

Abstract
Human transmissible spongiform encephalopathies (TSEs) or prion diseases are neurodegenerative disorders of infectious, inherited or sporadic origin and include Creutzfeldt-Jakob disease (CJD), Gerstmann-Sträussler-Scheinker disease (GSS), kuru and fatal familial insomnia (FFI). Clinicopathologic features of FFI differ markedly from other human TSEs. Previous studies demonstrated selective neuronal vulnerability of parvalbumin positive (PV+) GABAergic inhibitory interneurons in sporadic CJD and experimental TSEs. In this report we show uniform severe loss of PV+ neurons also in other TSEs such as GSS, kuru, new variant and familial CJD. In contrast, these neurons are mostly well preserved, or only moderately reduced, in FFI. Only PV+ neurons surrounded by isolectin-B4 positive perineuronal nets were severely affected in TSEs, suggesting a factor residing in this type of extracellular matrix around PV+ neurons as modulator for the selective neuronal vulnerability.
Transmissible spongiform encephalopathies (TSEs) or prion diseases are neurodegenerative disorders of infectious, inherited, or sporadic origin (for reviews see Refs. 1-3 ). Human TSEs include Creutzfeldt-Jakob disease (CJD), Gerstmann-Sträussler-Scheinker disease (GSS), kuru, and fatal familial insomnia (FFI). CJD is traditionally categorized into the most common sporadic as well as iatrogenic, familial (fCJD) and new variant (nvCJD) disease, which emerged in the past years in the United Kingdom. 4 Spongiform change, astrogliosis, and neuronal loss are the classical neuropathological triad of tissue lesions in TSEs. Whereas CJD, GSS, and kuru display overlapping clinical and neuropathological features, the clinical presentation and neuropathological hallmarks of FFI significantly differ from all other human TSEs. 5
Recent studies have observed selective vulnerability of distinct neuronal subsets in sporadic CJD, experimental CJD, and experimental scrapie. These reports showed early, severe and selective loss of parvalbumin positive (PV+) neurons, 6,7 which are a subset of GABAergic interneurons; correlation between tissue pathology and distribution of PV+ neurons, 8 aberrant neuropeptide Y mRNA induction, and decrease of neuropeptide Y Y2 receptor binding sites in the hippocampus of scrapie-infected mice. 9 These subset specific changes were suggested as possible basis for some of the clinical symptoms in TSEs. However, subset-specific pathology has not been investigated in human TSEs apart from sporadic CJD, such as GSS, kuru, FFI, fCJD, and nvCJD. In this study, we investigate PV+ neurons in these less common types of human TSEs.
Materials and Methods
We used formol-fixed, paraffin-embedded human brain tissues obtained at autopsy. Five FFI (three male and two female patients, age range 26–56 years, prion protein (PrP) gene (PRNP) genotype D178N; four homozygous at the polymorphic PRNP codon 129 for methionine (M), one unknown, including four cases from a new Austrian family reported previously 10,11 ), four GSS (two male and two female patients, age range 37–62 years, with PRNP genotype P102L (4/4) and M129M (3/4, one unknown); including two cases from the original Austrian family reported previously 12 ), four fCJD with the PRNP mutation E200K (two male and two female patients, age range 50–66 years, including one case with codon V129V reported previously 13 ), 1 kuru (16-year-old male, V129V; NIH case kindly provided by Dr. P. Liberski, Lodz, Poland, reported previously 14 ), and two nvCJD cases (51-year-old male and 27-year-old female, M129M; kindly provided by Dr. J. Ironside, Edinburgh, UK; reported previously 4 ). The following regions were investigated: frontal cortex, cerebellum, hippocampus, and adjacent temporal cortex. Hippocampal blocks from two nvCJD, two fCJD, and one FFI cases were not available for investigation. As controls, eight formol-fixed human autopsy brains (four male and four female patients, age range 27–66 years) with normal histology were used.
We performed immunohistochemistry for parvalbumin (PV) and lectin staining with isolectin-B4 (ILB4) to decorate perineuronal nets (PNNs) of extracellular matrix around PV+ neurons. 15 Two monoclonal antibodies against PV (PV-235, PARV-19, both diluted 1:5000, Sigma Chemical Co., St. Louis, MO) were used. Sections for PV labeling were boiled 10 minutes in target retrieval solution (pH 9.9) (Dako, Glostrup, Denmark). Lectin staining (ILB4-peroxidase labeled, 10 μg/ml Tris-buffered saline, 1 hour; Sigma) was performed on sections boiled for 10 minutes in citrate-buffered saline (pH 6.0). Both anti-PV antibodies showed a similar distribution of PV-like immunoreactivity, but the PV-235 immunostained sections had a better signal- to-background ratio and thus were mainly used for evaluation.
Quantification was made blindly by two of us (M. G. and J. W.). Counting by both showed very similar results; mean values were entered into final statistical evaluation. PV+ neurons were assessed in the same regions (frontal and temporal cortex) of control, FFI, GSS, fCJD, and nvCJD cases by counting immunopositive cell bodies in 3 representative fields with a ×20 objective. The mean of all 3 fields was entered into statistical evaluation. The total numbers of neurons were evaluated in H&E-stained sections of control, FFI, GSS, fCJD, and nvCJD cases by counting unequivocal neuronal cell bodies in the 3 fields examined with a ×40 objective. The mean of all 3 fields was entered into statistical evaluation. Neuronal cell bodies were well distinguishable from the homogeneously red cytoplasm of reactive astrocytes, which are abundant in TSEs. The mean regional numbers of all and of PV+ neurons were calculated and tested for significant differences in all groups (control, FFI, and other TSEs) using a Kruskal-Wallis one-way followed by a two-tailed Mann-Whitney U-test.
Results
Controls
Controls had a similar profile of region-specific distribution of PV+ neurons, with highest numbers in pre- and parasubiculum, frontal (Figure 1A) ▶ and temporal cortex, moderate numbers in entorhinal cortex and subiculum, and low numbers in hippocampal sectors CA1–4. In the cerebellum, Purkinje cells were strongly positive for parvalbumin (Figure 2C) ▶ . Neurons with ILB4+ PNNs were distributed in the cortex and hippocampal formation; in contrast, ILB4+ PNNs were not detectable around Purkinje cells (Figure 2A) ▶ . The number of ILB4+ PNNs was higher in the frontal cortex than in the temporal cortex.
Figure 1.
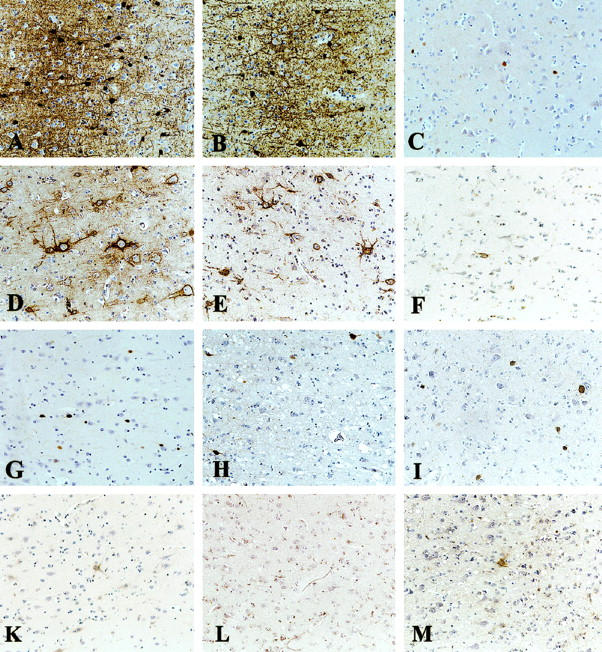
Parvalbumin immunostaining (A–C, G–I) and isolectin-B4 staining for perineuronal nets of extracellular matrix (D–F, K–M) in frontal cortex in control (A, D), FFI (B, E), GSS (C, F), nvCJD (G, K), fCJD (H, L), and kuru (I, M) brains. Severe loss of PV+ neurons and PNNs in GSS, nvCJD, fCJD, and kuru. In FFI, these neurons are better preserved than in all other TSEs. Original magnification, ×128.
Figure 2.
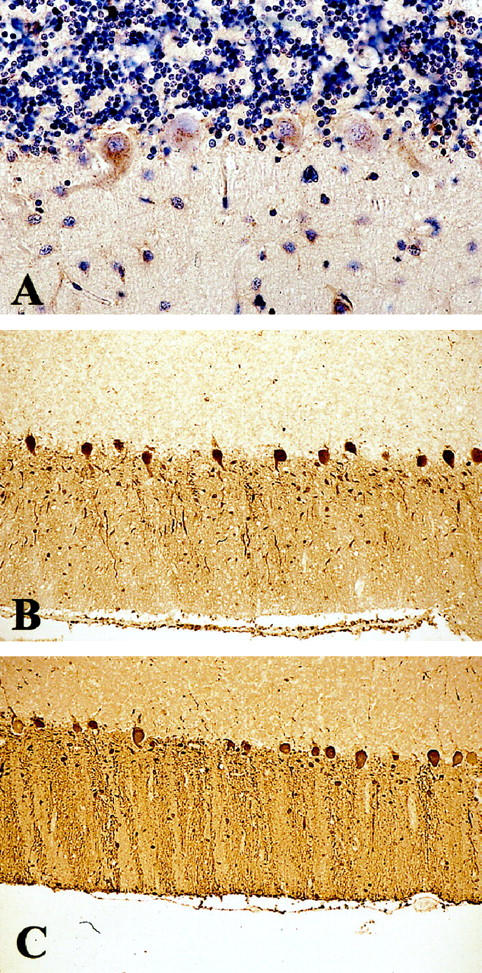
ILB4 staining (A) of a control cerebellum and parvalbumin immunostaining of a kuru (B) and control (C) cerebellum. There is lack of ILB4+ PNNs around PV+ Purkinje cells in normal brain; Purkinje cells are preserved in kuru. Original magnification: A, ×64; B and C, ×128.
Gerstmann-Sträussler-Scheinker Disease, Kuru, Familial, and New Variant Creutzfeldt-Jakob Disease
All cases showed TSE-specific pathology: spongiform change, astrogliosis, neuronal loss (in the frontal cortex ∼22% ± 2.3 SEM as compared to controls) and PrP deposition. These different TSE types showed a uniform subtotal to total loss of PV+ neurons and severe loss of PV neuropil staining in the frontal (∼71% ± 5.0 SEM, P ≤ 0.001; Figure 1C, G, H, I ▶ ; Figure 3 ▶ ), temporal (∼57.4% ± 16.7 SEM, P ≤ 0.02) and entorhinal cortex, pre- and parasubiculum and all regions of the hippocampus, irrespective of the local severity of spongiform change or astrogliosis. Only one GSS case showed a normal number of PV+ neurons in the temporal cortex. The loss of PV+ neuropil and synaptic buttons appeared more severe than the loss of PV+ cell bodies (Figure 1C, G, H, I) ▶ . The numbers and morphology of cerebellar PV+ neurons (Purkinje cells) and neuropil appeared well preserved (Figure 2B) ▶ . Neurons with ILB4+ PNNs were severely depleted in all investigated regions (Figure 1F, K, L, M) ▶ , confirming the loss of PV+ cells. The loss of ILB4+ PNNs seemed to be more severe than the loss of PV+ neurons (Figure 1F, K, L, M) ▶ .
Figure 3.

Graphic representation of the numbers of PV+ neurons in the frontal cortex. The columns depict the average numbers of PV+ neurons in the frontal cortex (P ≤ 0.001, Kruskal-Wallis test; *P < 0.02, compared to controls; **P < 0.01, compared to FFI or controls, respectively; Mann-Whitney U-test). The numbers of PV+ neurons in FFI differ from all other TSEs. Scale bars represent SEM; the y axis shows the average numbers of PV+ neurons in 3 representative fields (with a ×20 objective) of the frontal cortex.
Fatal Familial Insomnia
All cases showed slight but variable TSE-specific pathology: spongiform change (restricted to thalamus and one cortical focus each in three of four brains), astrogliosis, neuronal loss (in the frontal cortex ∼14% ± 1.0 SEM as compared to controls), and in two cases focal PrP deposition. All cases showed moderate loss (∼38% ± 8.1 SEM; P ≤ 0.02) of PV+ neurons in the frontal cortex (Figures 1B and 3) ▶ ▶ where the neuropil staining appeared only slightly reduced (Figure 1B) ▶ . There was a statistically significant difference between frontal cortical numbers of PV+ neurons in FFI and all other TSEs (P ≤ 0.005) (Figure 3) ▶ . In temporal (∼16.6% ± 7.25 SEM loss as compared to controls, P = 0.7) and entorhinal cortex, pre- and parasubiculum, all regions of the hippocampus and the cerebellum, PV+ neurons were well preserved. Morphology of PV+ neurons in all investigated regions was normal (Figure 1B) ▶ . ILB4+ PNNs appeared to be slightly reduced in the frontal cortex (Figure 1E) ▶ but were dense in all other investigated regions, especially in the temporal cortex.
Discussion
Neuronal subsets demarcated by calcium binding proteins (eg, parvalbumin, calbindin, calretinin) have been recently demonstrated to be selectively vulnerable or resistant in neurodegenerative disorders such as Alzheimer’s disease, 16,17 Parkinson’s disease, 18 Pick’s disease, 19 and others. Previous reports by our and other groups have shown a severe and selective loss of PV+ neurons in sporadic CJD, 6-8 which developed already very early in experimental TSEs. 7 This was suggested as a possible substrate of typical symptoms in these disorders such as myoclonus and characteristic EEG pattern. 6-8 The subset-specific neuronal vulnerability was recently corroborated by severe loss of PNNs in sporadic CJD 20 and experimental TSEs. 7 Here we describe loss of the PV+ subset of GABAergic neurons in other types of TSEs. The decrease of a second marker for the PV+ subset of GABAergic neurons, ILB4, confirms the loss of this neuronal subset. PV is localized in most cortical GABAergic neurons 21 (20 to 25% of all neurons in the frontal cortex are GABAergic 22 ), and the total neuronal loss varies from 14 to 22% in our counts. This result suggests that the PV+ subset forms the main group of neurons lost in TSEs. The disproportionate loss of PV+ neurons to the total neuronal loss reported here and in previous reports 7,8 suggests that damage of this particular subset is not consequence of the local severity of the pathological process, but more likely a primary event.
Clinically and neuropathologically, FFI clearly differs from all other human TSEs. Tissue pathology in FFI focuses on the thalamus, and there is little or no detectable tissue pathology in the cortex. 11 Surprisingly, FFI shows only moderate (as compared to all other human TSEs in our series) loss of PV+ neurons and neuropil staining in the frontal cortex, appearing to be the only unequivocal diffuse pathological change in the FFI cortex. This moderate loss of cortical PV+ neurons likely accounts for the slight total neuronal cortical loss in FFI. However, this neuronal subset is well preserved in the temporal cortex and adjacent hippocampus, in striking contrast to all other TSE types. This adds another element to the exceptional position of FFI among human TSEs. It is possible that some differences in clinical presentation of FFI, as compared with other TSEs, might be due to the better preservation of PV+ cortical neurons.
Cortical but not cerebellar PV+ neurons (Purkinje cells) 7 are selectively vulnerable in TSEs. Two attributes discriminate Purkinje cells from all other PV+ neurons: co-expression of another calcium binding protein, calbindin, 7 and lack of ILB4+ PNNs. The fact that PV+ neurons devoid from ILB4+ PNNs such as Purkinje cells are usually well preserved in TSEs and that loss of ILB4+ PNNs appeared to be even more severe than loss of PV+ neurons 7,20 suggests a factor residing in the extracellular matrix around cortical or hippocampal PV+ neurons as modulator for their vulnerability. It remains to be established how calcium binding proteins and the extracellular matrix interact to confer such selective neuronal vulnerability or resistance in TSEs, respectively.
Acknowledgments
We thank especially Dr. James Ironside, Edinburgh, UK for allowing us to use slides from two nvCJD brains. Ms. C. Karner is kindly acknowledged for excellent technical assistance, Dr. Peter Birner for helping with statistics, and Dr. Johannes Hainfellner for help and support.
Footnotes
Address reprint requests to Dr. H. Budka, Institute of Neurology, University of Vienna, AKH 04J, Währinger Gürtel 18–20, POB 48, A-1097 Wien, Austria. E-mail: h.budka@akh-wien.ac.at.
Supported by European Union Biomed-2 Concerted Action “Human Transmissible Spongiform Encephalopathies (Prion Diseases): Neuropathology and Phenotypic Variation” (Project Leader: H. Budka) and European Union Biomed-2 Shared Cost Action “Molecular Biology of Prion Diseases” (Project Leader: J. Collinge, London).
M. Guentchev and J. Wanschitz contributed equally to this work.
References
- 1.Horwich AL, Weissman JS: Deadly conformations–protein misfolding in prion disease. Cell 1997, 89:499-510 [DOI] [PubMed] [Google Scholar]
- 2.Prusiner SB, Scott MR, DeArmond SJ, Cohen FE: Prion protein biology. Cell 1998, 93:337-348 [DOI] [PubMed] [Google Scholar]
- 3.Prusiner SB: The prion diseases. Brain Pathol 1998, 8:499-513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Will R, Ironside J, Zeidler M, Cousens S, Estibeiro K, Alperovitch A, Poser S, Pocchiari M, Hofman A, Smith P: A new variant of Creutzfeldt-Jakob disease in the UK. Lancet 1996, 347:921-926 [DOI] [PubMed] [Google Scholar]
- 5.Parchi P, Petersen RB, Chen SG, Autilio-Gambetti L, Capellari S, Monari L, Cortelli P, Montagna P, Lugaresi E, Gambetti P: Molecular pathology of fatal familial insomnia. Brain Pathol 1998, 8:539-548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferrer I, Casas R, Rivera R: Parvalbumin-immunoreactive cortical neurons in Creutzfeldt-Jakob disease. Ann Neurol 1993, 34:864-866 [DOI] [PubMed] [Google Scholar]
- 7.Guentchev M, Groschup MH, Kordek R, Liberski PP, Budka H: Severe, early, and selective loss of a subpopulation of GABAergic inhibitory neurons in experimental transmissible spongiform encephalopathies. Brain Pathol 1998, 8:615-623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guentchev M, Hainfellner JA, Trabattoni GR, Budka H: Distribution of parvalbumin-immunoreactive neurons in brain correlates with hippocampal and temporal cortical pathology in Creutzfeldt-Jakob disease. J Neuropathol Exp Neurol 1997, 56:1119-1124 [DOI] [PubMed] [Google Scholar]
- 9.Diez M, Koistinaho J, DeArmond SJ, Groth D, Prusiner SB, Hokfelt T: Marked decrease of neuropeptide Y Y2 receptor binding sites in the hippocampus in murine prion disease. Proc Natl Acad Sci USA 1997, 94:13267-13272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Budka H, Almer G, Hainfellner JA, Brücke T, Jellinger K: The Austrian FFI cases. Brain Pathol 1998, 8:554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Almer G, Hainfellner JA, Brücke T, Jellinger K, Kleinert R, Bayer G, Windl O, Kretzschmar HA, Hill A, Sidle K, Collinge J, Budka H: Fatal familial insomnia: a new Austrian family. Brain 1999, 122:5-16 [DOI] [PubMed] [Google Scholar]
- 12.Hainfellner JA, Brantner-Inthaler S, Cervenakova L, Brown P, Kitamoto T, Tateishi J, Diringer H, Liberski PP, Regele H, Feucht M, Budka H: The original Gerstmann-Sträussler-Scheinker family of Austria: divergent clinicopathological phenotypes but constant PrP genotype. Brain Pathol 1995, 5:201-211 [DOI] [PubMed] [Google Scholar]
- 13.Hainfellner J, Parchi P, Kitamoto T, Jarius C, Gambetti P, Budka H: A novel phenotype in familial Creutzfeldt-Jakob disease: prion protein gene E200K mutation coupled with valine at codon 129 and type 2 protease resistant prion protein. Ann Neurol 1999, 45:812-816 [PubMed] [Google Scholar]
- 14.Hainfellner JA, Liberski PP, Guiroy DC, Cervenakova L, Brown P, Gajdusek DC, Budka H: Pathology and immunocytochemistry of a kuru brain. Brain Pathol 1997, 7:547-553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Celio MR: Perineuronal nets of extracellular matrix around parvalbumin-containing neurons of the hippocampus. Hippocampus 1993, 3:55-60 [PubMed] [Google Scholar]
- 16.Brady DR, Mufson EJ: Parvalbumin-immunoreactive neurons in the hippocampal formation of Alzheimer’s diseased brain. Neuroscience 1997, 80:1113-1125 [DOI] [PubMed] [Google Scholar]
- 17.Solodkin A, Veldhuizen S, Van-Hoesen G: Contingent vulnerability of entorhinal parvalbumin-containing neurons in Alzheimer’s disease. J Neurosci 1996, 16:3311-3321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ito H, Goto S, Sakamoto S, Hirano A: Calbindin-D28k in the basal ganglia of patients with Parkinsonism. Ann Neurol 1992, 32:543-550 [DOI] [PubMed] [Google Scholar]
- 19.Arai H, Noguchi I, Makino Y, Kosaka K, Heizmann CW, Iizuka R: Parvalbumin-immunoreactive neurons in the cortex in Pick’s disease. J Neurol 1991, 238:200-202 [DOI] [PubMed] [Google Scholar]
- 20.Belichenko P, Miklossy J, Belser B, Budka H, Celio M: Early destruction of the extracellular matrix around parvalbumin-immunoreactive interneurones in sporadic cases of Creutzfeldt-Jakob Disease. Neurobiol Dis 1999, 6:269-279 [DOI] [PubMed] [Google Scholar]
- 21.Celio MR: Parvalbumin in most γ-aminobutyric acid-containing neurons of the rat cerebral cortex. Science 1986, 231:995-997 [DOI] [PubMed] [Google Scholar]
- 22.Hornung J, Tribolet N: Chemical organization of the human cerebral cortex. Tracey D eds. Neurotransmitters in the Human Brain. 1995, :pp 41-60 Plenum Press, New York and London [Google Scholar]