

Abstract
Oligodendrocytes are a major target of the purported autoimmune response in multiple sclerosis (MS) lesions, but little is known about the mechanisms underlying their demise. Despite the expression of proapoptotic receptors, these cells are rarely seen to undergo apoptosis in situ. On the other hand, cytotoxic mediators present in MS lesions, such as tumor necrosis factor-α, are known to generate survival signals through the activation of the transcription factors NF-κB and c-jun. The aim of this study was to investigate in chronic active and silent MS lesions and control white matter the expression of c-jun, its activating molecule, JNK, as well as NF-κB complex and its inhibitor, IκB. By immunohistochemistry we found negligible reactivity for these molecules in control white matter and silent MS plaques. In active MS lesions, double-label immunohistochemistry with oligodendrocyte markers showed up-regulation of the nuclear staining for both NF-κB and JNK on a large proportion of oligodendrocytes located at the edge of active lesions and on microglia/macrophages throughout plaques. Oligodendrocytes showed no reactivity for IκB, which was predominantly confined to the cytoplasm of microglia/macrophages. We hypothesize that activation of these transcriptional pathways may be one mechanism accounting for the paucity of oligodendrocyte apoptosis reported in MS.
Multiple sclerosis (MS) is a demyelinating disease of the central nervous system (CNS) in which myelin and myelin-forming cells (oligodendrocytes) become the target of an inflammatory response, resulting in their depletion from the MS plaque. However, the mechanisms which regulate these events are poorly understood. 1-3 Among the cytotoxic mediators present in MS lesions and claimed to be involved in oligodendrocyte pathology, the cytokines tumor necrosis factor-α (TNF-α) and interleukin-1 (IL-1) are of particular interest because, in addition to their proinflammatory role and apoptotic effect on oligodendrocytes, 4 they are capable of activating alternative intracellular pathways (viz. the transcription factors NF-κB and c-jun), which may have cytoprotective effects on target cells. 5-10
The transcription factor NF-κB is formed by at least five cytoplasmic proteins (p50, p52, p65 (RelA), c-Rel, and RelB) located in the cytoplasm of most cells. The complex is physiologically inhibited by IκB molecules; activation of NF-κB involves the cytoplasmic degradation of IκB, allowing the translocation of NF-κB into the nucleus where Rel subunits can bind DNA sequences of target genes and initiate their transcription. 6,7 Genes regulated by NF-κB include major histocompatibility proteins, adhesion molecules, and the cytokines IL-2, IL-6, interferon-β, transforming growth factor-β, and TNF-α. 7,8 The nuclear protein and transcription factor c-jun represents a pivotal element in regulating the neuronal and glial response to injury. Induction of c-jun and its transcriptional activity involves a molecular cascade of kinases leading to phosphorylation in the nucleus of c-jun N-terminal kinases (JNK), which in turn activates its target, c-jun. 9,10 Recent studies have investigated the expression of NF-κB and its inhibitor in MS and in experimental autoimmune encephalomyelitis on the effector elements of local inflammation. 11,12 However, little is known about the role of these transcription factors in oligodendrocytes, the purported target of the autoimmune response in MS.
The aim of this study was to investigate whether the above transcription factors are involved in oligodendrocyte pathology in MS lesions. In particular, we examined the expression of molecules belonging to the NF-κB and c-jun pathways to assess whether their nuclear translocation and activation occurred on oligodendrocytes within MS lesions, a process that might explain the reported lack of apoptosis in these glial cells during chronic MS. 13
Materials and Methods
Tissue Samples
Early postmortem (4 to 12 hours) CNS tissue was studied from 11 subjects (mean age, 46 years) with a clinical diagnosis of chronic progressive MS. In total, 19 blocks containing lesions and normal appearing white matter were examined. Histopathologically, 4 cases displayed a predominance of chronic active lesions with hypercellular margins, ongoing demyelination with macrophage and lymphocyte infiltration, and an hypocellular demyelinated center with oligodendroglial cell depletion. In the remaining 7 cases, the majority of lesions were defined as chronic silent, based on an absence of inflammation and the presence of demyelinated centers. Brain tissue from 4 subjects (mean age, 57.9 years) with other neurological diseases (OND) was available for control purposes (1 case each of Alzheimer’s disease, stroke, amyotrophic lateral sclerosis, and olivopontocerebellar atrophy). Normal CNS tissue with no evident pathology came from 3 subjects (mean age, 59 years) succumbing to non-neurological conditions. All tissues were snap-frozen and embedded in OCT medium and stored at −80°C until use.
Immunohistochemistry
Frozen sections were air-dried and fixed in cold acetone for 10 minutes. After blocking with normal serum, sections were incubated with primary antibodies overnight at 4°C. Polyclonal antisera were used to identify the subunit p65 (C-20) of NF-κB and its inhibitor IκBα (C-15) (1:1600; Santa Cruz Biotechnology); in addition, a monoclonal antibody to the activated p65 subunit (ie, recognizing an epitope of p65 exposed when it is not complexed to IκB) was used (1:500; Boehringer Mannheim). The active, phosphorylated form of JNK was identified by the monoclonal antibody G-7, recognizing the pThr-183/pTyr-185 epitope of human JNK1 (1:800; Santa Cruz); c-jun was identified by the polyclonal antiserum c-jun/AP-1, recognizing a DNA binding domain of the protein (1:100; Santa Cruz). Oligodendrocytes were identified with antisera to the phenotypic markers myelin basic protein (MBP) (1:1000; Dakopatts), or carbonic anhydrase II (1:800; Chemicon), while resident microglia and astrocytes were reacted with monoclonal antibodies for CD68 and glial fibrillary acid protein (Dakopatts), respectively. Appropriate secondary biotinylated antibodies followed by the avidin-biotin-complex Elite reagent (Vector Labs) were applied and the reaction visualized with 3,3′-diaminobenzidine (DAB). For evaluation of the phenotype of JNK- and NF-κB-positive cells, double-staining with an oligodendrocyte marker was performed. For this, after the peroxidase immunoreaction for JNK or NF-κB was developed with DAB, antiserum to MBP was applied and detected with an anti-rabbit antibody coupled to alkaline phosphatase. Red substrate (Vector Labs) was used to visualize the immunoreaction. Negative controls included omission of the primary antibody and isotype-specific, irrelevant antibody.
Apoptosis Assay
Apoptotic events in active and silent MS sections were assessed using the terminal deoxynucleotidyl transferase-mediated fluorescein-conjugated deoxyuridine triphosphate nick end-labeling (TUNEL) technique (Boehringer Mannheim), as previously described. 13 After fixation with paraformaldehyde and permeabilization with Triton X-100, reaction mixture was added and the samples incubated for 60 minutes at 37°C. The reaction was visualized with an anti-fluorescein Ig conjugated with peroxidase and DAB. As negative controls, the enzyme or secondary antibody was omitted. The phenotype of TUNEL-positive cells was assessed by double immunostaining, as described above.
Results
In normal white matter from control cases, using monoclonal antibodies to the activated forms of JNK and NF-κB, immunoreactivity was not seen (Figure 1A) ▶ or showed very faint staining in scattered astrocytes. Polyclonal antibody to the inactivated form of p65 gave low level constitutive staining on most cell types, including oligodendrocytes. In parallel sections, IκB was localized to the cytoplasm of scattered microglia and astrocytes, but not to oligodendrocytes. Immunostaining for IκB and activated JNK and NF-κB subunits in white matter from OND cases was similar to that observed in normal CNS, while faint c-jun reactivity was evident on astrocytes (data not shown).
Figure 1.
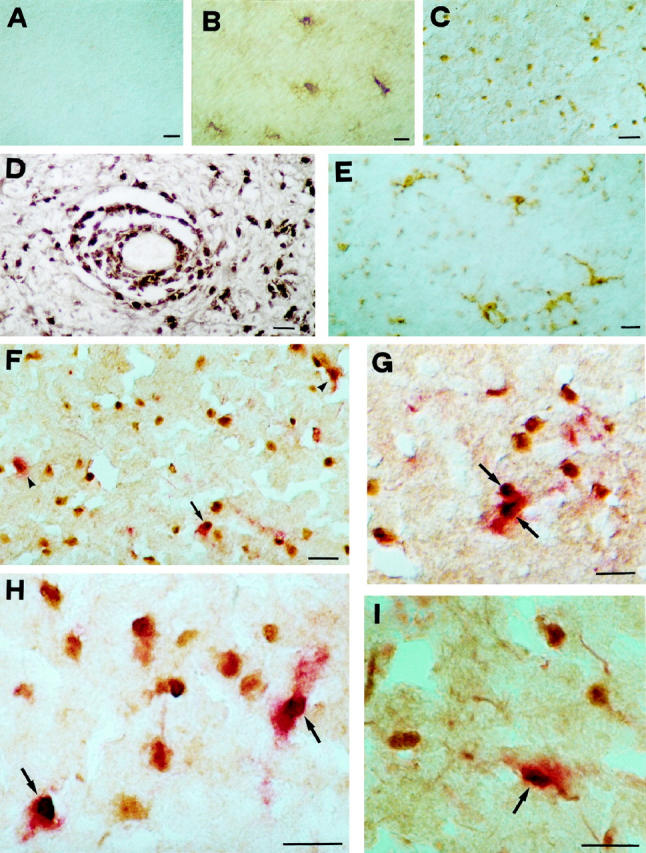
A to C: Immunoperoxidase staining for the activated NF-κB p65 subunit in normal CNS and MS lesions. A: No reactivity on glial cells is seen in white matter from control cases. B: Signal for NF-κB is visible in hypertrophic astrocytes in chronic silent MS lesions. C: At the edge of chronic active MS lesions, immunostaining for activated NF-κB shows nuclear reactivity on glial cells morphologically resembling oligodendrocytes or microglia. D: In addition to parenchymal cells, intense immunoperoxidase staining is detectable on perivascular, inflammatory elements. E: At variance with the pattern obtained for NF-κB, staining for its inhibitor, IκB, is detectable in the cytoplasm of microglia in the white matter surrounding MS plaques. F and H: Double-staining for activated p65 subunit of NF-κB (peroxidase, brown) and MBP (alkaline phosphatase, red) in chronic active MS lesions, reveals that a small proportion of NF-κB-positive nuclei present at the edge are surrounded by MBP-reactive cytoplasm (arrows), whereas most cells display no staining for MBP, probably representing microglial elements. About half of the oligodendrocyte population shows no NF-κB nuclear translocation (arrowheads). G and I: A pattern similar to NF-κB is detectable also for activated JNK at the edge of active MS lesions, where a proportion of MBP-positive oligodendrocytes (alkaline phosphatase, red; arrows) shows nuclear reactivity for JNK (peroxidase, brown). Scale bar, 30 μm.
At the edge of chronic silent MS lesions and in adjacent white matter, NF-κB reactivity was evident most commonly on hypertrophic astrocytes which displayed cytoplasmic and, occasionally, nuclear staining (Figure 1B) ▶ . In addition, at the edge of lesions, scattered cells with nuclear reactivity were present. By double immunohistochemistry with anti-MBP antiserum, no oligodendrocytes displaying NF-κB reactivity were found (data not shown). Immunoreactivity for c-jun and activated JNK molecules was similar to that observed for NF-κB, while IκB revealed a distinct signal in the cytoplasm of hypertrophic astrocytes and scattered microglia.
Within chronic active MS lesions and adjacent white matter, both NF-κB and c-jun/JNK reactivity was markedly up-regulated on glial cells and inflammatory elements (Figure 1, C and D) ▶ . Staining for the NF-κB inhibitor, IκB, was detectable in the cytoplasm of macrophages within plaques and on ramified microglial cells away from lesions (Figure 1E) ▶ . IκB staining was not evident on cells resembling oligodendrocytes. A positive correlation was observed between degree of inflammation and extent of glial cells with nuclear staining for NF-κB. As previously described for NF-κB proteins, 11 with antibodies for activated NF-κB and JNK subunits, microglia/macrophages were the predominant cell type showing nuclear reactivity throughout MS plaques. In this study, we focused on the area encompassing the outer lesion edge and on white matter immediately adjacent to active lesions where staining for the activated isoforms of both NF-κB and JNK was evident on cells morphologically resembling oligodendrocytes or microglia, appearances consistent with chronic signaling in this narrow zone. The pattern obtained for activated NF-κB, c-jun and phosphorylated JNK was similar both in terms of signal intensity and cellular distribution, and positively reactive cells displayed a prevalence of nuclear staining for molecules of the two pathways. With regard to NF-κB, results using a polyclonal antiserum to p65 and a monoclonal antibody recognizing its active form were comparable. However, the former showed both cytoplasmic and nuclear staining, while reactivity for the active form of NF-κB was preferentially intranuclear.
Due to the prevalence of nuclear staining of NF-κB and JNK proteins, the identification of reactive cells by purely morphological criteria was often difficult. For this reason, we performed double immunohistochemistry with anti-MBP antiserum to identify the phenotype of cells showing nuclear translocation of NF-κB or JNK (Figure 1, F–I) ▶ . At the outer lesion edge and in white matter immediately adjacent to active MS lesions, about 15 to 30% of cells positive for either NF-κB (Figure 1, F and H) ▶ or JNK (Figure 1, G and I) ▶ gave positive cytoplasmic staining for oligodendrocyte phenotypic markers and were identified morphologically as oligodendrocytes, while smaller MBP-negative cells were presumably microglial elements. About half of the MBP-positive oligodendrocytes around active lesions showed concomitant nuclear translocation of both NF-κB (Figure 1G) ▶ and JNK (Figure 1I) ▶ .
Regarding the other pathway that TNF-α may trigger, ie, cell death by apoptosis, we confirmed results previously obtained in chronic MS lesions, 13 whereby oligodendrocytes showed no evidence of DNA fragmentation, while scattered microglial and inflammatory elements were TUNEL-positive (data not shown).
Discussion
The aim of this study was to evaluate the expression in situ of molecules of the NF-κB and c-jun pathways in chronic MS lesions to determine whether these transcription factors might play a role in oligodendrocyte pathology in this purported autoimmune disease. Activation of both NF-κB and c-jun involves a cascade of intracellular events which lead activated isoforms in the nucleus to regulate the expression of several genes involved in cell survival. 5-10 By immunohistochemistry, evidence for up-regulation of both NF-κB and c-jun/JNK signals was found on microglia and oligodendrocytes in chronic active MS plaques, in comparison to silent MS lesions and control cases (OND and non-neurological conditions). Signal for both c-jun/JNK and NF-κB proteins was detected in the cytoplasm of astrocytes in silent MS lesions and control cases, while molecules of both pathways were up-regulated and localized by double immunohistochemistry in the nuclei of many oligodendrocytes and microglia at the outer edge of active MS lesions. No detectable levels of NF-κB inhibitor were found on these cells. In fact, IκB was up-regulated in the cytoplasm of microglia and scattered astrocytes at the edge of active MS lesions and in the surrounding white matter. Thus, the nuclear pattern for activated p65 NF-κB and JNK molecules, together with the cytoplasmic reactivity of IκBα observed in MS lesions, strongly suggested activation of these transcriptional pathways. Previous studies have underscored a role for NF-κB in MS and its animal model, experimental autoimmune encephalomyelitis 11,12 and in this regard, the present study confirmed the findings of Gveric et al, 11 who showed NF-κB activation in resident and infiltrating effector cells in MS lesions.
Although activation of NF-κB and c-jun/JNK pathways in glial cells may be triggered by a number of mechanisms, 7-10 the correlation observed herein between extent of transcriptional activation and degree of inflammation in MS plaques suggested that inflammatory mediators play a major role. Among elements present within active MS lesions which may trigger these pathways, IL-1 and TNF-α are probably prime candidates, since both cytokines and their receptors have been documented in MS plaques. 13-16 In this context, investigations in vitro have shown that TNF-α in concert with interferon-γ is capable of activating NF-κB transcriptional activity. 17 With regard to the pathogenesis of MS, these observations once again raise intriguing questions about the role of TNF-α, a matter of some debate over recent years. In fact, both TNF-α and its cognate, TNF-β (or lymphotoxin), are known to be major inducers of apoptotic cell death in several cell types, including oligodendrocytes. 4,18-20 In normal human CNS tissue, oligodendrocytes and microglia constitutively express the type II receptor of TNF-α 18 and both type I and II receptors are present on these glial cells in chronic MS plaques. 13 Thus, since both ligands and receptors of the TNF system are in place in MS lesions, one could hypothesize that apoptosis may play a major role in oligodendrocyte pathology in MS. However, morphological evidence of significant oligodendrocyte apoptosis has not yet been found in MS, 1,21 and search for DNA fragmentation in these cells has yielded conflicting results. 2,13,22,23 Therefore, although oligodendrocytes display pro-apoptotic receptors and are susceptible to TNF-mediated apoptotic death in vitro, there is no compelling evidence that they are depleted through apoptosis in MS. Several studies have demonstrated that, in addition to cytotoxicity, TNF-α can exert anti-apoptotic activity and can even prevent apoptosis through the NF-κB complex. 24-26 Overall, these observations support the dual ability of the TNF system to induce either apoptotic cell death or survival signals through the NF-κB pathway. In this context, our results appear to indicate that the MS plaque microenvironment is able to activate the NF-κB pathway in oligodendrocytes and microglia. We propose that in concert with other protective mechanisms such as bcl-2 13 and ciliary neurotrophic factor, 19,20 activation of NF-κB may exert anti-apoptotic effects and contribute to the absence of an apoptotic response by oligodendrocytes in MS.
As for the other TNF-inducible transcription factor, c-jun is known to play a critical role in neuronal and glial responses to injury, 10 although its final effect on glial cell fate remains controversial, with some investigators showing that induction of c-jun by nerve growth factor or TNF-α in oligodendrocytes correlates with apoptosis, 27,28 and others demonstrating that activation of c-jun by TNF-α has no effect on the apoptotic process. 5 Clearly, further experiments are needed to address the precise role of c-jun/JNK in oligodendrocyte biology. In the present study, we have found up-regulation of c-jun together with high signals for activated JNK in the nuclei of oligodendrocytes in chronic active MS lesions. The concomitant absence of oligodendroglial cell death would speak against a direct role of c-jun in the apoptotic process of these glial cells. In this regard, activation of JNK without apoptosis by TNF-α has been reported in astrocyte and oligodendrocyte cultures. 29
Taken in concert, we propose that up-regulation of the transcription factor NF-κB, and possibly c-jun, may help to explain the lack of convincing evidence of oligodendrocyte apoptosis in chronic MS lesions, despite the presence of proapoptotic molecules.
Acknowledgments
We thank Everett Swanson for expert technical assistance.
Footnotes
Address reprint requests to Dr. Bruno Bonetti, Sezione di Neurologia Clinica, Dipartimento di Scienze Neurologiche e della Visione, Universita’ di Verona, Ospedale Policlinico Borgo Roma, 37134 Verona, Italy. E-mail: bonetti@borgoroma.univr.it.
Supported in part by the Fondazione Italiana Sclerosi Multipla (97/R/17) to B.B., and the following to C.S.R.: NS 08952 and NS 11920 from the NINDS; NMSS RG 1001-I-9 from the National Multiple Sclerosis Society; the Sol Goldman Charitable Trust; and the Wollowick Family Foundation.
References
- 1.Raine CS: Demyelinating diseases. 3rd ed. Davis RL Robertson DM eds. Textbook of Neuropathology, 1997, :pp 627-714 Williams & Wilkins, Baltimore [Google Scholar]
- 2.Lucchinetti CF, Bruck W, Rodriguez M, Lassmann H: Distinct patterns of multiple sclerosis pathology indicate heterogeneity in pathogenesis. Brain Pathol 1996, 6:259-274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raine CS: The Dale E. McFarlin Memorial Lecture: The immunology of the multiple sclerosis lesion. Ann Neurol 1994, 36:561-572 [DOI] [PubMed] [Google Scholar]
- 4.Selmaj KW, Raine CS: Tumor necrosis factor mediates myelin and oligodendrocyte damage in vitro. Ann Neurol 1988, 23:339-346 [DOI] [PubMed] [Google Scholar]
- 5.Liu ZG, Hsu H, Goeddel DV, Karin M: Dissection of TNF receptor 1 effector functions: JNK is not linked to apoptosis while NFκB activation prevents cell death. Cell 1996, 87:565-576 [DOI] [PubMed] [Google Scholar]
- 6.Grilli MG, Chiu JJ, Lenardo MJ: NFκB, and Rel: participants in a multiform transcriptional regulatory system. Int Rev Cytol 1993, 143:1-62 [DOI] [PubMed] [Google Scholar]
- 7.Bauerle PA, Baltimore D: NFκB: ten years later. Cell 1996, 87:13-20 [DOI] [PubMed] [Google Scholar]
- 8.O’Neill LAJ, Kaltschmidt C: NFκB: a crucial transcription factor for glial and neuronal cell function. Trends Neurol Sci 1997, 20:252-258 [DOI] [PubMed] [Google Scholar]
- 9.Kyriakis JM, Banerjee P, Nikolakaki E, Dai T, Rubie E, Ahmad MF, Avruch J, Woodgett JR: The stress-activated protein kinase subfamily of c-jun kinases. Nature 1994, 369:156-160 [DOI] [PubMed] [Google Scholar]
- 10.Herdegen T, Skene P, Bahr M: The c-jun transcription factor: bipotential mediator of neuronal death, survival and regeneration. TINS 1997, 20:227-231 [DOI] [PubMed] [Google Scholar]
- 11.Gveric D, Kaltschmidt C, Cuzner ML, Newcombe J: Transcription factor NFκB and inhibitor IκBα are localized in macrophage in active MS lesions. J Neuropathol Exp Neurol 1998, 57:168-178 [DOI] [PubMed] [Google Scholar]
- 12.Kaltschmidt C, Kaltschmidt B, Lannes-Vieira J, Kreutzberg GW, Wekerle H, Bauerle PA: Transcription factor NF-κB is activated in microglia during experimental encephalomyelitis. J Neuroimmunol 1994, 55:99-106 [DOI] [PubMed] [Google Scholar]
- 13.Bonetti B, Raine CS: Multiple sclerosis: oligodendrocytes display cell death-related molecules in situ but do not undergo apoptosis. Ann Neurol 1997, 42:74-84 [DOI] [PubMed] [Google Scholar]
- 14.Hofman FM, Hinton DR, Johnson K, Merrill JE: Tumor necrosis factor identified in multiple sclerosis brain. J Exp Med 1989, 170:607-612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Selmaj KW, Raine CS, Cannella B, Brosnan CF: Identification of lymphotoxin and tumor necrosis factor in multiple sclerosis lesions. J Clin Invest 1991, 87:949-954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Woodroofe MN, Cuzner ML: Cytokine mRNA expression in inflammatory multiple sclerosis lesions: detection by non-radioactive in situ hybridization. Cytokine 1993, 5:583-588 [DOI] [PubMed] [Google Scholar]
- 17.Agresti C, Bernardo A, Del Russo N, Marziali G, Battistini A, Aloisi F, Levi G, Coccia EM: Synergistic stimulation of major histocompatibility complex class I and interferon regulatory factor 1 gene expression by interferon γ and tumor necrosis factor α in oligodendrocytes. Eur J Neurosci 1998, 10:2975-2983 [DOI] [PubMed] [Google Scholar]
- 18.Wilt SG, Milward E, Zhou JM, Nagasato K, Patton H, Rusten R, Griffin DE, O’Connor M, Dubois-Dalcq M: In vitro evidence for a dual role of tumor necrosis factor-α in human immunodeficiency virus type-1 encephalopathy. Ann Neurol 1995, 37:381-394 [DOI] [PubMed] [Google Scholar]
- 19.Louis JC, Megal E, Takayama S, Varon S: CNTF protection of oligodendrocytes against natural and tumor necrosis factor-induced death. Science 1993, 259:689-692 [DOI] [PubMed] [Google Scholar]
- 20.D’Souza S, Alinauskas K, Mc Crea E, Goodyer C, Antel JP: Differential susceptibility of human CNS-derived cell populations to TNF-dependent and independent immune-mediated injury. J Neurosci 1995, 15:7293-7300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prineas JW: The neuropathology of multiple sclerosis. Handbook of Clinical Neurology, vol. 47: Demyelinating Diseases. Edited by Koetsier JC. Amsterdam, Elsevier Science, 1985, pp 213–257
- 22.D’Souza SD, Bonetti B, Balasingam V, Cashman NR, Barker PA, Troutt AB, Raine CS, Antel JP: Multiple sclerosis: fas signalling in oligodendrocyte cell death. J Exp Med 1996, 184:2361-2370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bauer J, Wekerle H, Lassmann H: Apoptosis in brain-specific autoimmune disease. Curr Opin Immunol 1995, 7:839-843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Amer A, Baltimore D: An essential role for NFκB in preventing TNFα-induced cell death. Science 1996, 274:782-784 [DOI] [PubMed] [Google Scholar]
- 25.Wang CY, Mayo M, Baldwin A: TNF-, and cancer therapy-induced apoptosis potentiation by inhibition of NFκB. Science 1996, 274:784-787 [DOI] [PubMed] [Google Scholar]
- 26.Van Antwerp D, Martin S, Kafri T, Green DR, Verma IM: Suppression of TNFα-induced apoptosis by NFκB. Science 1996, 274:787-789 [DOI] [PubMed] [Google Scholar]
- 27.Yoon SO, Casaccia-Bonnefil P, Carter B, Chao MV: Competitive signaling between trkA and p75 nerve growth factor receptors determines cell survival. J Neurosci 1998, 18:3273-3281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ladiwala U, Lachance C, Simoneau S, Bhakar A, Barker PA, Antel JP: p75 neurotrophin receptor expression on adult human oligodendrocytes: signaling without cell death in response to NGF. J Neurosci 1998, 18:1297-1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang P, Miller BS, Rosenzweig SA, Bhat NR: Activation of c-jun N-terminal kinase/stress-activated protein kinase in primary glial cultures. J Neurosci Res 1996, 46:114-121 [DOI] [PubMed] [Google Scholar]