

Abstract
Bcl-xL, a prosurvival member of the Bcl-2 family that is expressed in many tumors, represses apoptosis induced by chemotherapeutic drugs in vitro. However, the contribution of apoptosis and prosurvival Bcl-2-related proteins to chemotherapy resistance in vivo is unknown and has been challenged by recent results with clonogenic survival assays. To test the ability of Bcl-xL to provide chemotherapy resistance to tumors, we transfected the mouse bcl-xL gene into the tumorigenic SCK mammary cell line and assessed the response of tumor cells to chemotherapeutic drugs in clonogenic assays and in a syngeneic mouse model. Bcl-xL conferred protection on SCK cells against methotrexate at certain drug concentrations, but not at all against 5-fluorouracil in clonogenic survival assays in vitro. Injection of SCK cells transfected with Bcl-xL or control plasmid in the mammary fat pads of syngeneic recipient mice resulted in tumors of similar size. However, although the volume of control tumors regressed up to 80% after 4 to 5 days of chemotherapy, SCK tumors expressing Bcl-xL did not regress and continued to grow in the presence of methotrexate or 5-fluorouracil. In addition, numbers of apoptotic cells were significantly higher in control tumors as compared to Bcl-xL-expressing tumors in animals treated with methotrexate or 5-fluorouracil. These results provide evidence that inhibition of apoptosis through Bcl-xL overexpression can promote resistance to chemotherapy in tumors in vivo.
The treatment of cancer cells with chemotherapy and irradiation is limited by the emergence of cancer cells resistant to these therapies. Frequently, many tumors respond to chemotherapy during initial treatment but develop a multidrug resistance phenotype with continued therapy. The molecular events responsible for this resistance remain largely unknown. Overexpression of the P-glycoprotein and other members of the ATP-binding cassette (ABC) superfamily has been shown to induce multidrug resistance. 1-3 However, development of multidrug resistance is often observed in the absence of ABC protein overexpression, 4,5 suggesting that other mechanisms play a role in drug resistance. Another mechanism responsible for this pleotropic drug resistance may involve expression of antiapoptotic proteins, as multiple chemotherapy drugs have been shown to kill tumor cells by apoptosis. 6,7 Members of the Bcl-2 family of proteins are important regulators of apoptosis induced by a wide array of stimuli, including chemotherapeutic agents. 8-10 Two members of this family, Bcl-2 and Bcl-xL, function as repressors of cell death and are expressed in a wide variety of human tumors derived from epithelial, hematopoietic, and soft tissue lineages. 11 Expression levels of Bcl-2-related proteins change as tumors become less differentiated, or after treatment, 12-15 suggesting that expression of Bcl-2 family members may play an important role in tumor progression and/or resistance to therapy. Although the precise mechanism by which Bcl-2 and Bcl-xL inhibit apoptosis is controversial, it is thought that these prosurvival proteins act by interfering with the activation of initiator (upstream) caspases. 16 Because chemotherapeutic drugs induce caspase activation, 17,18 Bcl-2 and Bcl-xL may inhibit chemotherapy-induced apoptosis, at least in part, by repressing chemotherapy-induced caspase activity. However, it remains to be determined whether inhibition of apoptosis in tumor cells could lead to resistance to chemotherapy in vivo.
Because Bcl-2 and Bcl-xL can inhibit chemotherapy-induced apoptosis in vitro, it has been suggested that the expression of these proteins plays a role in chemotherapy resistance. However, the contribution of Bcl-2 and Bcl-xL to tumor chemotherapy resistance remains unclear. Bcl-2 expression has been associated with poor response to chemotherapy in acute myeloid leukemia 19,20 and large-cell lymphoma. 21,22 Yet the significance of Bcl-2 or Bcl-xL expression is complicated by the observation that these survival proteins inhibit chemotherapy- and irradiation-induced apoptosis in short-term assays, but in certain systems do not affect clonogenic survival of tumor cells in vitro. 23-25 These observations suggest that the antiapoptotic effect of Bcl-2 may not necessarily translate into increased survival of tumor cells in vivo, as the capacity of Bcl-2 or Bcl-xL to modulate chemotherapy-induced apoptosis in growing tumors remains to be determined. In the present study, we have expressed Bcl-xL in the murine mammary adenocarcinoma cell line SCK and determined the ability of Bcl-xL to regulate the response of tumor cells to chemotherapy in clonogenic assays in vitro and in vivo using a syngeneic mouse model.
Materials and Methods
Cell Culture and Transfection
SCK mouse mammary carcinoma cells, 26 kindly provided by Dr. C. W. Song of the University of Minnesota, were cultured in RPMI 1640 medium supplemented with 10% fetal calf serum, 2 mmol/L L-glutamine, and 100 μg/ml streptomycin. SCK cells (5 × 106) were transfected using lipofectamine (Gibco BRL, Rockville, MD) with 6 μg of the pSFFV-m-Bcl-xL to produce Flag-tagged mouse Bcl-xL 27 or control pSFFV plasmid (Invitrogen, Carlsbad, CA). pcDNA-3-m-Bcl-xL was constructed by ligation of Flag-tagged mouse Bcl-xL cDNA into the plasmid pcDNA-3 (Invitrogen). Authenticity of the constructs was confirmed by dideoxy sequencing. Individual cell clones were selected for growth in the presence of G418 (0.5 mg/ml) by limiting dilution. Expression of Flag-Bcl-xL in single cell clones was analyzed by flow cytometry using anti-Flag and protein expression was confirmed by Western blot analysis as described below.
Clonogenic Survival Assay
Cells were seeded in rows of 750, 250, and 100 cells/well in 6-well plates (Costar, Corning, NY) and further incubated for 18 hours. Concentrated stock solutions of methotrexate and 5-fluorouracil were serially diluted and added to each of the rows. After a 6-hour exposure, the medium was aspirated and the wells were washed in drug-free medium for 1 hour, followed by a 10-day incubation in drug-free medium to allow colony formation. At the end of this incubation, medium was aspirated and the cells were fixed and stained by the addition of 0.5% methylene blue in 50% ethanol for 45 minutes at room temperature. The plates were gently washed with water and allowed to air-dry. Visible colonies were counted to determine the percent colony formation of plated cells for each drug treatment. Colony formation percentages for each drug treatment were compared to colony-formation values of untreated controls. Values were expressed as the mean ± SE from triplicate experiments.
Western Blot
The expression of Flag-murine Bcl-xL was determined by Western blot analysis as described previously 28 using anti-Flag mAb (5 μg/ml). After incubation with rabbit anti-mouse IgG secondary antibody, the reaction was developed by enhanced chemiluminescense using the ECL kit (Amersham, Arlington Heights, IL) and exposed to film (Eastman Kodak).
Apoptosis Assays
For in vitro apoptosis assays, cells were seeded at 1 × 10 5 cells in triplicate wells in media containing methotrexate 1 μg/ml (Immunex, Seattle, WA) or 5-fluorouracil 1 μg/ml (Hoffman-LaRoche, Nutley, NJ). The percentage of apoptotic cells was determined at different time points in triplicate cultures by nuclear propidium iodide staining followed by flow cytometric analysis as described previously. 28 Results were based on the analysis of at least 5 × 10 4 events from each triplicate culture. Values were expressed as the mean ± SE from triplicate cultures. Apoptosis in tumors was evaluated by analysis of tumor sections stained with hematoxylin and eosin using histological criteria as described. 29 Apoptosis was quantitated in 10 consecutive high power fields (HPF; 40× lens objective) in each tumor sample. Number of apoptotic cells was evaluated in viable tumor at the outer cortex of the tumor mass in a blinded fashion. Each HPF consisted of solid sheets of tumor cells, with the same approximate number of cells noted in both control and Bcl-xL expressing tumors. Number of mitotic figures per HPF were quantitated in a similar fashion.
Establishment of Tumors and Measurement of Tumor Volume
SCK tumors were established in syngeneic female A/J mice as described above. The incision was closed with wound clips and tumors were allowed to grow for 9 to 10 days. Methotrexate (0.9 mg/kg/day) or 5-fluorouracil (23 mg/kg/day) diluted in sterile normal saline was administered intraperitoneally (i.p.) q.d. × 3 to 4 days. Control animals received sterile normal saline alone, and tumor measurements were taken in parallel. Tumor volume was measured every day with linear calipers and calculated in cubic millimeters as (a × b2/2), where a is the larger diameter and b the smaller diameter of the tumor.
Statistical Analysis
For in vitro chemotherapy-induced death assays, statistical significance was calculated by two-way analysis of variance using SYSTAT software (Chicago, IL). For in vivo chemotherapy response assays, statistical significance was calculated using a general linear model with two-way analysis of variance. Post hoc comparisons of mean values from different groups using Bonferroni adjustment were performed for both in vitro and in vivo assays.
Results
Bcl-xL Inhibits Apoptosis of SCK Breast Carcinoma Cells Treated with Methotrexate and 5-Fluorouracil in Vitro
We selected the SCK mammary mouse tumor model to assess the ability of Bcl-xL to promote chemotherapy resistance in vivo, as SCK carcinoma cells were derived spontaneously and established as tumors in the absence of any exposure to chemotherapeutic drugs. 26 Furthermore, SCK form tumors in female syngeneic A/J hosts, which mimics the clinical setting more closely than xenograft models. Before determining the ability of Bcl-xL to regulate chemotherapy resistance of tumors in the animal, we determined if Bcl-xL provides protection to SCK carcinoma cells against chemotherapy-induced apoptosis in vitro. In these experiments, we used one of two different Bcl-xL expression plasmids, pSFFV- Bcl-xL or pcDNA3-Bcl-xL, to overexpress Flag-tagged mouse Bcl-xL. As a control, we transfected SCK cells with the corresponding empty vectors. After selection in G418, three independently derived SCK clones that stably overexpress Bcl-xL were identified (Figure 1A) ▶ . All three Bcl-xL clones exhibited reduced apoptosis after incubation 5-fluorouracil (Figure 1B) ▶ and methotrexate (Figure 1C) ▶ , two drugs commonly used to treat patients with breast cancer, when compared with SCK clones transfected with control plasmids. These results are in agreement with a large body of evidence indicating that Bcl-xL inhibits or delays chemotherapy-induced apoptosis of tumor cells in short-term assays in vitro. 8-10
Figure 1.
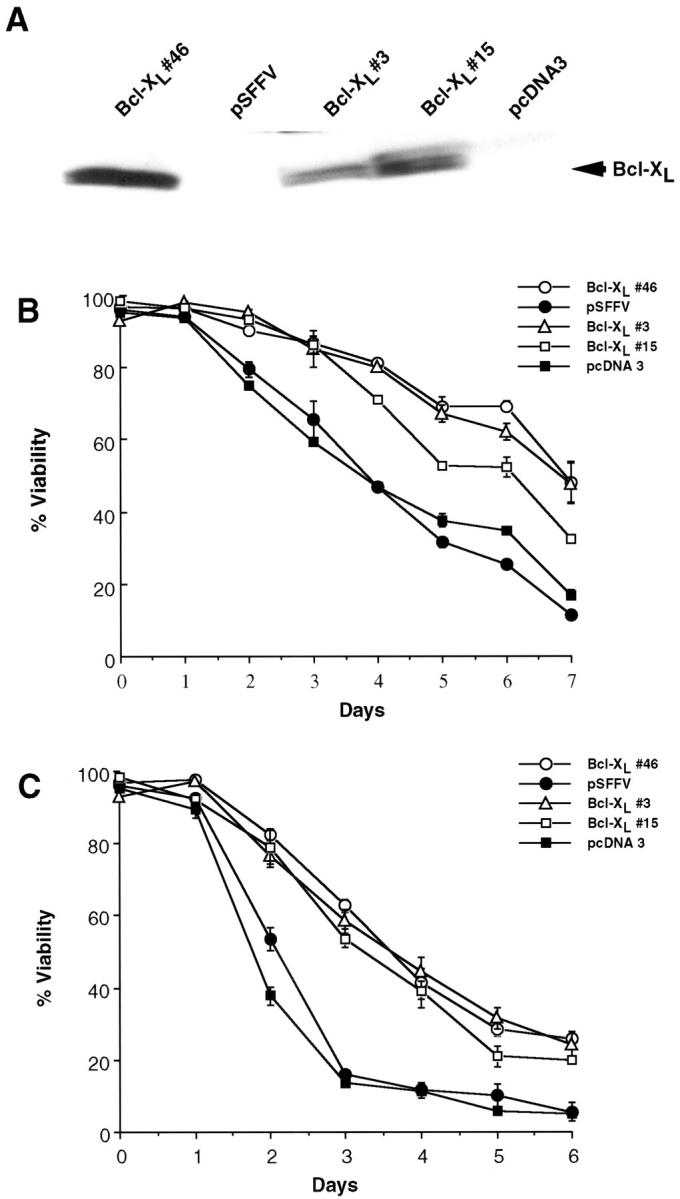
Effects of Bcl-xL expression on viability of SCK cells exposed to chemotherapy. A: Western blot analysis of Flag-tagged murine Bcl-xL. Clone 46 was derived after transfection with pSFFV-Bcl-xL and clones 3 and 15 with pcDNA3-Bcl-xL. Lysates from clones from SCK transfected with the corresponding control plasmids pSFFV and pcDNA3 are shown as controls. Protein lysates were immunoblotted for Flag. B: Viability analysis of SCK clones expressing Bcl-xL and control plasmids after continuous exposure to 5-fluorouracil (1 μg/ml). C: Viability analysis of SCK clones expressing Bcl-xL and control plasmids after continuous exposure to methotrexate (1 μg/ml). Percent viability represents the percentage of nonapoptotic cells at each time point after addition of each chemotherapy drug as measured by flow cytometric analysis of nuclei stained with propidium iodide. All values represent the mean ± SE from triplicate cultures. The experiments shown are representative of at least three individual experiments. Differences in viability between SCK-Bcl-xL and SCK control clones were significant (P < 0.00001).
Bcl-xL Has Little or No Significant Effect on the Clonogenic Survival of SCK Cells Treated with Methotrexate or 5-Fluorouracil
The clonogenic ability of tumor cells is thought to be a more reliable assay than short-term survival in predicting the response of tumor cells to chemotherapy in vivo. We determined next whether Bcl-xL would provide increased clonogenic survival in vitro after transient exposure to chemotherapeutic drugs. SCK cells expressing Bcl-xL, or control cells stably transfected with control plasmid, were treated with increasing concentrations of methotrexate, 5-fluorouracil, or vehicle control for 6 hours, then washed and cultured in drug-free medium to permit colony formation. Colonies that formed in triplicate cultures after 10 days were stained with methylene blue and counted. The ability of both SCK-Bcl-xL and control SCK-pSFFV cells to form colonies declined after treatment with methotrexate or 5-fluorouracil in a dose-dependent manner when compared to untreated cells (Figure 2, A and B) ▶ . In three separate experiments, Bcl-xL-expressing cells demonstrated significantly increased colony formation after exposure to certain concentrations of methotrexate when compared to control cells (Figure 2A) ▶ , but Bcl-xL did not have any significant effect on colony formation at any 5-fluorouracil concentration (Figure 2B) ▶ .
Figure 2.

Clonogenic ability of SCK cells expressing Bcl-xL and control. SCK-Bcl-xL (clone 46) and SCK-pSFFV cells were seeded in rows of 750, 250, and 100 cells/well in 6-well plates and incubated with increasing concentrations of methotrexate (A) or 5-fluorouracil (B). After a 6-hour exposure, the medium was aspirated and the wells were washed in drug-free medium for 1 hour, followed by a 10-day incubation in drug-free culture medium to allow colony formation. Colonies were fixed and stained by the addition of 0.5% methylene blue. Visible colonies were counted to determine the percent colony formation of plated cells for each drug treatment. Colony formation percentages for each drug treatment were compared to colony formation values of untreated controls. Values are expressed as the mean ± SE from triplicate cultures. Differences in colony formation between SCK-Bcl-xL and SCK-pSFFV clones were significant only at concentrations of 5, 10, and 25 μg/ml (P < 0.01). There was no statistical difference in colony formation between SCK-Bcl-xL and SCK-pSFFV control after treatment with any concentration of 5-fluorouracil (P > 0.1).
Bcl-xL Confers Chemotherapy Resistance to SCK Tumors in Vivo
To determine whether expression of Bcl-xL would confer resistance to chemotherapy in vivo, we used SCK murine mammary adenocarcinoma cells grown in female syngeneic A/J recipient mice as an in vivo tumor model. Dose-response experiments revealed that i.p. injections of methotrexate at 0.9 mg/kg/day or 5-fluorouracil at 23 mg/kg/day were optimal to assess the response of SCK tumors to chemotherapy (data not shown). We injected 1 × 10 6 SCK cells stably transfected with Bcl-xL or control plasmid in the right and left mammary fat pads of the same mice to adequately control for the amount of chemotherapy given to both groups of tumors. Tumors averaging 60 mm 3 were established by day 8 to 9 after injection of 1 × 10 6 SCK cells transfected with Bcl-xL or control plasmid into the mammary fat pads. No significant difference in volume was observed between tumors established from SCK-Bcl-xL and SCK-control plasmid (59.5 ± 28 versus 61.9 ± 27 mm3, n = 27). Mice bearing SCK tumors expressing Bcl-xL (one mammary pad) and control plasmid (contralateral mammary pad) were treated with daily i.p. injections of 5-fluorouracil or methotrexate, two chemotherapeutic drugs commonly used to treat mammary cancers. Tumors derived from SCK cells transfected with Bcl-xL (clone 46) were significantly larger than control tumors after treatment with daily injections of 5-fluorouracil (Figure 3A) ▶ . Quantitative analysis revealed that tumors derived from SCK transfected with control plasmid decreased to 20 to 30% of the original tumor volume by day 2, and to less than 20% by day 4 after daily i.p. administration of 5-fluorouracil (Figure 3B) ▶ . In contrast, tumors derived from SCK cells expressing Bcl-xL cells were resistant to 5-fluorouracil in that they continued to increase in volume over 200% in the presence of the drug (Figure 3B) ▶ . Mice harboring SCK tumors expressing Bcl-xL had to be sacrificed by day 6 or 7 after administration of chemotherapy due to large tumor burden.
Figure 3.

Response of SCK tumors expressing Bcl-xL to 5-fluorouracil. A: Tumors were established by injection of 1 × 10 6 SCK-Bcl-xL cells (clone 46) or 1 × 10 6 SCK-pSFFV cells (control) into the left or right mammary fat pads of syngeneic recipient mice. After 8 days, mice bearing tumors were injected i.p. with 23 mg/kg/day of 5-fluorouracil daily. Tumors were excised from 5 representative mice and photographed. B: Growth of tumors established from clone Bcl-xL clone 46 and pSFFV in the presence and absence of 5-fluorouracil. Female A/J mice were treated with i.p. 5-fluorouracil (5-FU) daily. Tumor measurements were taken daily (after initiation of chemotherapy) with calipers, and tumor volumes were calculated as indicated in Materials and Methods. To control for variation in tumor volume before chemotherapy, tumor volume at day 1 of chemotherapy was considered as 100%. Values represent the mean ± SE from 9 recipient mice, each bearing a SCK-Bcl-xL and a SCK-pSFFV control tumor. The difference in volume between SCK-Bcl-xL and SCK-pSFFV control tumors was significant (P < 0.05).
To determine whether the ability of Bcl-xL to confer resistance of tumors to 5-fluorouracil could be extended to other chemotherapeutic drugs, we analyzed the susceptibility of Bcl-xL-expressing and control SCK tumors to methotrexate. Tumors from SCK cells transfected with Bcl-xL or control plasmid grew at the same rate in vivo in the absence of methotrexate (Figure 4A) ▶ . As we observed with 5-fluorouracil, tumors derived from SCK cells transfected with Bcl-xL (clone 46) continued to increase in volume, whereas control SCK tumors decreased by day 2 and 3 after drug administration (Figure 4, A and B) ▶ . To further verify these observations, we assessed the susceptibility to methotrexate of tumors derived from two additional SCK-Bcl-xL clones (clones 3 and 15) and two SCK control clones (clones 3.3 and 3.15). Tumors established from SCK-Bcl-xL clones were resistant to chemotherapy when compared with SCK clones transfected with empty vector (Figure 4B) ▶ . These results indicate that these observations with Bcl-xL are not due to clonal variation in that they could be reproduced in three independently derived clones.
Figure 4.

Response of SCK tumors expressing Bcl-xL to methotrexate. A: Response to chemotherapy of SCK tumors established from Bcl-xL (clone 46) and control pSFFV plasmid. Female A/J mice recipients were treated with i.p. methotrexate 0.9 mg/kg/day for 3 days with sterile saline alone as a control. Tumor measurements were taken daily (after initiation of chemotherapy) with calipers, and tumor volumes were calculated as indicated in Materials and Methods. Values represent the mean ± SE from 10 recipient mice (treated with methotrexate) or 5 recipient mice (untreated), each bearing a SCK-Bcl-xL and a SCK-pSFFV control tumor. The difference in volume between SCK-Bcl-xL and SCK-pSFFV control tumors was significant (P < 0.05). There was no statistical difference between SCK-Bcl-xL tumors treated with methotrexate and untreated SCK-pSFFV control tumors. B: Response to chemotherapy of SCK tumors established from Bcl-xL clones (clones 3 and 15) and plasmid control clones (clones pcDNA 3.3 and 3.15). Female A/J mice were treated with i.p. methotrexate at 0.9 mg/kg/day for 4 days. Values represent the mean ± SE from 3 recipient mice in each group, each bearing a SCK-Bcl-xL and a SCK-pcDNA3 control tumor. The difference in volume between SCK-Bcl-xL and SCK-pcDNA3 control tumors was significant (P < 0.001).
Bcl-xL Inhibits Apoptosis in Vivo
To determine whether expression of Bcl-xL would inhibit chemotherapy-induced apoptosis in vivo or if the increase in volume seen in Bcl-xL-expressing tumors was due to increased mitotic activity, we assessed the number of apoptotic cancer cells in histological sections of SCK tumors. 1 × 10 6 SCK cells transfected with Bcl-xL or control plasmid in the right and left mammary fat pads of the same syngeneic A/J mice were established, and the mice were treated with daily injections of i.p. methotrexate or 5-fluorouracil. Tumors were excised at day 0 and day 5 or 6 of treatment. Numbers of apoptotic cells were significantly higher in control tumors by day 5 or 6 of treatment as compared to Bcl-xL-expressing tumors in animals treated with methotrexate or 5-fluorouracil (Figure 5, A and B) ▶ . Numbers of mitotic figures in control tumors were not significantly different from those in Bcl-xL-expressing tumors at day 0 and day 5 or 6 (data not shown).
Figure 5.

Effects of Bcl-xL expression on apoptosis in SCK tumors. A: Response to chemotherapy of SCK tumors established from Bcl-xL (clone 46) and control pSFFV plasmid. Female A/J mice recipients were treated with i.p. 5-fluorouracil 23 mg/kg/day for 6 days with sterile saline alone as a control. Values represent the mean ± SE from 3 recipient mice at day 0 and from 7 recipient mice at day 6, each bearing a SCK-Bcl-xL and a SCK-pSFFV control tumor. The difference in number of apoptotic cells/HPF between SCK-Bcl-xL and SCK-pSFFV control tumors after chemotherapy was significant (P < 0.001). B: Response to chemotherapy of SCK tumors established from Bcl-xL (clone 46) and control pSFFV plasmid. Female A/J mice recipients were treated with i.p. methotrexate 0.9 mg/kg/day for 5 days with sterile saline alone as a control. Values represent the mean ± SE from 3 recipient mice at day 0 and from 5 recipient mice at day 5, each bearing a SCK-Bcl-xL and a SCK-pSFFV control tumor. The difference in number of apoptotic cells/HPF between SCK-Bcl-xL and SCK-pSFFV control tumors was significant (P < 0.001).
Discussion
The results presented here demonstrate that overexpression of Bcl-xL promotes resistance of mammary cancer cells to chemotherapy in a syngeneic mouse model, suggesting that this protein may confer a similar function on primary tumors overexpressing Bcl-xL in humans. A large variety of human tumors derived from different tissues including the breast express Bcl-xL. 30 Significantly, expression of Bcl-xL in primary breast tumors has been associated with poor prognosis, 30 suggesting that Bcl-xL plays a role in tumor progression or response to therapy. To our knowledge, this is the first demonstration that a prosurvival Bcl-2 family member can promote resistance of tumor cells to chemotherapy in vivo. The results obtained here with Bcl-xL may also apply to Bcl-2, as both of these structurally related proteins are thought to share a common mechanism that inhibits apoptosis. 16 Significantly, expression of Bcl-2 and Bcl-xL in untreated primary tumors is often focal, with only a subset of the tumor cells expressing high levels of these proteins. 12-15,30 A prediction from our results is that chemotherapy would select for tumor clones that overexpress Bcl-2 or Bcl-xL. Consistent with this hypothesis, the percentage of tumor cells that express Bcl-2 and/or the intensity of Bcl-2 staining increases after chemotherapy in primary tumors, 12,13,19,20 and selection of SCC 25 squamous carcinoma cells for resistance to chemotherapy in vitro is associated with expression of Bcl-xL. 31
An important observation derived from these studies is the lack of correlation between the ability of Bcl-xL to affect clonogenic survival in vitro and tumor growth in vivo after chemotherapy. This was particularly observed with the response of SCK tumor cells to 5-fluorouracil. In vivo, tumor cells receive signals from locally produced growth and survival factors, as well as from stromal and cell-to-cell interactions. Because proteins like Bcl-xL are thought to regulate an intracellular survival threshold critical for the induction of apoptosis, these tissue signals are likely to play an important role in determining the apoptotic response in vivo. In addition, clonogenic ability in vitro and tumor growth in vivo require signals that promote both survival and cell cycle progression. Methotrexate and 5-fluorouracil induce both cell cycle arrest and apoptosis in tumor cells. 32 Bcl-xL is thought to function primarily to block apoptotic signals. 16 Indeed, we have found that Bcl-xL-expressing tumors exhibit diminished apoptosis as compared to control tumors in animals treated with methotrexate and 5-fluorouracil. By inhibiting apoptosis, Bcl-xL may allow tumor cells to receive signals required to overcome cell cycle arrest induced by chemotherapy drugs. Consistent with this thesis, CD40 ligation and interleukin-4 have been shown to act with Bcl-2 to increase clonogenicity of lymphoma cells. 33 Thus, Bcl-xL may cooperate with signals provided by the tumor microenvironment to promote tumor growth in the presence of chemotherapy. Alternatively, to function, Bcl-xL may require signals that are provided in vivo, but not in vitro. These signals may include phosphorylation or other posttranscriptional modifications of Bcl-xL itself or that of Bcl-xL regulatory proteins. 16 For example, phosphorylation of BAD, an inhibitor of Bcl-xL, is regulated by growth factors and this regulation might be lacking or diminished in vitro. 34,35
Chemotherapy resistance in tumor cells is complex and involves many mechanisms that depend in part on the specific drug being used. The contribution of the apoptotic pathway to chemotherapy resistance in tumors is unclear. Our results with Bcl-xL suggest that inhibition of the apoptotic process is an important cause of chemotherapy resistance in vivo. The mechanism by which Bcl-xL and Bcl-2 promote chemotherapy resistance is different from classical drug-target interactions in that these survival proteins provide a multidrug resistance phenotype. The broad-range drug resistance effect of Bcl-2 and Bcl-xL can be explained by their ability to act at a common step in the apoptotic pathway. 16 In addition to prosurvival Bcl-2 family members, several proteins have been identified that inhibit apoptosis and are expressed in primary tumor cells. They include cellular FLIP 36 and survivin, 37,38 proteins that may inhibit apoptosis by selectively targeting caspases. 36-38 Thus, in addition to Bcl-2-related proteins such as Bcl-xL, it is likely that several antiapoptotic proteins contribute to chemotherapy resistance in primary tumors. Further work should provide insight into the contribution of these apoptosis inhibitors to clinical drug resistance, which may lead to the development of novel approaches to counter such resistance.
Acknowledgments
We thank M. Benedict, M. Clarke, S. Ethier, and L. del Peso for help and suggestions during these studies and C. W. Song for the gift of SCK cells.
Footnotes
Address reprint requests to Gabriel Nuñez, M.D., University of Michigan Medical School, 1500 East Medical Center Drive, 4215 CCGC 0938, Ann Arbor, MI 48109. E-mail: gabriel.nunez@umich.edu.
Supported in part by grant R01 CA64556–01 from the National Institutes of Health. G. N. was supported by a Research Career Development Award K04 CA64421–01 from the National Institutes of Health.
References
- 1.Endicott JA, Ling V: The biochemistry of P-glycoprotein-mediated multidrug resistance. Annu Rev Biochem 1989, 58:137-171 [DOI] [PubMed] [Google Scholar]
- 2.Ling V: Multidrug resistance: molecular mechanisms and clinical relevance. Cancer Chemother Pharmacol 1997, 40:3-8 [DOI] [PubMed] [Google Scholar]
- 3.Grant CE, Valdimarsson G, Hipfner DR, Almquist KC, Cole SPC, Deeley RG: Overexpression of multidrug resistance-associated protein (MRP) increases resistance to natural product drugs. Cancer Res 1994, 54:357-361 [PubMed] [Google Scholar]
- 4.Futscher BW, Abbaszadegan MR, Domann F, Dalton WS: Analysis of MRP mRNA in mitoxantrone-selected, multidrug-resistant human tumor cells. Biochem Pharmacol 1994, 47:1601-1606 [DOI] [PubMed] [Google Scholar]
- 5.Zaman GJR, Versantvoort CHM, Smit JJM, Eijdems EWHM, deHaas M, Smith AJ, Broxterman HJ, Mulder NH, de Vries EGE, Baas F, Borst P: Analysis of the expression of MRP, the gene for a new putative transmembrane drug transporter, in human multidrug resistant lung cancer cell lines. Cancer Res 1993, 53:1747-1750 [PubMed] [Google Scholar]
- 6.Barry MA, Behnke CA, Eastman A: Activation of programmed cell death (apoptosis) by cisplatin, other anticancer drugs, toxins, and hyperthermia. Biochem Pharmacol 1990, 40:2343-2362 [DOI] [PubMed] [Google Scholar]
- 7.Kaufmann SH: Induction of endonucleolytic DNA cleavage in human acute myelogenous leukemia cells by etoposide, camptothecin, and other cytotoxic anticancer drugs: a cautionary note. Cancer Res 1989, 49:5870-5878 [PubMed] [Google Scholar]
- 8.Miyashita T, Reed JC: Bcl-2 oncoprotein blocks chemotherapy-induced apoptosis in a human leukemia cell line. Blood 1993, 81:151. [PubMed] [Google Scholar]
- 9.Minn AJ, Rudin CM, Boise LH, Thompson CB: Expression of bcl-xL can confer a multidrug resistance phenotype. Blood 1995, 86:1903. [PubMed] [Google Scholar]
- 10.Simonian PL, Grillot DAM, Nuñez G: Bcl-2 and Bcl-xL can differentially block chemotherapy-induced cell death. Blood 1997, 90:1208-1216 [PubMed] [Google Scholar]
- 11.Yang E, Korsmeyer SJ: Molecular thanatopsis: a discourse on the Bcl-2 family and cell death. Blood 1996, 88:386-401 [PubMed] [Google Scholar]
- 12.Castle VP, Heidelberger KP, Bromberg J, Ou X, Dole M, Nuñez G: Expression of the apoptosis-suppressing protein bcl-2, in neuroblastoma is associated with unfavorable histology and N-myc amplification. Am J Pathol 1993, 143:1543-1550 [PMC free article] [PubMed] [Google Scholar]
- 13.Weller M, Malipiero U, Aguzzi A, Reed JC, Fontana A: Protooncogene bcl-2 gene transfer abrogates Fas/APO-1 antibody-mediated apoptosis of human malignant glioma cells and confers resistance to chemotherapeutic drugs and therapeutic irradiation. J Clin Invest 1995, 95:2633-2643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krajewska M, Krajewski S, Epstein JI, Shabaik A, Sauvageot J, Song K, Kitada S, Reed JC: Immunohistochemical analysis of bcl-2, bax, bcl-x, and mcl-1 expression in prostate cancers. Am J Pathol 1996, 148:1567-1576 [PMC free article] [PubMed] [Google Scholar]
- 15.Tu Y, Renner S, Xu F, Fleishman A, Taylor J, Weisz J, Vescio R, Rettig M, Berenson J, Krajewski S, Reed JC, Lichtenstein A: Bcl-x expression in multiple myeloma: possible indicator of chemoresistance. Cancer Res 1998, 58:256-262 [PubMed] [Google Scholar]
- 16.Adams JM, Cory S: The Bcl-2 protein family: arbiters of cell survival. Science 1998, 281:1322-1326 [DOI] [PubMed] [Google Scholar]
- 17.Eischen CM, Kottke TJ, Martins LM, Basi GS, Tung JS, Earnshaw WC, Leibson PJ, Kaufmann SH: Comparison of apoptosis in wild-type and Fas-resistant cells: chemotherapy-induced apoptosis is not dependent on Fas/Fas ligand interactions. Blood 1997, 90:935-943 [PubMed] [Google Scholar]
- 18.Sun X-M, MacFarlane M, Zhuang J, Wolf BB, Green DR, Cohen GM: Distinct caspase cascades are initiated in receptor-mediated and chemical-induced apoptosis. J Biol Chem 1999, 274:5053-5060 [DOI] [PubMed] [Google Scholar]
- 19.Campos L, Rouault JP, Sabido O, Oriol P, Roubi N, Vasselon C, Archimbaud E, Magaud JP, Guyotat D: High expression of bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Blood 1993, 81:3091. [PubMed] [Google Scholar]
- 20.Maung ZT, MacLean FR, Reid MM, Pearson AD, Proctor SJ, Hamilton PJ, Hall AG: The relationship between bcl-2 expression and response to chemotherapy in acute leukaemia. Br J Haematol 1994, 88:105-109 [DOI] [PubMed] [Google Scholar]
- 21.Hill ME, MacLennan KA, Cunningham DC, Vaughan Hudson B, Burke M, Clarke P, Di Stefano F, Anderson L, Vaughan Hudson G, Mason D, Selby P, Linch DC: Prognostic significance of Bcl-2 expression and bcl-2 major breakpoint region rearrangement in diffuse large cell non-Hodgkin’s lymphoma: a British National Lymphoma Investigation Study. Blood 1996, 88:1046-1051 [PubMed] [Google Scholar]
- 22.Hermine O, Haioun C, Lepage E, d’Agay MF, Briere J, Lavignac C, Fillet G, Salles G, Marolleau JP, Diebold J, Reyas F, Gaulard P: Prognostic significance of bcl-2 protein expression in aggressive non-Hodgkin’s lymphoma. Groupe d’Etude des Lymphomes de l’Adulte (GELA). Blood 1996, 87:265-272 [PubMed] [Google Scholar]
- 23.Lock RB, Stribiniskiene L: Dual modes of death induced by etoposide in human epithelial tumor cells allow Bcl-2 to inhibit apoptosis without affecting clonogenic survival. Cancer Res 1996, 56:4006-4012 [PubMed] [Google Scholar]
- 24.Yin DX, Schimke RT: Bcl-2 expression delays drug-induced apoptosis but does not increase clonogenic survival after drug treatment in HeLa cells. Cancer Res 1995, 55:4922-4928 [PubMed] [Google Scholar]
- 25.Kyprianou N, King ED, Bradbury D, Rhee JG: Bcl-2 overexpression delays radiation-induced apoptosis without affecting the clonogenic survival of human prostate cancer cells. Int J Cancer 1997, 70:341-346 [DOI] [PubMed] [Google Scholar]
- 26.Lin JC, Song SW: Influence of vascular thermotolerance on the heat-induced changes in blood flow, pO2, and cell survival in tumors. Cancer Res 1993, 53:2076-2080 [PubMed] [Google Scholar]
- 27.González-García M, Ballestero R, Ding L, Duan L, Boise L, Duan L, Boise LH, Thompson CB, Nuñez G: Bcl-xL is the major Bcl-x in RNA from expressed during murine development, and its product localizes to mitochondria. Development 1994, 120:3033-3042 [DOI] [PubMed] [Google Scholar]
- 28.Merino R, Grillot DAM, Simonian PL, Muthukkumar S, Fanslow WC, Bondada S, Nuñez G: Modulation of anti-IgM-induced B cell apoptosis by Bcl-xL and CD40: dissociation from cell cycle arrest and dependence on the avidity of the antibody-IgM receptor interaction. J Immunol 1995, 155:3830-3838 [PubMed] [Google Scholar]
- 29.Nishimura R, Nagao K, Miyayama H, Matsuda M, Baba K, Matsuoka Y, Yamashita H, Fukuda M, Higu Chi A: Apoptosis in breast cancer and its relationship to clinic pathological characteristics and prognosis. J Surg Oncol 1999, 71:226-234 [DOI] [PubMed] [Google Scholar]
- 30.Olopade OI, Adeyanju MO, Safa AR, Hagos F, Mick R, Thompson CB, Recant WM: Overexpression of Bcl-x protein in primary breast cancer is associated with high tumor grade and nodial metastases. Cancer J Sci Am 1997, 3:230-237 [PubMed] [Google Scholar]
- 31.Datta R, Manome Y, Taneja N, Boise LH, Weichselbaum R, Thompson CB, Slapak CA, Kufe D: Overexpression of Bcl-xL by cytotoxic drug exposure confers resistance to ionizing radiation-induced internucleosomal DNA fragmentation. Cell Growth Differ 1995, 6:363-370 [PubMed] [Google Scholar]
- 32.el Alaoui S, Lawry J, Griffin M: The cell cycle and induction of apoptosis in a hamster fibrosarcoma cell line treated with anti-cancer drugs: its importance to solid tumour chemotherapy. J Neurooncol 1997, 31:195-207 [DOI] [PubMed] [Google Scholar]
- 33.Walker A, Taylor ST, Hickman JA, Dive C: Germinal center-derived signals act with Bcl-2 to decrease apoptosis and increase clonogenicity of drug-treated human B lymphoma cells. Cancer Res 1997, 57:1939-1945 [PubMed] [Google Scholar]
- 34.del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nuñez G: Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science 1997, 278:687-689 [DOI] [PubMed] [Google Scholar]
- 35.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME: Akt phosphorylation of Bad couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91:231-241 [DOI] [PubMed] [Google Scholar]
- 36.Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer JL, Schroter M, Burns K, Mattmann C, Rimoldi D, French LE, Tschopp J: Inhibition of death receptor signals by cellular FLIP. Nature 1997, 388:190-195 [DOI] [PubMed] [Google Scholar]
- 37.Ambrosini G, Adida C, Altieri DC: A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med 1997, 3:917-921 [DOI] [PubMed] [Google Scholar]
- 38.Tamm I, Wang Y, Sausville E, Scudiero DA, Vigna N, Oltersdorf T, Reed JC: IAP-family protein survivin inhibits caspase activity and apoptosis induced by Fas (CD95), Bax, caspases, and anticancer drugs. Cancer Res 1998, 58:5315-5320 [PubMed] [Google Scholar]