mice of strain htau40-1 fell than heterozygous htau40-5 transgenic animals (Figure 2a) ▶ . Likewise, significantly more homozygous mice of strain htau40-1 lost hold of the inverted wire mesh grid, in comparison with wild-type and other transgenic mice that lack or express the transgene to a lower extent (Figure 2c) ▶ . These observations prove that htau40 transgenic mice displayed a reduced endurance, a postural instability, a loss of motor coordination and of maintenance of equilibrium, and a muscular weakness.
Figure 2.

Performance of transgenic and wild-type mice in three sensorimotor tasks. a: Number of mice that fell off the walking rod during a 3-minute test period, expressed relative to the number of tested in each group. Compared to WT mice, significantly more 1H, 1HH, and 5HH mice fell down. *WT-1H: P < 0.001, O.R. 13.5 (40–4.6); WT-1HH: P < 0.001, O.R. 90 (1000–4.5); WT-5HH: P < 0.001, O.R. 5.4 (20.4–1.4). This motor impairment was shown to be gene dosage dependent. **5H-1HH: P < 0.001, O.R. 38.4 (1.8–1000); 5HH-1HH: P < 0.05, O.R. 17 (0.8–500); ***5H-1H: P < 0.001, O.R. 5.8 (1.6–20.8). b: Swimming speed defined as distance traveled in 1 minute. In the forced swimming test, 1HH mice (*) traversed a significantly shorter distance in 1 minute than wild-type and other transgenic mice (P < 0.001). 1H mice (**) performed even better than wild-type littermates (P < 0.05). c: Inverted wire grid hanging, expressed as number of mice that remained suspended for the entire 1-minute test period, relative to the number of mice tested in each group. Compared to wild-type mice, significantly more 1HH mice (*) failed to remain suspended on the inverted wire mesh grid. WT-1HH: P < 0.001, O.R. 22.2 (125–3.8). Number of mice tested is given in Materials and Methods. Asterisks do not denote significance levels, but indicate groups of mice to be compared. WT, wild-type mice; 1H and 1HH, heterozygous and homozygous htau40-1 mice, respectively; 5H and 5HH, heterozygous and homozygous htau40-5 mice, respectively. All mice were between 2 and 4 months old.
Surprisingly, heterozygous htau40-1 mice exhibited a somewhat higher swimming speed than wild-type mice did. The heterozygous htau40-1 mice were, however, agitated and stressed, and this appeared to compensate for their mild motor impairment (Figure 2b) ▶ .
Histochemical, Immunohistochemical, and Ultrastructural Analysis
The brain and spinal cord of 15 homozygous and heterozygous htau40-1, -2, and -5 transgenic mice, aged between 8 weeks and 8 months, were analyzed histochemically, immunohistochemically, and ultrastructurally to determine the neuropathological cause of their phenotype.
H&E staining did not show striking abnormalities. Bielschowsky’s silver impregnation did reveal, however, grossly dilated axons in brain and spinal cord. These dilations had mostly a rounded contour and were often as large as neighboring neuronal cell bodies. In brain, dilated axons were mainly present in the neocortex, hippocampus, and thalamus, but were rare in the subcortical white matter, corpus callosum, and internal capsule (Figure 3, a and b) ▶ . The proximal location of the dilated axons in the gray matter suggested a proximal type of axonopathy. In the spinal cord, the dilated axons were present mostly in the gray matter, as well as in some white matter fiber tracts. In addition to the dilated axons, silver staining revealed thickened and irregularly shaped, argyrophilic dystrophic neurites scattered throughout brain and spinal cord gray matter (Figure 3c) ▶ . However, staining with silver or thioflavine-S did not reveal neurofibrillary tangles.
Figure 3.

Silver staining of brain and spinal cord of homozygous htau40 transgenic mice at the age of 2.5 months. a: Multiple dilated axons or axonal spheroids (arrows) and some irregular dystrophic axons (arrowheads) in cortex (×360). b: Higher magnification of two dilated axons in thalamus. Note that the dilations approach the size of neuronal cell bodies (×920). c: Aspect of spinal cord gray matter (anterior horn) with a grossly dilated axon (arrow) and several irregularly thickened dystrophic axons (arrowheads) (×390).
Immunohistochemically, the expression of human tau protein was widespread in the brain of all htau40 transgenic mice analyzed. Monoclonal antibody HT-7 stained nerve cell bodies and their processes in hippocampus, cortex, and subcortical nuclei most strongly, especially in cortical layer V. Also in the spinal cord, the axons, dendrites, and perikaria were clearly stained. With antibodies directed to specified phosphorylated epitopes of human tau protein, ie, AT-8, AT-180, AT-270, and PHF-1, a widespread somatodendritic localization of phosphorylated human tau protein became visible (Figure 4a) ▶ . Pyramidal neurons in the cortex and hippocampus and nerve cells within the thalamus stained strongly, as did the axonal processes in corpus callosum and the mossy fibers of the hippocampal granular cells. In the spinal cord, nerve cell bodies and their processes were stained by these antibodies. A more limited set of neurons and the dystrophic neurites were strongly reactive with antibodies Alz-50 and MC-1, which recognize a conformational epitope of the tau protein present in paired helical filaments 20 (Figure 4, b and c) ▶ . Neurons immunoreactive for human tau protein and axonal dilations also stained with antibodies directed against the three neurofilament subunits, ie, NF-L (anti-NF-68), NF-M (anti-NF-160), and NF-H (SMI-31, SMI-32, NF-200) (Figure 4, d–f) ▶ . In brain, dilated SMI-32 immunoreactive axons were abundant in neocortex and amygdala. Western blot analyses of brain and spinal cord homogenates with phosphate-dependent tau antibodies confirmed that transgenic human tau was phosphorylated at different epitopes. As different antibodies exhibit different affinities for these epitopes, quantitation on Western blot was not possible. Nevertheless, the relative degree of phosphorylation of transgenic human tau varied in both compartments of the CNS: in spinal cord the AT-8 labeling was always lower than in brain (Figure 5) ▶ .
Figure 4.

Immunohistochemistry of brain and spinal cord of homozygous htau40 transgenic mice at 2.5 months. a: Low-power view of neocortex stained with monoclonal antibody AT-8, showing somatodendritic localization of tau protein in pyramidal neurons of layer V (×110). b: Reaction of monoclonal antibody MC-1 in neocortex with a neuron (arrowhead) and dystrophic neurites (arrows) (×800). c: Anterior horn of spinal cord, showing axonal dilations (arrows), dystrophic neurites (arrowheads), and a neuronal cell body staining with antibody MC-1 (×365). d: Neuron with axonal dilation (arrow), staining with the antibody SMI-32, directed to neurofilament (NF)-H. The proximal axon (small arrow) connecting the cell body (arrowhead) has a normal caliber (×600). e and f: Dilated axons (arrowheads) in thalamus immunostained for NF-L (e) and NF-M (f) (×305).
Figure 5.
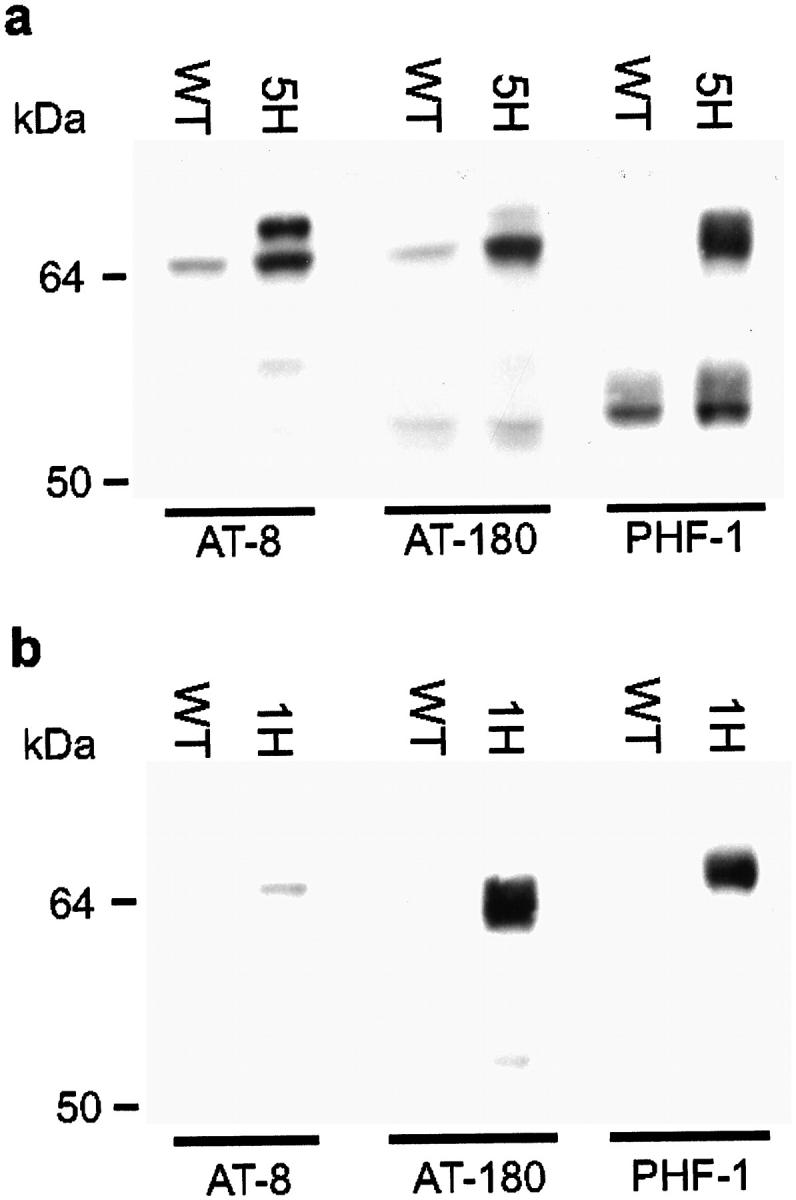
Western blotting of extracts of brain and spinal cord from wild-type and transgenic htau40 mice of 3 months with phosphorylation-dependent antibodies AT-8, AT-180, and PHF-1. Western blots of brain (a) and spinal cord (b) homogenates of wild-type (WT) and heterozygous htau40-5 (5H) and htau40-1 (1H) transgenic mice. Approximately 100 μg of protein was loaded per lane for incubation with AT-8 and AT-180, and 15 μg for detection with PHF-1.
The degree of axonopathy was gene-dosage-related. Quantitative analysis indicated a significant larger average number of dilated axons per section of spinal cord and cortex of homozygous htau40-1 mice compared to heterozygous littermates and homozygous mice of strain htau40-5, and of heterozygous htau40-1 mice compared to htau40-5 homozygous transgenic animals. Dilated axons were completely absent in wild-type mice, in both brain and spinal cord (Table 1) ▶ . The degree of astrogliosis in the affected regions of spinal cord and cortex correlated with the expression level of the human tau transgene as well (results not shown). GFAP immunostaining revealed activated astrocytes in cortex and spinal cord of homozygous htau40-1 mice, which were only rarely observed, if at all, in the other transgenic htau40 strains (Figure 6, a–d) ▶ . In addition, immunohistochemical staining for ubiquitin labeled some of the dilated axons in cortex and spinal cord (Figure 6, e and f) ▶ .
Table 1.
Quantification of Dilated Axons in Brain and Spinal Cord of Transgenic Mice
| Mouse type | n | Spinal cord | Cerebral cortex | |
|---|---|---|---|---|
| Mean no.* | Mean no. rostral** | Mean no. caudal*** | ||
| WT | 4 | 0 | 0 | 0 |
| 1H | 5 | 9.3 | 9.1 | 3.7 |
| 1HH | 3 | 22.0 | 29.9 | 11.5 |
| 5HH | 3 | 4.3 | 5.6 | 2.7 |
Numbers of dilated axons in entire transversal sections of the spinal cord (6 μm thick) and in coronal sections of the entire right hemispheric cortex (40 μm thick) are presented. Numbers of dilated axons in the rostral and caudal part of the cerebral neocortex from sections through the hippocampus were counted. Silver impregnation (nine sections per mouse type counted) and SMI-32 immunostaining (eight sections per mouse type counted) yielded similar results. Mice used were 3 months of age.
The Kruskal-Wallis analysis revealed a gene-dosage related pathology in the spinal cord and hemispheric cortex. *1H-5HH: P = 0.02; 1HH-5HH: P = 0.049; 1HH-1H: P = 0.02. **1H-5HH: P = 0.025; 1HH-5HH: P = 0.049; 1HH-1H: P = 0.025. ***1H-5HH: P = 0.23; 1HH-5HH: P = 0.049; 1HH-1H: P = 0.025.
WT, wild-type mice; 1H and 1HH, heterozygous and homozygous htau40-1 mice, respectively; 5HH, homozygous htau40-5 mice; n, number of mice analyzed.
Figure 6.
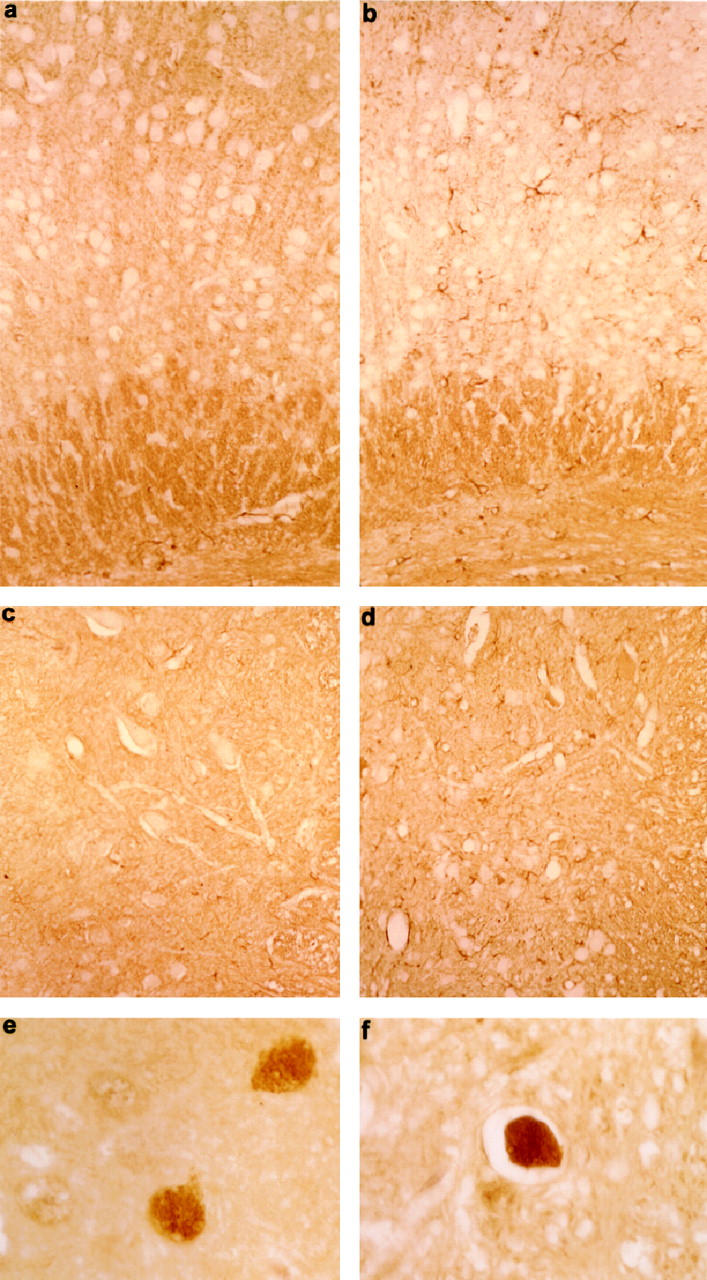
Immunohistochemistry of brain and spinal cord of homozygous htau40 transgenic mice of 2.5 months. a–d: GFAP immunostaining of cortex and spinal cord of homozygous htau40-1 mice showing astrogliosis in the cortex (b) and anterior horn (d). Compare with cortex (a) and anterior horn (c) of a wild-type mouse. Magnification: a and b, ×210; c and d, ×190. e and f: Ubiquitin-positive dilated axons in cortex (e) and spinal cord (f) of a homozygous htau40-1 mouse (×700).
Ultrastructural examination was performed on brain, spinal cord, and sciatic nerve from three homozygous htau40-1 mice and age-matched wild-type mice at the ages of 8 weeks and 8 months. TEM confirmed the presence of dilated axons with proportions similar to those of neuronal cell bodies (Figure 7a) ▶ . They showed prominent accumulation of neurofilaments, microtubules, mitochondria, endoplasmatic reticulum, and vesicles (Figure 7d) ▶ . Some dilated axons showed signs of degeneration. This varied from axons containing few degenerating mitochondria to axons with numerous dense and multivesicular bodies. Often the myelin sheat was thinned, detached, or completely retracted (Figure 7, b and c) ▶ . Also the thick dystrophic neurites seen with Bielschowsky’s silver staining corresponded to dilated axons. Morphologically, they were similar to the dystrophic axons described in Alzheimer’s disease (AD) and other neurodegenerative diseases. 39
Figure 7.
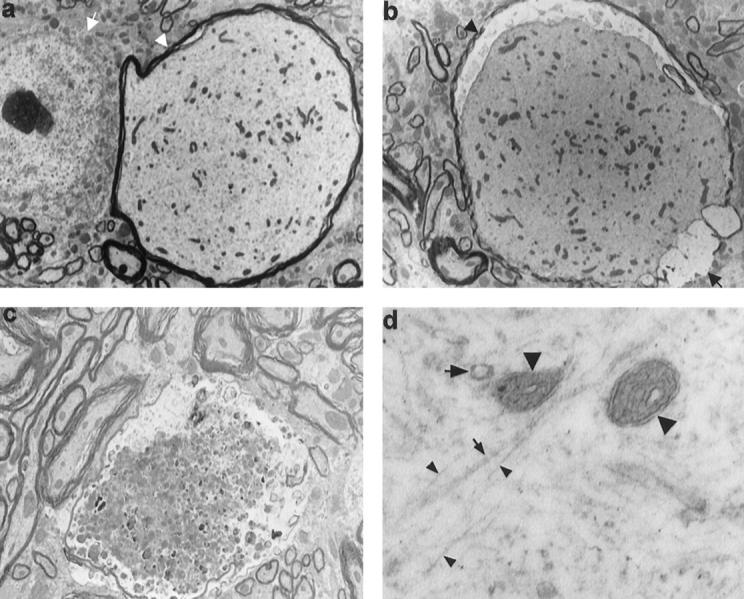
Ultrastructure of dilated axons with varying degrees of degeneration in spinal cord of 2- and 8-month-old homozygous htau40-1 mice. a: Dilated axon (arrowhead), larger in diameter than the neighboring neuronal cell body (arrow), is distended by accumulation of neurofilaments, microtubuli, mitochondria, and vesicles. Compare with several normal axons present in the lower left-hand corner of the section (×2915). b: Dilated axon with thinned (arrowhead) or absent (arrow) myelin sheath and with retracted axoplasm (×2915). c: Degenerating dystrophic axon filled with numerous dense and multivesicular bodies and with the myelin sheath thinned and disrupted (×4165). d: Higher magnification of dilated axon, showing neurofilaments (small arrowheads), microtubuli (small arrow), mitochondria (large arrowheads), and vesicles (large arrow) (×40,000).
Occasionally, microglia with phagocytosed myelin debris and myelin ovoids were present, indicating Wallerian degeneration. Ultrastructural signs of apoptosis were absent, confirming the negative tunel staining (results not shown).
Ultrastructurally, no filamentous aggregates or tangles were evident, neither directly by TEM nor after staining with AT-8 or Alz-50 and gold-labeled secondary antibodies. The latter method resulted in gold particles projecting on microtubuli or dispersed in the cytoplasm (Figure 8) ▶ .
Figure 8.

Immunogold staining of a dilated axon in the spinal cord of a transgenic htau40-1 mouse (homozygous, 8 month). Preembedding immunogold staining with AT-8 (a) and Alz-50 (b). Note that the silver-enhanced gold particles are equally dispersed in the axoplasm. The irregularity of the particles is due to the silver enhancement and osmium postfixation. Magnification: a, ×25,200; b, ×35,700.
The distal part of axons from lumbosacral motor neurons was examined in the sciatic nerve. These revealed affected axons and scattered macrophages with phagocytosed myelin, confirming Wallerian degeneration (Figure 9a) ▶ . Some axons had thinned myelin sheaths and contained axon-Schwann cell networks, as seen in chronic neuropathies (Figure 9b) ▶ . The functional impairment of the damaged axons was also reflected by the skeletal muscle fibers that they innervate. The quadriceps and the gastrocnemius skeletal muscles of homozygous htau40-1 transgenic mice showed grouping of atrophic fibers and fascicular atrophy, diagnostic for neurogenic atrophy (Figure 9c) ▶ . The degree of muscular atrophy correlated with the degree of CNS pathology, inasmuch as little muscular atrophy was observed in heterozygous htau40-1 mice and homozygous htau40-5 mice as opposed to homozygous htau40-1age-matched animals. The reduction in muscle mass contributed to a loss of body weight of about 30% in homozygous htau40-1 mice, even when the mice were only 2 months old. As muscle atrophy was demonstrated in homozygous htau40-1 mice, the possibility of neuronal loss in the ventral horn of the spinal cord was examined. No significant reduction in the number of neurons was recorded. In wild-type and homozygous htau40-1 mice, the average number of neurons was 49 ± 13 and 44 ± 12, respectively (P = 0.6310). Rare chromatolytic neurons, characterized by loss of Nissl substance and an excentric nucleus, were observed in the ventral horn of homozygous htau40-1 transgenic mice.
Figure 9.

Peripheral nerve and skeletal muscle of htau40 transgenic mice at the age of 2 and 8 months. a: Semithin section of sciatic nerve displaying macrophages filled with myelin debris (arrows), indicative of Wallerian degeneration. Note also a slightly dilated degenerating axon with a thinned myelin sheath (arrowhead) (×1300). b: Electron micrograph of two axons with prominent axon-Schwann cell networks (arrows) (×3735) as opposed to a normal axon (arrowhead). c: Quadriceps muscle with grouping of atrophic fibers (left side of field) and normal fibers (right side of field) (×210).
Discussion
Tauopathies, whether sporadic or inherited, differ in nerve cell type and brain regions that are affected, while sharing the presence of filamentous aggregates of hyperphosphorylated tau protein. 40 This common denominator has reinforced the view that events leading to the formation of tau protein filaments or the mere presence of tangles are sufficient to cause nerve cell degeneration. Nevertheless, it is not clear whether the formation of neurofibrillary tangles per se is needed to cause pathology. Only recently has the tau protein been linked, directly, to neurodegenerative diseases, 13 and, in combination with observations in different experimental systems, 41 tau protein has gained the status of a direct cause of neurodegeneration.
The current observations in htau40 transgenic mice constitute the first in vivo evidence for tau protein-mediated axonal damage. In these transgenic mice, the induced intraneuronal excess of tau protein caused axonopathy, evidenced by proximal axonal dilations with accumulation of neurofilaments, microtubuli, mitochondria, and vesicles, and a Wallerian type of degeneration of distal parts of the axons. Although no neuron loss was established, accumulation of ubiquitinated protein conjugates in some of the dilated axons and astrogliosis both in brain and spinal cord of homozygous htau40-1 transgenic mice reflect onset of neurodegeneration. Moreover, this model proves that excess normal tau protein was sufficient to cause neuronal injury, in the absence of intraneuronal neurofibrillary tangles. The degree of axonopathy, psychomotorical impairment, and muscle atrophy was related to the level of expression of the transgene. During the first 8 months, the longest period of observation at this moment, the pathology in the CNS of homozygous htau40-1 mice progressed only slightly. Detailed analysis of the neuropathology in aging mice up to 2 years will enable us to evaluate the progression of the pathology affecting the CNS of homozygous htau40-1 mice.
A two- to threefold augmentation of tau protein in the brain and spinal cord of homozygous htau40-5 transgenic mice resulted already in the somatodendritic redistribution and ensuing conformational alterations, evident from immunoreactivity with Alz-50 and MC-1. The 3′untranslated region of the tau mRNA is known to target the transcript to the cell body and proximal part of the axon 42 but is absent in the Thy1-htau40 construct. Similar signaling sequences in the Thy1 mRNA have not been reported, to our knowledge. Hence, the somatodendritic and axonal localization of the htau40 protein may be determined by specific tau protein transport or by diffusion. In addition, the documented phosphorylation of human tau protein may contribute to its presence in the somatodendritic compartment, which is presumed to be a pretangle stage phenomenon. 43 Similar redistribution and conformational alterations have also been observed in other transgenic mice in which the expression of the human tau protein was much lower, at about 10% 24 and 14% 25 of total tau protein. The absence of tangle-like inclusions in all transgenic models generated so far, either with four-repeat or three-repeat tau protein isoforms, could be explained by expression levels, although additional factors might be required. 24,25,44 This situation is not essentially different from the problems experienced in obtaining transgenic mice with amyloid plaques, in which the level of expression has been proved to be essential. 45 The analogy can be taken further: transgenic mice with amyloid plaques have indicated that plaques might not be essential for neuronal injury and that they are a late consequence of ongoing neurodegenerative processes. 27,29,45
Not reported before are the observed axonal dystrophy and axonal dilations in the current htau transgenic mice. The dilated axons resemble the axonal dilations seen proximal to an axon ligation or axotomy. 46 The stasis of axoplasm reflected by accumulation of neurofilaments and organelles is suggestive of a functional axotomy and defective axonal transport. Axonal spheroids, similar to the dilated axons in the htau40 transgenic mice, have been documented in neurological disorders such as amyotrophic lateral sclerosis, in which disturbed axonal transport is evident. 47-49 This finding lends support to the hypothesis that an excess of the four-repeat tau protein would saturate binding sites on the microtubules, thus interfering with kinesin-dependent transport as observed in cellular paradigms. 41 Since tau protein is considered to be a major component of the short cross-bridges between microtubules, 12 overexpression of tau protein may increase these cross-bridges and thereby hinder normal axonal transport. In this respect, we observed binding of the transgenic htau40 protein to isolated neuronal microtubules isolated from htau40-1 transgenic mice, by Western blotting on taxol-stabilized brain homogenates (results not shown). Likewise, the significantly reduced extension of the intermediate filaments caused by the four-repeat tau protein in CHO cells 41 might have an in vivo counterpart in the htau40 transgenic mice. The axonal dilations in the CNS of htau40 transgenic mice also stained with antibodies directed against the three neurofilament subunits, indicating that accumulation of neurofilaments in the proximal part of the axon might contribute to deteriorated axonal transport, as has been postulated in neurofilament-overexpressing transgenic mice. 49-51 Since the etiology of any tauopathy is unknown, our findings provide a model to gain insight into the pathological processes that are operating in different tauopathies and in tau protein-mediated axonal transport.
Dystrophic neurites, defined as thickened or irregular neuronal processes immunoreactive for tau protein, are a well-known feature of neuropathological disorders and are considered to mark widespread alteration of the neuronal cytoskeleton. In AD, dystrophic axons appear to be prominent and widespread, are particularly abundant in the hippocampal fiber systems originating from the subiculum, CA1, and the entorhinal cortex and may represent one of the main pathological lesions in AD. The morphological, immunohistochemical, and ultrastructural features of the dystrophic axons seen in brain of the htau40 mice resemble those in AD. 39,52
The accumulation of ubiquitin-protein conjugates in the axonal dilations 53 and the activation of astrocytes confirmed neuronal injury in cortex and spinal cord of transgenic mice. Astrogliosis was not observed in CNS of heterozygous htau40-1 mice that were between 2 and 4 months old, as opposed to homozygous htau40-1 mice of the same age. This suggests that astrocytes became activated subsequent to and likely as a consequence of neuronal injury. The activated astrocytes did not stain with tau protein-specific antibodies and were therefore clearly different from the tau-positive tufted astrocytes, typical for progressive supranuclear palsy. 54
The majority of patients suffering from FTDP-17 and other tauopathies develop tau protein deposits in both neurons and glial cells. The mouse thy-1 promoter used by us caused a widespread expression selectively in neurons of the CNS of htau40 transgenic mice. Therefore, these mice model not all but some of the pathological features of FTDP-17 subtypes, which are characterized by an increased level of four-repeat isoforms present in neuronal cytosol or sequestered in inclusions. At the moment four mutations have been described at positions +3, +13, +14, and +16 of the intron after exon 10 of the tau gene. They all cause overexpression of four-repeat isoforms and segregate with neurodegenerative illness. 13,14,55 Recently, tau pathology has been reported for familial multiple system tauopathy with presenile dementia (MSTD) 14,56 and disinhibition-dementia-parkinsonism-amyotrophy complex (DDPAC), 13,20,57 caused by the +3 and +14 mutations, respectively. In MSTD, the proven preponderance of the four-repeat tau protein isoform caused axonal swellings in the spinal cord and tangle formation in brain and spinal cord gray matter. In DDPAC, excess of four-repeat tau protein provoked anterior horn pathology and muscle wasting, in the absence of neurofibrillary tangles. Thus, the prominent axonal swellings in the spinal cord, the neurogenic atrophy of muscles, and, in addition, the causal relationship between these pathological characteristics and their genotype make these htau40 transgenic mice interesting animal models that recapitulate features of MSTD and DDPAC. The pathological findings in the CNS of the current transgenic mouse models and in DDPAC patients indicate that increased levels of tau protein or dysregulation of tau protein expression are sufficient to injure neurons in the CNS.
In human brain, the ratio of tau isoforms with four and three repeats (4R/3R) increases from nearly 1 in healthy individuals to 2, 1.8, and 1.6 in patients suffering from DDPAC, 20 MSTD (M. G. Spillantini, personal communication), and progressive subcortical gliosis (PSG), 55 respectively. The amount of soluble tau remains constant since the level of 3R tau isoforms decreases proportionally. In murine brain, on the contrary, only 4R tau isoforms are expressed during adulthood. 58 One might assume that the 4R/3R balance in adult human neurons is more vulnerable to changes relative to the 4R situation in adult mice, requiring an absolute increase in 4R tau isoforms to initiate neuropathology in the later. Homozygous htau40-5 transgenic mice, estimated to express 4R tau protein two to three times more compared to wild-type mice, already exhibit axonopathy and psychomotorical impairments. In descendants of strains htau40-1 and htau40-2 the same neuropathological features are more prominent. Therefore we believe that the level of overexpression obtained in this study is within the pathophysiological range, reminiscent of the relative twofold increase in 4R tau isoforms in some FTDP-17 subtypes.
As each tauopathy strikes selectively specific groups of neurons, further work should be directed at identifying specific biological triggers or genetic factors that lead to a specific pattern of neuronal injury and neuron degeneration, including tangle formation, thereby provoking the corresponding clinical phenotype. In AD the evident candidates are amyloid precursor protein, presenilin, and ApoE4. Others to be considered are suspected tau protein kinases, 59-62 excitotoxins, 63 ischemia, and resulting radicals 64,65 and even sulfated glycosaminoglycans. 66-68 These factors can now be tested by incorporation into the current model, which also offers the potential to screen for drugs and to test therapeutic strategies.
Acknowledgments
The intellectual, material, and technical contributions of the following scientists are gratefully acknowledged: J. P. Brion, H. Van Der Putten, M. Goedert, P. Davies, E. Van Mechelen, A. Van de Voorde, R. Vlietinck, B. Van der Schueren, C. Armée, R. Renwart, A. Vandormael, and T. Boon. We thank C. Vochten for the help with administration.
Footnotes
Address reprint requests to Dr. Fred Van Leuven, Experimental Genetics Group, Vlaams Instituut voor Biotechnologie, Center for Human Genetics, Katholieke Universiteit Leuven, Campus Gasthuisberg, O&N 06, B-3000 Leuven, Belgium. E-mail: fredvl@med.kuleuven.ac.be.
Supported by FWO-Vlaanderen, by the Interuniversity Network for Fundamental Research (IUAP), by the Biotechnology Program of the Flemish Government (IWT/VLAB/COT-008), by NFWO-Lotto, by the Rooms Fund, by the Janssen Research Foundation, and by Leuven Research and Development.
The first three authors contributed equally to this work.
References
- 1.Wiche G, Oberkanins C, Himmler A: Molecular structure and function of microtubule-associated proteins. Int Rev Cytol 1991, 124:217-273 [DOI] [PubMed] [Google Scholar]
- 2.Billingsley ML, Kincaid RL: Regulated phosphorylation and dephosphorylation of tau protein: effects on microtubule interaction, intracellular trafficking and neurodegeneration. Biochem J 1997, 323:577-591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mandelkow EM, Mandelkow E: Tau in Alzheimer’s disease. Trends Cell Biol 1998, 8:125-127 [DOI] [PubMed] [Google Scholar]
- 4.Goedert M, Spillantini MG, Crowther RA: Cloning of a big tau microtubule-associated protein characteristic of the peripheral nervous system. Proc Natl Acad Sci USA 1992, 89:1983-1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andreadis A, Brown WM, Kosik KS: Structure and novel exons of the human tau gene. Biochemistry 1992, 31:10626-10633 [DOI] [PubMed] [Google Scholar]
- 6.Boyne LJ, Tessler A, Murray M, Fischer I: Distribution of big tau in the central nervous system of the adult and developing rat. J Comp Neurol 1995, 358:279-293 [DOI] [PubMed] [Google Scholar]
- 7.Shea TB, Beermann ML, Nixon RA, Fischer I: Microtubule-associated protein tau is required for axonal neurite elaboration by neuroblastoma cells. J Neurosci Res 1992, 32:363-374 [DOI] [PubMed] [Google Scholar]
- 8.Knops J, Kosik KS, Lee G, Pardee JD, Cohen-Gould L, McConlogue L: Overexpression of tau in a nonneuronal cell induces long cellular processes. J Cell Biol 1991, 144:725-733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Esmaeli-Azad B, McCarty JH, Feinstein SC: Sense and antisense transfection analysis of tau function: tau influences net microtubule assembly, neurite outgrowth and neuritic stability. J Cell Sci 1994, 107:869-879 [DOI] [PubMed] [Google Scholar]
- 10.Brandt R, Léger J, Lee G: Interaction of tau with the neural plasma membrane mediated by tau’s amino-terminal projection domain. J Cell Biol 1995, 131:1327-1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caceres A, Kosik KS: Inhibition of neurite polarity by tau antisense oligonucleotides in primary cerebellar neurons. Nature 1990, 343:461-463 [DOI] [PubMed] [Google Scholar]
- 12.Harada A, Oguchi K, Okabe S, Kuno J, Terada S, Ohshima T, Sato-Yoshitake R, Takei Y, Noda T, Hirokawa N: Altered microtubule organization in small-caliber axons of mice lacking tau protein. Nature 1994, 369:488-491 [DOI] [PubMed] [Google Scholar]
- 13.Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P: Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998, 393:702-705 [DOI] [PubMed] [Google Scholar]
- 14.Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B: Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA 1998, 95:7737-7741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dumanchin C, Camuzat A, Campion D, Verpillat P, Hannequin D, Dubois B, Saugier-Veber P, Martin C, Penet C, Charbonnier F, Agid Y, Frebourg T, Brice A: Segregation of a missense mutation in the microtubule-associated protein tau gene with familial frontotemporal dementia and parkinsonism. Hum Mol Genet 1998, 7:1825-1829 [DOI] [PubMed] [Google Scholar]
- 16.Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Andreadis A, Wiederholt WC, Raskind M, Schellenberg GD: Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol 1998, 43:815-825 [DOI] [PubMed] [Google Scholar]
- 17.Clark LN, Poorkaj P, Wszolek Z, Geschwind DH, Nasreddine ZS, Miller B, Li D, Payami H, Awert F, Markopoulou K, Andreadis A, D’Souza I, Lee VM-Y, Reed L, Trojanowski JQ, Zhukareva V, Bird T, Schellenberg G, Wilhelmsen KC: Pathogenic implications of mutations in the tau gene in pallido-ponto-nigral degeneration and related neurodegenerative disorders linked to chromosome 17. Proc Natl Acad Sci USA 1998, 95:13103-13107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rizzu P, Van Swieten JC, Joosse M, Hasegawa M, Stevens M, Tibben A, Niermeijer MF, Hillebrand M, Ravid R, Oostra BA, Goedert M, van Duijn CM, Heutink P: High prevalence of mutations in the microtubule-associated protein tau in a population study of frontotemporal dementia in the Netherlands. Am J Hum Genet 1999, 64:414-421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iijima M, Tabira T, Poorkaj P, Schellenberg GD, Trojanowski JQ, Lee VM-Y, Schmidt ML, Takahashi K, Nabika T, Matsumoto T, Yamashita Y, Yoshioka S, Ishino H: A distinct familial dementia with a novel missense mutation in the tau gene. NeuroReport 1999, 10:497-501 [DOI] [PubMed] [Google Scholar]
- 20.Hong M, Zhukareva V, Vogelsberg-Ragaglia V, Wszolek Z, Reed L, Miller BI, Geschwind DH, Bird TD, McKeel D, Goate A, Morris JC, Wilhelmsen KC, Schellenberg GD, Trojanowski JQ, Lee MYV: Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science 1998, 282:1914-1917 [DOI] [PubMed] [Google Scholar]
- 21.Hasegawa M, Smith MJ, Goedert M: Tau proteins with FTDP-17 mutations have a reduced ability to promote microtubule assembly. FEBS Lett 1998, 437:207-210 [DOI] [PubMed] [Google Scholar]
- 22.Hasegawa M, Smith MJ, Iijima M, Tabira T, Goedert M: FTDP-17 mutations N279K and S305N in tau produce increased splicing of exon 10. FEBS Lett 1999, 443:93-96 [DOI] [PubMed] [Google Scholar]
- 23.D’Souza I, Poorkaj P, Hong M, Nochlin D, Lee VM-Y, Bird TD, Schellenberg GD: Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism-chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc Natl Acad Sci USA 1999, 96:5598-5603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Götz J, Probst A, Spillantini MG, Schäfer T, Jakes R, Bürki K, Goedert M: Somatodendritic localization and hyperphosphorylation of tau protein in transgenic mice overexpressing the longest human brain tau isoform. EMBO J 1995, 14:1304-1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brion JP, Tremp G, Octave JN: Transgenic expression of the shortest human tau affects its compartmentalization and its phosphorylation as in the pretangle stage of Alzheimer’s disease. Am J Pathol 1999, 154:255-270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA: Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 1989, 3:519-526 [DOI] [PubMed] [Google Scholar]
- 27.Moechars D, Lorent K, De Strooper B, Dewachter I, Van Leuven F: Expression in brain of amyloid precursor protein mutated in the α-secretase site causes disturbed behavior, neuronal degeneration and premature death in transgenic mice. EMBO J 1996, 15:1265-1274 [PMC free article] [PubMed] [Google Scholar]
- 28.Lamberty Y, Gower AJ: Age-related changes in spontaneous behaviour and learning in NMRI mice from middle to old age. Phys Behav 1991, 51:81-88 [DOI] [PubMed] [Google Scholar]
- 29.Moechars D, Dewachter I, Lorent K, Reversé D, Baekelandt V, Naidu A, Tesseur I, Spittaels K, Van den Haute C, Checler F, Godaux E, Cordell B, Van Leuven F: Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J Biol Chem 1999, 274:6483-6492 [DOI] [PubMed] [Google Scholar]
- 30.Brion JP, Hanger DP, Bruce MT, Couck A-M, Flament-Durand J, Anderton BH: Tau in Alzheimer neurofibrillary tangles. Biochem J 1991, 273:127-133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mercken M, Vandermeeren M, Lübke U, Six J, Boons J, Van de Voorde A, Martin J-J, Gheuens J: Monoclonal antibodies with selective specificity for Alzheimer tau are directed against phosphatase-sensitive epitopes. Acta Neuropathol 1992, 84:265-272 [DOI] [PubMed] [Google Scholar]
- 32.Biernat J, Mandelkow E-M, Schröter C, Lichtenberg-Kraag B, Steiner B, Berling B, Meyer H, Mercken M, Vandermeeren A, Goedert M, Mandelkow E: The switch of tau protein to an Alzheimer-like state includes the phosphorylation of two serine-proline motifs upstream of the microtubule binding region. EMBO J 1992, 11:1593-1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goedert M, Jakes R, Crowther A, Cohen P, Vanmechelen E, Vandermeeren M, Cras P: Epitope mapping of monoclonal antibodies to the paired helical filaments of Alzheimer’s disease: identification of phosphorylation sites in tau protein. Biochem J 1994, 301:871-877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Otvos L, Jr, Feiner L, Lang E, Szendrei GI, Goedert M, Lee VM-Y: Monoclonal antibody PHF-1 recognizes tau protein phosphorylated at serine residues 396 and 404. J Neurosci Res 1994, 39:669-673 [DOI] [PubMed] [Google Scholar]
- 35.Ksiezak-Reding H, Davies P, Yen S-H: Alz-50, a monoclonal antibody to Alzheimer’s disease antigen, cross-reacts with tau proteins from bovine and normal human brain. J Biol Chem 1988, 263:7943-7947 [PubMed] [Google Scholar]
- 36.Carmel G, Mager EM, Binder LI, Kuret J: The structural basis of monoclonal antibody Alz50’s selectivity for Alzheimer’s disease pathology. J Biol Chem 1996, 271:32789-32795 [DOI] [PubMed] [Google Scholar]
- 37.Jicha GA, Bowser R, Kazam IG, Davies P: Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res 1997, 48:128-132 [DOI] [PubMed] [Google Scholar]
- 38.Cox G: Bancroft JD Stevens A eds. Neuropathological Techniques: Theory and Practice of Histological Techniques. 1977, :pp 249-273 Churchill Livingstone, Edinburgh, London, [Google Scholar]
- 39.Gibson PH: Ultrastructural abnormalities in the cerebral neocortex and hippocampus associated with Alzheimer’s disease and aging. Acta Neuropathol 1987, 73:86-91 [DOI] [PubMed] [Google Scholar]
- 40.Spillantini MG, Goedert M: Tau protein pathology in neurodegenerative disease. Trends Neurosci 1998, 21:428-433 [DOI] [PubMed] [Google Scholar]
- 41.Ebneth A, Godemann R, Stamer K, Illenberger S, Trinczek B, Mandelkow EM, Mandelkow E: Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmatic reticulum: implications for Alzheimer’s disease. J Cell Biol 1998, 143:777-794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Behar L, Marx R, Sadot E, Barg J, Ginzburg I: cis-acting signals and trans-acting proteins are involved in tau mRNA targeting into neurites of differentiating neuronal cells. Int J Dev Neurosci 1995, 13:113-127 [DOI] [PubMed] [Google Scholar]
- 43.Braak E, Braak H, Mandelkow EM: A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol 1994, 87:554-567 [DOI] [PubMed] [Google Scholar]
- 44.Hall GF, Yao J, Lee G: Human tau becomes phosphorylated and forms filamentous deposits when overexpressed in lamprey central neurons in situ. Proc Natl Acad Sci USA 1997, 94:4733-4738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Duff K: Recent work on Alzheimer’s disease transgenics. Curr Opin Biotechnol 1998, 9:561-564 [DOI] [PubMed] [Google Scholar]
- 46.Midroni G, Bilbao JM: Midroni G Bilbao JM eds. Biopsy Diagnosis of Peripheral Neuropathy. 1995, :pp 45-74 Butterworth-Heinemann, Boston, [Google Scholar]
- 47.Hirano A, Donnenfeld H, Sasaki S, Nakano I: Fine structural observations of neurofilamentous changes in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 1984, 34:461-470 [DOI] [PubMed] [Google Scholar]
- 48.Côté F, Collard JF, Julien JP: Progressive neuronopathy in transgenic mice expressing the human neurofilament heavy gene: a mouse model of amyotropic lateral sclerosis. Cell 1993, 73:35-46 [DOI] [PubMed] [Google Scholar]
- 49.Collard JF, Côté F, Julien JP: Defective axonal transport in a transgenic mouse model of amyotrophic lateral sclerosis. Nature 1995, 375:61-64 [DOI] [PubMed] [Google Scholar]
- 50.Xu Z, Cork LC, Griffin JW, Cleveland DW: Increased expression of neurofilament subunit NF-L produces morphological alterations that resemble the pathology of human motor neuron disease. Cell 1993, 73:23-33 [DOI] [PubMed] [Google Scholar]
- 51.Lee MK, Marszlek JR, Cleveland DW: A mutant neurofilament subunit causes massive, selective motor neuron death: implications for the pathogenesis of human motor neuron disease. Neuron 1994, 13:975-988 [DOI] [PubMed] [Google Scholar]
- 52.Su JH, Cummings BJ, Cotman CW: Identification and distribution of axonal dystrophic neurites in Alzheimer’s disease. Brain Res 1993, 625:228-237 [DOI] [PubMed] [Google Scholar]
- 53.Alves-Rodrigues A, Gregori L, Figueiredo-Pereira ME: Ubiquitin, cellular inclusions and their role in neurodegeneration. Trends Neurosci 1998, 21:516-520 [DOI] [PubMed] [Google Scholar]
- 54.Bigio EH, Brown DF, White CL: Progressive supranuclear palsy with dementia: cortical pathology. J Neuropathol Exp Neurol 1999, 58:359-364 [DOI] [PubMed] [Google Scholar]
- 55.Goedert M, Spillantini MG, Crowther RA, Chen SG, Parchi P, Tabaton M, Lanska DJ, Markesbery WR, Wilhelmsen KC, Dickson DW, Petersen RB, Gambetti P: Tau gene mutation in familial progressive subcortical gliosis. Nature Med 1999, 5:454-457 [DOI] [PubMed] [Google Scholar]
- 56.Spillantini MG, Goedert M, Crowther RA, Murrell JR, Farlow MR, Ghetti B: Familial multiple system tauopathy with presenile dementia: a disease with abundant neuronal and glial tau filaments. Proc Natl Acad Sci USA 1997, 94:4113-4118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lynch T, Sano M, Marder KS, Bell KL, Foster NL, Defendini RF, Sima AAF, Keohane C, Nygaard TG, Fahn S, Mayeux R, Rowland LP, Wilhelmsen KC: Clinical characteristics of a family with chromosome 17-linked disinhibition-dementia-parkinsonism-amyotrophy complex. Neurology 1994, 44:1878-1884 [DOI] [PubMed] [Google Scholar]
- 58.Kampers T, Pangalos M, Geerts H, Wiech H, Mandelkow E: Assembly of paired helical filaments from mouse tau: implications for the neurofibrillary pathology in transgenic mouse models for Alzheimer’s disease. FEBS Lett 1999, 451:39-44 [DOI] [PubMed] [Google Scholar]
- 59.Hong M, Chen DCR, Klein PS, Lee VMY: Lithium reduces tau phosphorylation by inhibition of glycogen synthase kinase-3. J Biol Chem 1997, 272:25326-25332 [DOI] [PubMed] [Google Scholar]
- 60.Drewes G, Ebneth A, Preuss U, Mandelkow EM, Mandelkow E: MARK, a novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule distribution. Cell 1997, 89:297-308 [DOI] [PubMed] [Google Scholar]
- 61.Ishiguro K, Takamatsu M, Tomizawa K, Omori A, Takahashi M, Arioka M, Uchida T, Imahori K: Tau protein kinase I converts normal tau protein into A68-like component of paired helical filaments. J Biol Chem 1992, 267:10897-10901 [PubMed] [Google Scholar]
- 62.Yamaguchi H, Ishiguro K, Uchida T, Takashima A, Lemere CA, Imahori K: Preferential labeling of Alzheimer neurofibrillary tangles with antisera for tau protein kinase (TPK) I/glycogen synthase kinase-3β and cyclin-dependent kinase 5, a component of TPK II. Acta Neuropathol 1996, 92:232-241 [DOI] [PubMed] [Google Scholar]
- 63.Masliah E, Alford M, DeTeresa R, Mallory M, Hansen L: Deficient glutamate transport is associated with neurodegeneration in Alzheimer’s disease. Ann Neurol 1996, 40:759-766 [DOI] [PubMed] [Google Scholar]
- 64.Good PF, Werner P, Hsu A, Olanow CW, Perl DP: Evidence of neuronal oxidative damage in Alzheimer’s disease. Am J Pathol 1996, 149:21-28 [PMC free article] [PubMed] [Google Scholar]
- 65.Dewar D, Graham DI, Teasdale GM, McCulloch J: Alz-50 and ubiquitin immunoreactivity is induced by permanent focal cerebral ischaemia in the cat. Acta Neuropathol 1993, 86:623-629 [DOI] [PubMed] [Google Scholar]
- 66.Goedert M, Jakes R, Spillantini MG, Hasegawa M, Smith MJ, Crowther RA: Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 1996, 383:550-553 [DOI] [PubMed] [Google Scholar]
- 67.Perry G, Siedlak SL, Richey P, Kawai M, Cras P, Kalaria RN, Galloway PG, Scardina JM, Cordell B, Greenberg BD, Ledbetter SR, Gambetti P: Association of heparan sulphate proteoglycan with the neurofibrillary tangles of Alzheimer’s disease. J Neurosci 1991, 11:3679-3683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Su JH, Cummings BJ, Cotman CW: Localization of heparan sulphate glycosaminoglycan and proteoglycan core protein in aged brain and Alzheimer’s disease. Neuroscience 1992, 51:801-813 [DOI] [PubMed] [Google Scholar]