

Abstract
Neuronal loss is prominent in Alzheimer’s disease (AD), and its mechanisms remain unresolved. Apoptotic cell death has been implicated on the basis of studies demonstrating DNA fragmentation and an up-regulation of proapoptotic proteins in the AD brain. However, DNA fragmentation in neurons is too frequent to account for the continuous neuronal loss in a degenerative disease extending over many years. Furthermore, the typical apoptotic morphology has not been convincingly documented in AD neurons with fragmented DNA. We report the detection of the activated form of caspase-3, the central effector enzyme of the apoptotic cascade, in AD and Down’s syndrome (DS) brain using an affinity-purified antiserum. In AD and DS, single neurons with apoptotic morphology showed cytoplasmic immunoreactivity for activated caspase-3, whereas no neurons were labeled in age-matched controls. Apoptotic neurons were identified at an approximate frequency of 1 in 1100 to 5000 neurons in the cases examined. Furthermore, caspase-3 immunoreactivity was detected in granules of granulovacuolar degeneration. Our results provide direct evidence for apoptotic neuronal death in AD with a frequency compatible with the progression of neuronal degeneration in this chronic disease and identify autophagic vacuoles of granulovacuolar degeneration as possible means for the protective segregation of early apoptotic alterations in the neuronal cytoplasm.
Nerve cell loss is extensive in brains of Alzheimer’s disease (AD) patients, and little is known about its cause, time course, and mechanisms. Recently, programmed cell death or apoptosis has been implicated as a mode of cell death in AD. Evidence stems mainly from studies linking AD-associated genes such as amyloid precursor protein (APP), presenilin 1, and presenilin 2 to the control of cell death. 1-4 Exposure of neuronal cultures to βA4, the amyloidogenic cleavage product of APP, induces apoptosis. 5,6 Apoptotic neuronal cell death has been observed in primary cultures of Down’s syndrome (DS) neurons, 7 and altered expression of apoptosis related proteins, such as Par-4, bak, bad, bax, bcl-2, p53, CPP32, and fas in AD brains was reported. 8-14 Histochemical techniques for the demonstration of fragmented DNA revealed large numbers of positive neurons in postmortem AD brains. 15-22 However, the majority of neurons with DNA fragmentation did not display the typical morphological features of apoptosis. 16,17,20,22 Furthermore, AD is a chronic disease with a protracted course over many years, but only few neurons can be expected to die at a given time point. Thus, it is likely that only extremely few neurons in an AD brain (less than 1:4000) show acute changes of apoptosis, yet other cells may present with alterations indicative of partial or compensatory damage. 23,24
To further investigate the extent and mode of neuronal death in AD, we performed immunohistochemical studies applying an antiserum against activated caspase-3. 25 Caspase-3 is considered the central apoptotic effector enzyme responsible for many of the biochemical and morphological features of apoptosis. 26,27 Activation of caspase-3 represents an irreversible step in the cell death pathway, and cells containing activated caspase-3 are prone to die. We found cytoplasmic immunoreactivity for activated caspase-3 in single apoptotic neurons in AD and DS, but not in controls. In addition, activated caspase-3 was found in cytoplasmic granules of granulovacuolar degeneration (GVD). 28 This may indicate that the activation of the apoptotic cascade in affected neurons is counteracted by the seclusion of damaged areas into autophagic vacuoles.
Materials and Methods
Tissue
Brain tissues from nine cases of clinically diagnosed and neuropathologically confirmed AD, four cases of DS, and seven age-matched controls without neurological disease were obtained at autopsy, fixed in buffered formalin, and routinely embedded in paraffin. In addition, one similarly processed case of infantile pontosubicular neuron necrosis was included as positive control for apoptotic neuronal cell death in routinely processed human autopsy tissue. 20,29,30 All AD cases fulfilled the quantitative neuropathological criteria for the diagnosis of AD according to Khatchaturian and the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). 31,32 For immunohistochemistry, temporal lobe sections including temporal isocortex, hippocampus, and entorhinal cortex were used.
Immunohistochemistry
Immunohistochemistry was performed applying an affinity-purified rabbit polyclonal antiserum reactive against human activated caspase-3 (CM1; IDUN Pharmaceuticals, La Jolla, CA). 25 In Western blots, this antiserum recognizes only the large (p18) subunit of processed caspase-3, but not the unprocessed zymogen or the processed small (p12) subunit. It is thus specific for the cleaved and thereby activated form of caspase-3. CM1 has previously been used to specifically detect apoptotic cells in vitro and in vivo. 25,33 Briefly, after deparaffinization and blocking with 10% fetal calf serum (FCS)/phosphate buffered saline (PBS), we applied the primary antibody CM1 at a concentration of 0.1 μg/ml in 10% FCS/PBS and permitted it to bind over night at 4°C. Control sections were incubated without primary antibody or with polyclonal antisera against irrelevant antigens. A standard avidin-biotin-peroxidase method with DAB as the chromogenic substrate was used to visualize antibody binding. 20 Alternatively, an alkaline phosphatase anti-alkaline phosphatase system (Dako, Glostrup, D) was used with Fast Red TR salt (Sigma) as chromogen. Pretreatment of the sections by microwaving 3× 5 minutes in 10 mmol/L citric acid buffer, pH 6.0, enhanced the sensitivity of CM1 for apoptotic cells but did not change the overall staining pattern. To assess the relationship between neuronal cell death and tau pathology, double-labeling experiments applying the mouse monoclonal antibody AT8 (1:1000; Innogenetics, Ghent, Belgium) against paired helical filament (PHF)-tau were performed. 34-36 An alkaline phosphatase anti-alkaline phosphatase technique with Fast Blue BB salt (Sigma, St. Louis, MO) as chromogen was used for AT8 stainings.
Results
Apoptotic Neurons in Pontosubicular Neuron Necrosis Selectively Contain Activated Caspase-3
In pontosubicular neuron necrosis, a disease condition resulting from perinatal hypoxia-ischemia, a large number of apoptotic neurons were present predominantly in pons and hippocampus. 20,29,30 Of these apoptotic neurons, 96.6% contained activated caspase-3. Caspase-3 immunoreactivity was found only in cells with chromatin condensation, nuclear fragmentation, and cytoplasmic condensation (Figure 1a) ▶ . No other cells or structures were stained with the CM1 antiserum in these sections.
Figure 1.
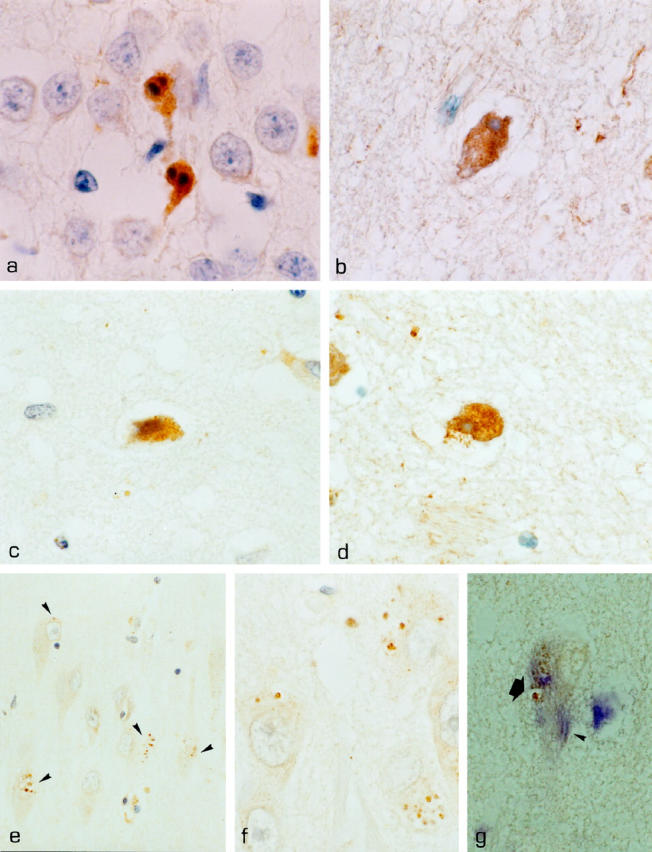
Activation of caspase-3 in AD and DS. a: Activated caspase-3 in apoptotic hippocampal granule cells in pontosubicular neuron necrosis. b: Subicular neuron of case DS 1 immunoreactive for activated caspase-3 with a condensed nucleus and shrunken cytoplasm indicative of apoptotic cell death. c: CM1-positive neuron in layer 2 of temporal isocortex in case AD 9. d: CM1-positive subicular neuron in case DS 1 displaying a condensed nucleus and cytoplasm. e and f: CA1 neurons of case DS 3; granules of GVD are immunolabeled with the CM1 antibody against activated caspase-3 (arrowheads), whereas other cytoplasmic compartments remain unstained. g: CA1 neuron in case DS 3 double-labeled with AT8; a small tangle (arrowhead) and diffuse AT8 positivity in a neuron with CM1-positive GVD (arrow). Activated caspase-3 is depicted in brown (DAB), AT8 in blue (Fast Blue). Nuclear counterstaining with hematoxylin, except for g, where no counterstaining was performed.Original magnifications, ×1000 (a-d, f, g) and ×400 (e).
Apoptotic Neuronal Death with Diffuse Cytoplasmic Activation of Caspase-3 Is an Exceptional Event in AD and DS
Temporal lobe sections of AD, DS, and age-matched controls were analyzed for the presence of CM1 immunopositive cells indicating the presence of activated caspase-3. Sections contained hippocampal allocortex, entorhinal cortex, and temporal isocortex. A total area per section of 171.18 ± 16.62 mm 2 in AD, 219.14 ± 33.76 mm 2 in DS, and 197.77 ± 22.09 mm 2 in age-matched controls was screened and the number of cells with distinct and strong cytoplasmic labeling with CM1 was determined. In 2/9 cases of AD and 1/4 cases of DS, cytoplasmic immunoreactivity for activated caspase-3 was detected in single CA1, subicular, and isocortical neurons: two CM1-positive neurons in the CA1 region were found in case AD 1, two CM1 positive cells in layer 2 of temporal isocortex in case AD 9, and three CM-1-immunopositive cells in the subiculum and in CA1 in case DS 1 (Figure 1, b ▶ -d, and Table 1 ▶ ). Unlike unstained neurons, neurons containing activated caspase-3 had shrunken, condensed, and sometimes fragmented nuclei and a condensed cytoplasm, thus displaying features typical for apoptosis (Figure 1, b ▶ -d). No CM1-immunoreactive neurons were found in age-matched controls. Apart from neurons, few apoptotic white matter cells identified as oligodendrocytes and few intra- and perivascular apoptotic leukocytes were immunoreactive for activated caspase-3 in AD, DS, and controls (Table 1) ▶ . The occurrence of apoptotic neurons in AD and DS was not related to postmortem time and cause of death in the respective cases (Table 1) ▶ .
Table 1.
Immunoreactivity for Activated Caspase-3 in AD, DS, and Controls
| Patient | Age/ sex | Cause of death | PMT (h) | Braak stage | No. of sections analyzed | Area per section (mm2) | No. of neurons immunopositive for activated caspase-3 | No. of white matter cells immunopositive for activated caspase-3 | % of neurons containing GVD (CA1 and subiculum) | GVD-containing neurons with GVD immunoreactive for activated caspase-3* |
|---|---|---|---|---|---|---|---|---|---|---|
| AD 1 | 65f | pulmonary embolism | 34 | V, C | 3 | 109.38 | 2 (CA1) | − | 19.7 | 22.4 (55/246) |
| AD 2 | 79m | pulmonary embolism | 16 | VI, C | 4 | 162.5 | − | − | 16.9 | 45.6 (52/114) |
| AD 3 | 75m | pneumonia | 45 | II, C | 4 | 106.25 | − | 1 | 12.55 | 45.6 (26/57) |
| AD 4 | 77f | myocardial infarction | 36 | II, B | 2 | 181.25 | − | − | 1.4 | 57.1 (4/7) |
| AD 5 | 79f | decubital ulcers | 27 | IV, C | 3 | 243.75 | − | − | 16.3 | 90.6 (250/276) |
| AD 6 | 87f | pulmonary embolism | 72 | V, C | 3 | 153.13 | − | − | 13 | 81.6 (208/255) |
| AD 7 | 70f | decubital ulcers | 37 | V, C | 2 | 196.88 | − | 1 | 0.65 | 82.4 (14/17) |
| AD 8 | 58f | sepsis | 27 | ammon’s horn sclerosis | 2 | 146.88 | − | 1 | 13 | 14.3 (4/28) |
| AD 9 | 86f | pulmonary embolism | 14 | IV, C | 2 | 240.63 | 2 neurons (layer 2 of | − | − (no HC) | − (no HC) |
| temporal isocortex) | ||||||||||
| DS 1 | 67f | pulmonary embolism | 6 | VI, C | 4 | 178.13 | 3 neurons (2 in | − | 23.1 | 2.1 (3/140) |
| subiculum; 1 in CA1) | ||||||||||
| DS 2 | 51f | pulmonary embolism | 3 | IV, C | 2 | 262.5 | − | 3 | 1.8 | 11.5 (6/52) |
| DS 3 | 64m | pulmonary embolism | n/a | VI, C | 1 | 146.87 | − | − | 31.5 | 90.9 (150/165) |
| DS 4 | 54m | myocardial infarction | n/a | VI, C | 1 | 289.06 | − | − | 30.4 | 52.9 (54/102) |
| CO 1 | 57m | pneumonia | 34 | − | 2 | 165.63 | − | − | 0 | 0/0 |
| CO 2 | 70m | respiratory failure (ALS) | 44 | − | 2 | 240.63 | − | − | 0 | 0/0 |
| CO 3 | 71f | myocardial infaction | 18 | − | 2 | 296.88 | − | 2 | 0/0 | |
| CO 4 | 73m | myocardial infarction | 6 | − | 2 | 193.75 | − | − | 1 | 0/1 |
| CO 5 | 84m | pulmonary edema | 10 | − | 3 | 175 | − | − | 2 | 2/2 |
| CO 6 | 65m | liver cirrhosis | 6 | − | 3 | 112.5 | − | − | 2 | 1/2 |
| CO 7 | 84f | myocardial infarction | 11 | few NFTs | 3 | 200 | − | − | − (no HC) | − (no HC) |
*Data for AD and DS cases are given as percentages, followed by the ratio of neurons containing caspade-3-immunoreactive GVD to neurons containing GVD. Figures for controls are absolute numbers of neurons (no percentages given). AD, Alzheimer’s disease; DS, Down’s syndrome; CO, control; HC, hippocampus.
We then sought to determine the frequency of CM1-positive apoptotic neurons per total number of neurons in cases AD 1, AD 9, and DS 1 by direct counting at a magnification of 400×. In case AD 1, 11,397 neurons were available for analysis; in case AD 9, 5746, and in case DS 1, 3415. The CM-1-positive neurons found in the respective AD and DS cases thus occurred at ratios of approximately 1:1100 to 1:5000 neurons showing changes characteristic of the apoptotic process. Double-labeling experiments revealed no neurons double-labeled for AT8 (PHF-tau) and cytoplasmic CM1.
Activated Caspase-3 Is Present in Granules of GVD
In addition to the cytoplasmic immunoreactivity observed in apoptotic neurons in AD and DS, activated caspase-3 was detected in granules of GVD (Figure 1, e ▶ -g). GVD was present in 11.69 ± 2.48% (all percentages are ± SE) of subicular and CA1 neurons in AD, and in 21.7 ± 6.89% of subicular and CA1 neurons in DS (Table 1) ▶ . Neurons harbored from one to more than 20 vacuoles with the characteristic basophilic core. GVD was scarce in subiculum and CA1 of age-matched controls, and a total of 5 neurons with GVD were found in the 7 control cases. Direct counting of CM1 immunoreactivity in GVD-positive neurons revealed that in AD, 54.87 ± 9.99% of GVD-positive neurons contained CM1 immunopositive granules. In DS, 39.4 ± 20.42% of GVD-positive neurons revealed granules stained with CM1. Neurons were counted positive if at least one of the granules was stained; however, in most cases, more than 50% of granules were immunoreactive. We noted a marked variability in the number of neurons containing CM1-immunopositive GVD, ranging from 2.14 to 90.9% in different cases of AD and DS. This may be attributable to different stages of GVD formation found in individual cases of AD and DS. CM1 immunoreactivity was restricted to the granulovacuoles of GVD and not present in other cytoplasmic compartments of the corresponding neurons. In particular, lipofuscin granules and neurofibrillary tangles were not labeled (Figure 1, e and f) ▶ . Furthermore, neurons with CM1-positive GVD did not display nuclear alterations indicative of apoptosis. Some of the few GVD vacuoles found in age matched controls equally contained immunoreactivity for activated caspase-3. Double-labeling experiments with AT8 revealed neurons harboring AT8-positive neurofibrillary tangles and CM1-positive GVD (Figure 1g) ▶ .
Neurons Bearing Abnormally Phosphorylated Tau Are More Likely to Also Harbor GVD
Neurons containing neurofibrillary tangles (NFTs) have been suggested to be prone to cell death in AD. 16,37 Given the presence of activated caspase-3 in GVD, we examined the relationship between AT8 positive inclusions (tangles and pretangle changes 38,39 ) and the concomitant presence of GVD in subicular and CA1 neurons in cases AD 1 to 8 and DS 1 to 4. Of neurons with abnormally phosphorylated cytoskeletal components as demonstrated by AT8 immunoreactivity, 25.99 ± 5.25% also harbored GVD as compared to only 4.69 ± 1.47% of neurons without AT8-positive inclusions (Figure 2) ▶ . GVD was much more likely to occur in AT8-positive (78.25% ± 7.76) than in AT8-negative (21.75% ± 7.76) neurons. The granules of GVD are not labeled by the AT8 antibody (Figure 1g) ▶ . 34
Figure 2.

Frequency of GVD in hippocampal neurons with and without AT8 positive cytoskeletal alterations (neurofibrillary tangles and pretangle changes). CA1 and subicular neurons were analyzed in cases AD 1–8 and DS 1–4. Among AT8-positive neurons, 25.99 ± 5.25% were found to contain GVD as compared to 4.69 ± 1.47% of AT8-negative neurons (P = 0.0012; Mann-Whitney U test), and 78.25 ± 7.76% of GVD was found in AT8-positive neurons.
Discussion
Apoptosis has been proposed as a dominant pathway of neuronal destruction in Alzheimer’s disease. 40 Familial AD-associated mutations of the presenilin 1 gene have been shown to sensitize neural cells to apoptotic cell death and render neurons from mutant mice susceptible to various inducers of cell death. 41-43 Cleavage of the presenilin 1 and 2 proteins generates anti-apoptotic C-terminal fragments. 44-46 Transfection of neuronal cells with mutants of the APP gene induces DNA fragmentation. 1 Furthermore, neuropathological studies in AD brains point toward a disturbed balance of pro- and antiapoptotic proteins: up-regulation of bax, c-jun, p53, fas, and Par-4 indicate the presence of a proapoptotic environment. 8,9,11,12,47 In addition, the local expression of cell cycle-related antigens indicates incomplete cell cycle activation in postmitotic AD neurons, possibly leading to their elimination by apoptosis. 48-52 A major argument for apoptotic cell death is further seen in the significantly elevated number of cells with DNA fragmentation in AD brains compared to normal controls. 15-22
There are, however, reservations about the notion that cells with DNA fragmentation are necessarily apoptotic. The incidence of cells with DNA fragmentation is, by several magnitudes, higher than would be expected in a disease with an average duration of 10 years. 16,23,24 Furthermore, cells with DNA fragmentation in AD do not show the classical morphological changes of apoptosis, ie, chromatin condensation and nuclear fragmentation, although these changes are readily detectable in human autopsy conditions where apoptosis occurs at a high frequency. 16,17,20,22,29 In a recent study, we found that the expression of an apoptosis-specific protein in AD is restricted to exceptional neurons in AD, which actually show typical alterations of apoptosis, and is absent in all other cells with DNA fragmentation. 20
In the present study, we used an antiserum against activated caspase-3 to unequivocally identify cells where activation of the apoptotic cell death program has taken place. Caspase activation is considered specific to the apoptotic process and defines an irreversible stage in the cell death process. Caspase-3 activation leads to the cleavage of so-called cell death substrates, such as fodrin, gelsolin, lamin, ICAD/DFF45, DNA-PK, PKδ, PARP, and many others. 26 This contributes to the disassembly of cell structures, the reorganization of the cytoskeleton, deficits in DNA repair and replication, and the cleavage of DNA into 180- to 200-bp fragments. Caspases have been shown to cleave the presenilins and APP during apoptotic cell death. 53-58 Recently, Masliah et al reported the presence of nonactivated caspase-3 in AD and control brains and established a correlation between the number of neurons with DNA fragmentation and the intensity of caspase-3 immunolabeling. 59 Gervais et al observed numerous labeled nonapoptotic CA3 neurons in AD using their antiserum against activated caspase-3. 58 We found activated caspase-3 in the cytoplasm of the vast majority of apoptotic neurons in pontosubicular neuron necrosis, demonstrating that detection of apoptotic neurons in human autopsy tissue is feasible, specific, and sensitive. In AD and DS, only exceptional neurons revealed strong cytoplasmic labeling for activated caspase-3; neurons immunopositive for activated caspase-3 displayed the full morphological spectrum of apoptosis. This is in contrast to studies using DNA fragmentation techniques 15-22 or immunohistochemistry for nonactivated caspase-3, 59 where numbers of positive neurons are high and concordance with apoptotic morphology is absent. Our data suggest that, indeed, some cells (though in exceptionally low numbers) die in the brains of patients with AD and DS at a given time point by apoptosis.
The incidence of approximately one apoptotic neuron in 1100 to 5000 neurons, found in this study, matches the recent estimate by Perry et al 23,24 and seems fairly realistic, given the short time required to complete apoptosis and the protracted course of AD. On the other hand, the significantly increased incidence of cells with DNA fragmentation, together with the proapoptotic phenotype of neurons in AD brains in comparison to age-matched controls, indicates that, overall, neurons in AD may be more vulnerable 20,42,43 and generally subject to a proapoptotic (micro)environment.
The finding of activated caspase-3 in the granulovacuoles of neurons with GVD, which were abundant in AD and DS but also present in age-matched controls in low incidence, may represent another footprint of the metabolic battle of neurons between proapoptotic signals and compensatory mechanisms. Granules of GVD are believed to be autophagic in origin. 60-63 The activation of caspase-3 in GVD could indicate the presence of cellular self-repair mechanisms, leading to the sequestration in autophagic vacuoles of cytoplasmic compartments where the potentially disastrous activation of proapoptotic enzymes has occurred. This notion is indirectly supported by elegant work on the ultrastructure of GVD by Okamoto et al demonstrating the sequestration of electron-dense cytoplasmic material by endoplasmic reticulum membranes, leading to the formation of vacuoles of GVD. 63 The presence of activated caspase-3 in granules of GVD may also be explained by an activation of caspases by lysosomal enzymes, 64-66 implying an activation of the apoptotic cascade in a subcellular compartment, thus providing relative safety to the affected neuron. It must also be considered that caspase-3 is activated during the process of lysosomal degradation of caspase-3-containing organelles or even exerts some function in the removal of damaged cellular constituents. However, evidence is accumulating that autophagy and apoptosis are, at least in some respect, related processes, and that autophagic forms of apoptosis exist. 67,68 Recently, a protein accumulating specifically in apoptotic cells was found to be related to a gene product essential for yeast autophagy. 69
NFTs have been considered a risk factor for neuronal death in AD. 16,37 In our study, no neurons double-labeled for diffuse cytoplasmic CM1 and AT8 (PHF-tau) could be identified. The number of CM1-positive neurons found in our cases of AD and DS, however, is too small to support any conclusions about the role of abnormally phosphorylated cytoskeletal components in neuronal death. The reported association between NFTs and neuronal death, together with our finding of activated caspase-3 in GVD, prompted us to examine the relationship between AT8 positivity and GVD in hippocampal AD and DS neurons. The 5.5 times higher risk that AT8-positive neurons (tangles and pretangle changes) will contain GVD and the vulnerability of certain brain regions (CA1, subiculum) to both NFTs and GVD 70,71 suggest an association between these two pathological alterations in AD neurons. So far, little is known about the relationship between neurofibrillary tangles and GVD. PHF-tau degradation (as well as formation) has been suggested to occur in GVD. 72,73 Immunohistochemical studies have shown the presence of neurofilament proteins, ubiquitin, tropomyosin, and various tau epitopes in both GVD and NFTs and have indicated the sequestration of undegraded tau in GVD.74 We found GVD in 26% of neurons with AT8 reactivity as compared to 4.7% without pathological tau protein. Together with our finding of activated caspase-3 in GVD, this supports the notion that relatively early damage, as manifested by abnormally phosphorylated cytoplasmic inclusions, may lead to the induction or activation of cell death-related compounds, eg, activated caspase-3, which are then sequestered into the membrane-bound vesicles of GVD, temporarily delaying neuronal demise.
Our work supports the notion that neuronal apoptosis occurs in the AD and DS brain, albeit at exceedingly low levels. The presence of a multitude of proapoptotic factors in AD together with the high amount of neurons with DNA fragmentation indicates, on the one hand, an ongoing proapoptotic challenge of AD neurons and, on the other hand, an enhanced vulnerability of AD neurons to a variety of noxious factors. The presence of activated caspase-3 in autophagic vacuoles of GVD may serve as evidence for neuronal mechanisms counteracting the apoptotic process in the AD brain.
Acknowledgments
We thank Helene Breitschopf, Marianne Leisser, Petra Tassotti, Angela Kury, and Jutta Wakley-Neuninger for expert technical assistance.
Footnotes
Address reprint requests to Prof. Dr. Hans Lassmann, Department of Neuroimmunology, Brain Research Institute, Schwarzspanierstrasse 17, A-1090 Vienna, Austria. E-mail: hans.lassmann@univie.ac.at.
Supported by European Community concerted action grant BMH4-CT96–0162.
References
- 1.Yamatsuji T, Matsui T, Okamoto T, Komatsuzaki K, Takeda S, Fukumoto H, Iwatsubo T, Suzuki N, Asami-Odaka A, Ireland S, Kinane B, Giambarella U, Nishimoto I: G-protein-mediated neuronal DNA fragmentation induced by familial Alzheimer’s disease-associated mutants of APP. Science 1996, 272:1349-1352 [DOI] [PubMed] [Google Scholar]
- 2.Guo Q, Sopher BL, Furukawa K, Pham DG, Robinson N, Martin GM, Mattson MP: Alzheimer’s presenilin mutation sensitizes neural cells to apoptosis induced by trophic factor withdrawal and amyloid β-peptide: involvement of calcium and oxyradicals. J Neurosci 1997, 17:4212-4222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vito P, Lacaná E, D’Adamio L: Interfering with apoptosis: Ca2+-binding protein ALG-2 and Alzheimer’s disease gene ALG-3. Science 1996, 271:521-524 [DOI] [PubMed] [Google Scholar]
- 4.Wolozin B, Iwasaki K, Vito P, Ganjei JK, Lacaná E, Sunderland T, Zhao B, Kusiak JW, Wasco W, D’Adamio L: Participation of presenilin 2 in apoptosis: enhanced basal activity conferred by an Alzheimer mutation. Science 1996, 274:1710-1713 [DOI] [PubMed] [Google Scholar]
- 5.Gschwind M, Huber G: Apoptotic cell death induced by β-amyloid 1–42 peptide is cell type dependent. J Neurochem 1995, 65:292-300 [DOI] [PubMed] [Google Scholar]
- 6.Li YP, Bushnell AF, Lee CM, Perlmutter LS, Wong SK: Beta-amyloid induces apoptosis in human-derived neurotypic SH-SY5Y cells. Brain Res 1996, 738:196-204 [DOI] [PubMed] [Google Scholar]
- 7.Busciglio J, Yankner BA: Apoptosis and increased generation of reactive oxygen species in Down’s syndrome neurons in vitro. Nature 1995, 378:776-779 [DOI] [PubMed] [Google Scholar]
- 8.Guo Q, Fu W, Xie J, Luo H, Sells SF, Geddes JW, Bondada V, Rangnekar VM, Mattson MP: Par-4 is a mediator of neuronal degeneration associated with the pathogenesis of Alzheimer’s disease. Nat Med 1998, 4:957-962 [DOI] [PubMed] [Google Scholar]
- 9.Su JH, Deng G, Cotman CW: Bax protein expression is increased in Alzheimer’s brain: correlations with DNA damage, bcl-2 expression, and brain pathology. J Neuropathol Exp Neurol 1997, 56:86-93 [DOI] [PubMed] [Google Scholar]
- 10.Su JH, Satou T, Anderson AJ, Cotman CW: Up-regulation of Bcl-2 is associated with neuronal DNA damage in Alzheimer’s disease. Neuroreport 1996, 7:437-440 [DOI] [PubMed] [Google Scholar]
- 11.Kitamura Y, Shimohama S, Kamoshima W, Matsuoka Y, Nomura Y, Taniguchi T: Changes of p53 in the brains of patients with Alzheimer’s disease. Biochem Biophys Res Commun 1997, 232:418-421 [DOI] [PubMed] [Google Scholar]
- 12.Nishimura T, Akiyama H, Yonehara S, Kondo H, Ikeda K, Kato M, Iseki E, Kosaka K: Fas antigen expression in brains of patients with Alzheimer-type dementia. Brain Res 1995, 695:137-145 [DOI] [PubMed] [Google Scholar]
- 13.Kitamura Y, Shimohama S, Kamoshima W, Ota T, Matsuoka Y, Nomura Y, Smith MA, Perry G, Whitehouse PJ, Taniguchi T: Alteration of proteins regulating apoptosis, Bcl-2, Bcl-x, Bax, Bak, Bad, ICH-1 and CPP32, in Alzheimer’s disease. Brain Res 1998, 780:260-269 [DOI] [PubMed] [Google Scholar]
- 14.Shimohama S, Tanino H, Fujimoto S: Changes in caspase expression in Alzheimer’s disease: comparison with development and aging. Biochem Biophys Res Commun 1999, 256:381-384 [DOI] [PubMed] [Google Scholar]
- 15.Dragunow M, Faull RL, Lawlor P, Beilharz EJ, Singleton K, Walker EB, Mee E: In situ evidence for DNA fragmentation in Huntington’s disease striatum, and Alzheimer’s disease temporal lobes. Neuroreport 1995, 6:1053-1057 [DOI] [PubMed] [Google Scholar]
- 16.Lassmann H, Bancher C, Breitschopf H, Wegiel J, Bobinski M, Jellinger K, Wisniewski HM: Cell death in Alzheimer’s disease evaluated by DNA fragmentation in situ. Acta Neuropathol (Berl) 1995, 89:35-41 [DOI] [PubMed] [Google Scholar]
- 17.Lucassen PJ, Chung WCJ, Kamphorst W, Swaab DF: DNA damage distribution in the human brain as shown by in situ end labeling; area-specific differences in aging and Alzheimer’s disease in the absence of apoptotic morphology. J Neuropathol Exp Neurol 1997, 56:887-900 [DOI] [PubMed] [Google Scholar]
- 18.Migheli A, Cavalla P, Marino S, Schiffer D: A study of apoptosis in normal and pathologic nervous tissue after in situ end-labeling of DNA strand breaks. J Neuropathol Exp Neurol 1994, 53:606-616 [DOI] [PubMed] [Google Scholar]
- 19.Smale G, Nichols NR, Brady DR, Finch CE, Horton Jr WE: Evidence for apoptotic cell death in Alzheimer’s disease. Exp Neurol 1995, 133:225–230 [DOI] [PubMed]
- 20.Stadelmann C, Brück W, Bancher C, Jellinger K, Lassmann H: Alzheimer disease: DNA fragmentation indicates increased neuronal vulnerability, but not apoptosis. J Neuropathol Exp Neurol 1998, 5:456-464 [DOI] [PubMed] [Google Scholar]
- 21.Su JH, Anderson AJ, Cummings BJ, Cotman CW: Immunohistochemical evidence for apoptosis in Alzheimer’s disease. Neuroreport 1994, 5:2529-2533 [DOI] [PubMed] [Google Scholar]
- 22.Troncoso JC, Sukhov RR, Kawas CH, Koliatsos VE: In situ labeling of dying cortical neurons in normal aging, and in Alzheimer’s disease: correlations with senile plaques and disease progression. J Neuropathol Exp Neurol 1996, 55:1134-1142 [DOI] [PubMed] [Google Scholar]
- 23.Perry G, Nunomura A, Smith MA: A suicide note from Alzheimer’s disease neurons? (commentary) Nat Med 1998, 4:897-898 [DOI] [PubMed] [Google Scholar]
- 24.Perry G, Nunomura A, Lucassen P, Lassmann H, Smith MA: Apoptosis and Alzheimer’s disease. (letter) Science 1998, 282:1268–1269 [DOI] [PubMed]
- 25.Srinivasan A, Roth KA, Sayers RO, Shindler KS, Wong AM, Fritz LC, Tomaselli K: In situ immunodetection of activated caspase-3 in apoptotic neurons in the developing nervous system. Cell Death Differ 1998, 5:1004-1016 [DOI] [PubMed] [Google Scholar]
- 26.Thornberry NA, Lazebnik Y: Caspases: enemies within. Science 1998, 281:1312-1316 [DOI] [PubMed] [Google Scholar]
- 27.Schulz JB, Weller M, Moskowitz MA: Caspases as treatment targets in stroke and neurodegenerative diseases. Ann Neurol 1999, 45:421-429 [DOI] [PubMed] [Google Scholar]
- 28.Simchowicz T: Histologische Studien über die senile Demenz. Histologische und histopathologische. Arbeiten über die Grosshirnrinde 1911, 4:267-444 [Google Scholar]
- 29.Brück Y, Brück W, Kretzschmar HA, Lassmann H: Evidence for neuronal apoptosis in pontosubicular neuron necrosis. Neuropathol Appl Neurobiol 1996, 22:23-29 [PubMed] [Google Scholar]
- 30.Friede RL: Ponto-subicular lesions in perinatal anoxia. Arch Pathol 1972, 94:343-354 [PubMed] [Google Scholar]
- 31.Khatchaturian ZS: Diagnosis of Alzheimer’s disease. Arch Neurol 1995, 42:1097-1105 [DOI] [PubMed] [Google Scholar]
- 32.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L: The consortium to establish a registry for Alzheimer’s disease (CERAD). II. Standardization of the neuropathological assessment of Alzheimer’s disease. Neurology 1991, 41:479-486 [DOI] [PubMed] [Google Scholar]
- 33.Chenghua G, Casaccia-Bonnefil P, Srinivasan A, Chao MV: Oligodendrocyte apoptosis mediated by caspase activation. J Neurosci 1999, 19:3043-3049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mercken M, Vandermeeren M, Lübke U, Six J, Boons J, Van de Voorde A, Martin J-J, Gheuens J: Monoclonal antibodies with selective specificity for Alzheimer tau are directed against phosphatase-sensitive epitopes. Acta Neuropathol (Berl) 1992, 84:265-272 [DOI] [PubMed] [Google Scholar]
- 35.Biernat J, Mandelkow EM, Schroter C, Lichtenberg-Kraag B, Steiner B, Berling B, Meyer H, Mercken M, Vandermeeren A, Goedert M, Mandelkow E: The switch of tau protein to an Alzheimer-like state includes the phosphorylation of two serine-proline motifs upstream of the microtubule binding region. EMBO J 1992, 11:1593-1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goedert M, Jakes R, Crowther RA, Six J, Lübke U, Vandermeeren M, Cras P, Trojanowski JQ, Lee VM-Y: The abnormal phosphorylation of tau protein at Ser-202 in Alzheimer disease recapitulates phosphorylation during development. Proc Natl Acad Sci USA 1993, 90:5066-5070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sheng JG, Mrak RE, Griffin WS: Progressive neuronal DNA damage associated with neurofibrillary tangle formation in Alzheimer disease. J Neuropathol Exp Neurol 1998, 57:323-328 [DOI] [PubMed] [Google Scholar]
- 38.Bancher C, Brunner C, Lassmann H, Budka H, Jellinger K, Wiche G, Seitelberger F, Grundke-Iqbal I, Iqbal K, Wisniewski HM: Accumulation of abnormally phophorylated τ precedes the formation of neurofibrillary tangles in Alzheimer’s disease. Brain Res 1989, 477:90-99 [DOI] [PubMed] [Google Scholar]
- 39.Braak E, Braak H, Mandelkow E-M: A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol (Berl) 1994, 87:554-567 [DOI] [PubMed] [Google Scholar]
- 40.Cotman CW, Su JH: Mechanisms of neuronal cell death in Alzheimer’s disease. Brain Pathol 1996, 6:493-506 [DOI] [PubMed] [Google Scholar]
- 41.Keller JN, Guo Q, Holtsberg FW, Bruce-Keller AJ, Mattson MP: Increased sensitivity to mitochondrial toxin-induced apoptosis in neural cells expressing mutant presenilin-1 is linked to perturbed calcium homeostasis and enhanced oxyradical production. J Neurosci 1998, 18:4439-4450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guo Q, Sebastian L, Sopher BL, Miller MW, Ware CB, Martin GM, Mattson MP: Increased vunerability of hippocampal neurons from presenilin-1 mutant knock-in mice to amyloid β-peptide toxicity: central roles of superoxide production and caspase activation. J Neurochem 1999, 72:1019-1029 [DOI] [PubMed] [Google Scholar]
- 43.Guo Q, Fu W, Sopher BL, Miller MW, Ware CB, Martin GM, Mattson MP: Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin-1 mutant knock-in mice. Nat Med 1999, 5:101-106 [DOI] [PubMed] [Google Scholar]
- 44.Vito P, Ghayur T, D’Adamio L: Generation of anti-apoptotic presenilin-2 polypeptides by alternative transcription, proteolysis, and caspase-3 cleavage. J Biol Chem 1997, 272:28315-28320 [DOI] [PubMed] [Google Scholar]
- 45.Walter J, Schindzielorz A, Grunberg J, Haass C: Phosphorylation of presenilin-2 regulates its cleavage by caspases and retards progression of apoptosis. Proc Natl Acad Sci USA 1999, 96:1391-1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vezina J, Tschopp C, Andersen E, Muller K: Overexpression of a C-terminal fragment of presenilin 1 delays anti-Fas induced apoptosis in Jurkat cells. Neurosci Lett 1999, 263:65-68 [DOI] [PubMed] [Google Scholar]
- 47.Anderson AJ, Su JH, Cotman CW: DNA damage and apoptosis in Alzheimer’s disease: colocalization with c-Jun immunoreactivity, relationship to brain area, and effect of postmortem delay. J Neurosci 1996, 16:1710-1719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arendt T, Holzer M, Gartner U: Neuronal expression of cycline dependent kinase inhibitors of the INK4 family in Alzheimer’s disease. J Neural Transm 1998, 105:949-960 [DOI] [PubMed] [Google Scholar]
- 49.McShea A, Harris PLR, Webster KR, Wahl AF, Smith MA: Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer’s disease. Am J Pathol 1997, 150:1933-1939 [PMC free article] [PubMed] [Google Scholar]
- 50.Nagy ZS, Esiri MM, Cato A-M, Smith AD: Cell cycle markers in the hippocampus in Alzheimer’s disease. Acta Neuropathol (Berl) 1997, 94:6-15 [DOI] [PubMed] [Google Scholar]
- 51.Ross ME: Cell division and the nervous system: regulating the cycle from neural differentiation to death. Trends Neurosci 1996, 19:62-68 [DOI] [PubMed] [Google Scholar]
- 52.Vincent I, Jicha G, Rosado M, Dickson DW: Aberrant expression of mitotic cdc2/cyclin B1 kinase in degenerating neurons of Alzheimer’s disease brain. J Neurosci 1997, 17:3588-3598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim TW, Pettingell WH, Jung YK, Kovacs DM, Tanzi RE: Alternative cleavage of Alzheimer-associated presenilins during apoptosis by a caspase-3 family protease. Science 1997, 277:373-376 [DOI] [PubMed] [Google Scholar]
- 54.Loetscher H, Deuschle U, Brockhaus M, Reinhardt D, Nelboeck P, Mous J, Grunberg J, Haass C, Jacobsen H: Presenilins are processed by caspase-type proteases. J Biol Chem 1997, 272:20655-20659 [DOI] [PubMed] [Google Scholar]
- 55.Barnes NY, Li L, Yoshikawa K, Schwartz LM, Oppenheim RW, Milligan CE: Increased production of amyloid precursor protein provides a substrate for caspase-3 in dying motoneurons. J Neurosci 1998, 18:5869-5880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weidemann A, Paliga K, Durrwang U, Reinhard FBM, Schuckert O, Evin G, Masters CL: Proteolytic processing of the Alzheimer’s disease amyloid precursor protein within its cytoplasmic domain by caspase-like proteases. J Biol Chem 1999, 274:5823-5829 [DOI] [PubMed] [Google Scholar]
- 57.van de Craen M, de Jonghe C, van den Brande I, Declercq W, van Gassen G, van Criekinge W, Vanderhoeven I, Fiers W, van Broeckhoven C, Hendriks L, Vandenabeele P: Identification of caspases that cleave presenilin-1 and presenilin-2: five presenilin-1 (PS1) mutations do not alter the sensitivity of PS1 to caspases. FEBS Lett 1999, 445:149-154 [DOI] [PubMed] [Google Scholar]
- 58.Gervais FG, Xu D, Robertson GS, Vaillancourt JP, Zhu Y, Huang JQ, LeBlanc A, Smith D, Rigby M, Shearman MS, Clarke EE, Zheng H, Van der Ploeg LHT, Ruffolo SC, Thornberry NA, Xanthoudakis S, Zamboni RJ, Roy S, Nicholson DW: Involvement of caspases in proteolytic cleavage of Alzheimer’s amyloid-β precursor protein and amyloidogenic Aβ peptide formation. Cell 1999, 97:395-406 [DOI] [PubMed] [Google Scholar]
- 59.Masliah E, Mallory M, Alford M, Tanaka S, Hansen LA: Caspase dependent DNA fragmentation might be associated with excitotoxicity in Alzheimer disease. J Neuropathol Exp Neurol 1998, 57:1041-1052 [DOI] [PubMed] [Google Scholar]
- 60.Wisniewski HM, Terry RD: Morphology of the aging brain, human and animal. Ford DM eds. Progress in Brain Research, 1973, vol 40.:pp 167-186 Elsevier, Amsterdam [DOI] [PubMed] [Google Scholar]
- 61.de Estable-Puig RF, Estable-Puig JF: Vacuolar degeneration in neurones of aging rats. Virchows Arch B Cell Pathol 1975, 17:337-346 [Google Scholar]
- 62.Mann DMA: Granulovacuolar degeneration in pyramidal cells of the hippocampus. Acta Neuropathol (Berl) 1978, 42:149-151 [DOI] [PubMed] [Google Scholar]
- 63.Okamoto K, Hirai S, Iizuka, Yanagisawa, Watanabe M: Reexamination of granulovacuolar degeneration. Acta Neuropathol (Berl) 1991, 82:340-345 [DOI] [PubMed] [Google Scholar]
- 64.Schotte P, van Criekinge W, van de Craen M, van Loo G, Desmedt M, Grooten J, Cornelissen M, de Ridder L, Vandekerckhove J, Fiers W, Vandenabeele P, Beyaert R: Cathepsin B-mediated activation of the proinflammatory caspase-11. Biochem Biophys Res Commun 1998, 251:379-387 [DOI] [PubMed] [Google Scholar]
- 65.Ishisaka R, Utsumi T, Yabuki M, Kanno T, Furuno T, Inoue M, Utsumi K: Activation of caspase-3 like protease by digitonin treated lysosomes. FEBS Lett 1998, 435:233-236 [DOI] [PubMed] [Google Scholar]
- 66.Monney L, Olivier R, Otter I, Jansen B, Poirier GG, Borner C: Role of an acidic compartment in tumor-necrosis-factor-α-induced production of ceramide, activation of caspase-3 and apoptosis. Eur J Biochem 1998, 251:295-303 [DOI] [PubMed] [Google Scholar]
- 67.Bursch W, Ellinger A, Török L, Parzefall W, Coulibaly S, Hochegger K, Schörkhuber M, Partik G, Marian B, Walker R, Sikorska M, Schulte-Hermann R: In vitro studies on subtypes, and regulation of active cell death. Toxicol In Vitro 1997, 11:579-588 [DOI] [PubMed] [Google Scholar]
- 68.Clarke PGH: Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol 1990, 181:195-213 [DOI] [PubMed] [Google Scholar]
- 69.Hammond EM, Brunet CL, Johnson GD, Parkhill J, Milner AE, Brady G, Gregory CD, Grand RJA: Homology between a human apoptosis specific protein and the product of APG5, a gene involved in autophagy in yeast. FEBS Lett 1998, 425:391-395 [DOI] [PubMed] [Google Scholar]
- 70.Tomlinson BE, Kitchener D: Granulovacuolar degeneration of hippocampal pyramidal cells. J Pathol 1972, 106:165-185 [DOI] [PubMed] [Google Scholar]
- 71.Ball MJ: Topographic distribution of neurofibrillary tangles and granulovacuolar degeneration in hippocampal cortex of aging and demented patients. A quantitative study. Acta Neuropathol (Berl) 1978, 42:73-80 [DOI] [PubMed] [Google Scholar]
- 72.Bondareff W, Wischik CM, Novak M, Roth M: Sequestration of tau by granulovacuolar degeneration in Alzheimer’s disease. Am J Pathol 1991, 139:641-647 [PMC free article] [PubMed] [Google Scholar]
- 73.Dickson DW, Ksiezwak-Reding H, Davies P, Yen SH: A monoclonal antibody that recognizes a phosphorylated epitope in Alzheimer neurofibrillary tangles, neurofilaments and tau proteins immunostains granulovacuolar degeneration. Acta Neuropathol (Berl) 1987, 73:254-258 [DOI] [PubMed] [Google Scholar]