

Abstract
The prion protein (PrP) has a central role in the pathogenesis of transmissible spongiform encephalopathies (TSE). Accumulating evidence suggests that normal cellular PrP (PrPc) may be involved in copper homeostasis and modulation of copper/zinc superoxide dismutase (Cu/ZnSOD) activity in neurons. Hydrogen peroxide (H2O2) is a toxic reactive oxygen species generated through normal cellular respiration, and neurons contain two important peroxide detoxifying systems (glutathione pathway and catalase). To determine whether PrP expression affects neuronal resistance to H2O2, we exposed primary cerebellar granule neuron cultures derived from PrP knockout (PrP−/−) and wild-type (WT) mice to H2O2 for 3, 6, and 24 hours. The PrP−/− neurons were significantly more susceptible to H2O2 toxicity than WT neurons after 6 and 24 hours’ exposure. The increased H2O2 toxicity may be related to a significant decrease in glutathione reductase activity measured in PrP−/− neurons both in vitro and in vivo. This was supported by the finding that inhibition of GR activity with 1,3-bis(2-chloroethyl)-1-nitrosurea (BCNU) increased H2O2 toxicity in WT neurons over the same exposure period. The PrP toxic peptide PrP106–126 significantly reduced neuronal glutathione reductase activity and increased susceptibility to H2O2 toxicity in neuronal cultures suggesting that PrP toxicity in vivo may involve altered glutathione reductase activity. Our results suggest the pathophysiology of prion diseases may involve perturbed PrPc function with increased vulnerability to peroxidative stress.
Creutzfeldt-Jakob disease (CJD) is a transmissible spongiform encephalopathy (TSE) with some variants (nvCJD) zoonotically linked to bovine spongiform encephalopathy. 1-3 Etiologically, TSEs are unique, developing from both genetic and infectious or transmissible origins. 4 The disease process is characterized neuropathologically by vacuolar neuronal degeneration and accumulation of altered forms of prion protein (PrP) in the brain. 5,6 Normal cellular PrP (PrPc) can be expressed in neurons as either a glycosylphosphotidylinositol-anchored sialoglycoprotein or as a transmembrane molecule. 7,8 The molecular and cellular events leading to PrP accumulation appear to involve transformation of the soluble low β-sheet-containing PrPc to the infectious, relatively protease-resistant form with a high β-sheet content (PrPres). 9,10 Infectivity and toxicity are dependent on the expression of PrPc because PrP knockout (PrP−/−) mice are resistant to scrapie prions in vivo. 11 In vitro studies have demonstrated that a synthetic peptide corresponding to human PrP residues 106–126 (PrP106–126) is toxic to neurons, 12,13 with toxicity simultaneously dependent on PrPc expression, 12 the presence of microglia, 14 and possibly NMDA receptor-associated Ca2+ channel activation. 15-18
Although the normal function of PrPc is unknown, a possible role for PrP in modulating neuronal oxidative stress has been recently identified. Studies using PrP−/− neuronal cultures have shown that a lack of PrP expression increases susceptibility to superoxide anions (O2−) 14 or copper-mediated toxicity. 19 This appears to be related to decreased copper/zinc superoxide dismutase (Cu/ZnSOD) activity in PrP−/− cultured neurons and brain. 20 The exposure of wild-type neurons to PrP106–126 also reduced Cu/ZnSOD activity and predisposed cells to increased O2− toxicity. If PrPc is associated with antioxidant activity, then the interaction between PrP106–126/PrPres and PrPc could impair these antioxidative defenses (through loss of function) and predispose neurons to increased free radical toxicity.
Another important antioxidant pathway in neurons involves glutathione reduction (GSH) and oxidation. The reduced form of GSH is critical for detoxification of cellular hydrogen peroxide (H2O2), prevention of lipid and protein oxidation, 21 and the transport, detoxification, and sequestration of copper. 22,23 GSH metabolism and peroxidation appear to play a central role in a number of neurodegenerative disorders. 24,25 We therefore studied the relationship between PrPc expression on H2O2 toxicity and the glutathione pathway. As H2O2 is a major metabolic and excitotoxic byproduct, we wished to determine whether PrP−/− neurons were more vulnerable to peroxide toxicity than WT neurons. Our studies revealed a significant increase in H2O2 toxicity in PrP−/− neurons. This may be related to the decreased glutathione reductase (GR) activity observed in PrP−/− neurons in vitro and in vivo. The toxic PrP106–126 peptide also induced a significant reduction in GR activity in neuronal cultures, suggesting that altered GR activity may be relevent to PrPres toxicity in vivo. These studies support the important extensive role PrPc has in antioxidant activity in neurons and that perturbations to PrP function in CJD may predispose neurons to peroxidative damage.
Materials and Methods
Materials
PolyL-lysine and 3,[4,5 dimethylthiazol-2yl]−2,5 diphenyltetrazolium bromide (MTT) were from Sigma Chemical Co. (Australia). Glutamine, glucose and gentamicin sulfate were obtained from Gibco BRL (Australia). Fetal calf serum (FCS) and horse serum were from the Commonwealth Serum Laboratories (Sydney, Australia). 1,3-bis(2-chloroethyl)-1-nitrosurea (BCNU) was from Bristol Myers Squibb (UK).
Primary Cultures
PrP−/− mice were obtained from Dr C. Weissmann, Institut fur Molekularbiologie I (Zurich, Switzerland). The absence of PrP expression in the PrP−/− mice has been previously reported 26 and was confirmed by immunoblot analysis in our laboratory (unpublished observations). Wild-type (WT) control mice (C57BL6J × 129/Sv) correspond to genetically matched mice from which the PrP−/− mice were derived 26 and are of the same background as those used in previous studies of PrP−/− mice. 14,19,20,27 Primary cultures of cerebellar granule neurons (CGN) were established from PrP−/− and WT mice using previously described methods. 28 Cerebellar granule neurons were plated at 350,000 cells/cm 2 in Basal Medium Eagle (BME) (Gibco BRL) with 10% (v/v) FCS. Cultures were maintained at 37°C in 5% CO2. Cytosine arabinofuranoside Ara C (10 μmol/L) was added at day 2.
Measurement of Neuronal Cell Viability and Cell Death
Cell viability was determined by measuring the redox potential of the cells using the MTT assay as previously described. 28,29 Culture medium was replaced with 0.6 mg/ml MTT in control salt solution (CSS) for 2 hours. The supernatant was removed and cells solubilized with dimethyl sulfoxide. Aliquots of 100 μl were measured with a spectrophotometer at 570 nm. The MTT assay compared well to other cell viability assays such as Alamar Blue (Serotec, Sydney, Australia) and provided an accurate indication of neuronal viability under the conditions used.
Induction of Oxidative Stress
Peroxide toxicity was induced in day 4 CGN cultures by adding H2O2 diluted from 30% stock solution to culture media for 3, 6, and 24 hours, followed immediately by MTT assay of cell viability. Data represents the mean and standard error of the mean (SE) of experiments performed in at least 3 to 4 cultures measured in triplicate. To determine the effect of BCNU on H2O2 toxicity, BCNU was added 1 hour before addition of H2O2. A BCNU stock solution (1 mmol/L) was prepared in absolute ethanol and stored at −70°C before use.
Exposure of Neuronal Cultures to Amyloidogenic Peptides
PrP106–126 and a scrambled version of this peptide were synthesized as previously described. 30 Peptides were dissolved in fresh, serum-free culture medium at a concentration of 2 mg/ml before use. PrP106–26 displayed fibrillogenic and neurotoxic properties consistent with previous reports. 18,28 CGN cultures were exposed to 80 μmol/L PrP106–126 or 80 μmol/L scrambled PrP106–126. These concentrations did not alter cell viability after 24 hours’ exposure.
Measurement of Glutathione Peroxidase and Glutathione Reductase Levels in Primary Cultures
Determination of glutathione peroxidase (GPx) and GR levels in CGN cultures and brain tissue were performed using the respective assay kits per the manufacturer’s instructions (Oxis International Inc., R&D Systems, Minneapolis, MN). Protein estimation was performed with a BCA protein assay kit (Pierce, Rockford, IL) on aliquots of homogenized cultures before centrifugation. For GPx activity, control and H2O2-treated CGN cultures and 2-week-old brain tissue were homogenized for 10 seconds in a polytron homogenizer in ice-cold 50 mmol/L Tris-HCl containing 1 mmol/L 2β-mercaptoethanol, pH 7.5, centrifuged at 8500 × g for 10 minutes at 4°C, and the supernatants were stored at −70°C. For GR activity, control, H2O2- and peptide-treated CGN cultures and 2-week-old brain tissue were homogenized as above in ice-cold 60 mmol/L KPO4 buffer, pH 7.5, centrifuged, and stored at −70°C. Determination of GR activity was based on the equation: (A340 minutes/6220 mol/L−1 cm−1) × 5 = mU/ml, where A340 = change in absorbance per minutes at 340 nm, 6220 mol/L−1 cm−1 = the molar extinction coefficient of NADPH, and 5 = assay dilution. Determination of GPx activity was achieved by the equation: (A340 minutes/6220 mol/L−1 cm−1) × 16 = mU/ml, where 16 = assay dilution. Results for both GR and GPx assays were then adjusted to give data as mU/mg protein. One GR unit reduces 1 μmol of oxidized glutathione (GSSG) per minute at 25°C and pH 7.6. One GPx unit consumes 1 μmol NADPH per minute at 25°C and pH 7.6.
Statistical Analysis
Data represent the mean and SE of experiments performed in at least 3 to 4 cultures measured in triplicate. In all cases, comparisons of data were performed with analysis of variance and Newman-Keuls tests.
Results
PrP−/− Cerebellar Granule Neurons Are More Susceptible to H2O2 Toxicity than WT Neurons
To determine whether PrPc expression affects neuronal survival under our basal culture conditions, CGN cultures were grown from PrP−/− and WT mice for up to 8 days and cell viability was determined with the MTT assay. PrP−/− and WT cultures grown with the astrocyte growth inhibitor, Ara C, added at day 2 showed no significant difference in cell viability between day 1 and day 8 in vitro. (Cell viability in PrP−/− cultures was 97 ± 4.0% of the WT value after 8 days in vitro). These data indicate that under the conditions used, PrPc expression did not affect basal cell survival in neuronal cultures.
Previous studies have shown that PrP−/− neurons are more susceptible to superoxide toxicity. 14,20 To determine whether PrP−/− neurons are more susceptible to other forms of oxidative stress, we tested PrP−/− neurons against peroxidative toxicity. Four-day-old cultures of PrP−/− and WT CGN (3.5 × 10 5 cells/cm2) were exposed to different concentrations of H2O2 for 3, 6, and 24 hours and cell viability was measured using the MTT assay. The cell density used is consistent with previous studies on CGN oxidative toxicity. 31-33 The MTT assay measures cellular redox potential and is a sensitive indicator of neuronal cell viability. 29 The MTT assay was used in preference to cell death assays (such as the lactate dehydrogenase assay) as nonlethal changes to neuronal viability are an important indicator of neuronal dysfunction and increased susceptibility to oxidative insults. 28,34,35 H2O2 induced a significant decrease in neuronal viability after 3 hours’ exposure (Figure 1A) ▶ . At this time point there was no difference in cell viability between PrP−/− and WT cultures. However, after a 6- or 24-hour treatment with H2O2, cell viability was significantly lower (*P < 0.05, **P < 0.01) in the PrP−/− cultures as compared to WT treated with ≥40 μmol/L H2O2 (Figure 1, B and C) ▶ . At 100 μmol/L H2O2, PrP−/− neurons revealed approximately 40 and 60% lower viability than WT neurons after 6 and 24 hours’ treatment, respectively (**P < 0.01, Figure 1, B and C ▶ ). These findings clearly demonstrate that, in vitro, neurons lacking PrPc are significantly more susceptible to peroxide toxicity than WT neurons.
Figure 1.
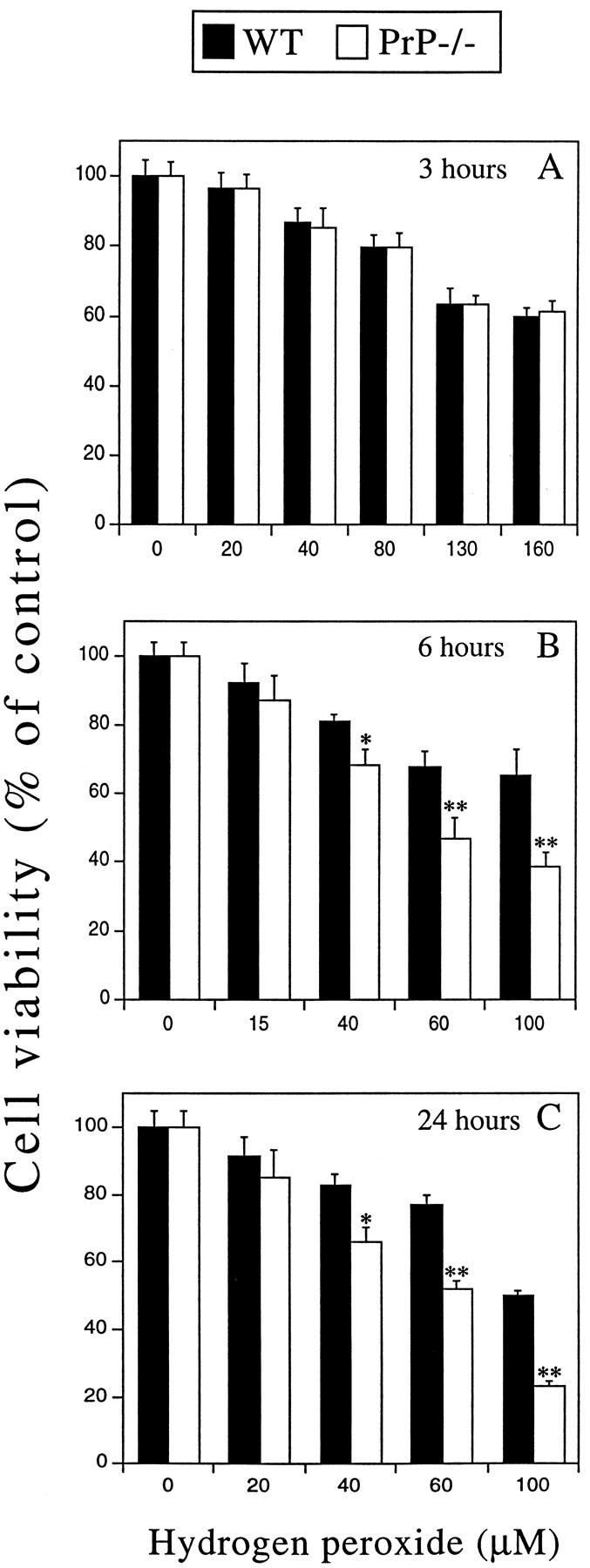
Effect of H2O2 on cell viability in PrP−/− and WT CGN cultures. A-C: Primary CGN cultures were grown for 4 days and exposed to H2O2 for 3 (A), 6 (B), and 24 hours (C). Untreated controls represent 100% cell viability. Cell viability was determined by MTT assay immediately after removal of H2O2. PrP−/− neurons revealed significantly lower cell viability than WT neurons exposed to H2O2 for 6 or 24 hours (*P < 0.05, **P < 0.01).
Glutathione Reductase Activity Is Significantly Reduced in PrP−/− CGN
Perturbations to the activity of the GSH-metabolizing enzymes GPx or GR can mediate increased sensitivity to H2O2 in neurons. 25 To determine whether increased susceptibility to H2O2 toxicity in PrP−/− neurons was related to perturbed GSH metabolism, we measured the activity of these two main GSH-associated enzymes, GPx and GR, in CGN cultures and cerebellum.
Assessment of GR levels in cultures under basal conditions revealed that PrP−/− neurons had approximately 17% lower GR activity than WT neurons at day 4 in vitro (*P < 0.05, Figure 2 ▶ , Control). To determine whether H2O2 exposure affected the GR activity in PrP−/− or WT neurons, 60 μmol/L H2O2 was added to cultures for 6 or 24 hours. In both WT and PrP−/− neurons, 60 μmol/L H2O2 did not significantly alter GR activity after 6 hours (Figure 2) ▶ . However, a 24-hour exposure to 60 μmol/L H2O2 caused a 31% lower GR activity in PrP−/− neurons compared to WT neurons (**P < 0.01, Figure 2 ▶ ). This treatment regimen (60 μmol/L H2O2 for 24 hours) resulted in 35% lower cell viability in PrP−/− compared to WT neurons (Figure 1C) ▶ . To determine whether these in vitro GR levels are consistent with in vivo GR levels, 2-week-old WT and PrP−/− mouse cerebella were assayed and GR activity was found to be approximately 30% lower in PrP−/− mice (**P < 0.01, Figure 2 ▶ ). These findings indicate that the difference in GR activity between WT and PrP−/− neurons in vitro is not a culture-derived artifact.
Figure 2.

GR activity in WT and PrP−/− neurons. Lysates from CGN cultures or 2-week-old cerebellum (cer) from WT or PrP−/− mice were used to determine GR activity. PrP−/− CGN revealed significantly lower GR activity than WT CGN under basal conditions (Control; *P < 0.05) and after 6 or 24 hours’ exposure to 60 μmol/L H2O2 (*P < 0.05, **P < 0.01). PrP−/− cerebellum revealed significantly lower GR activity than genetically and age-matched WT cerebellum (**P < 0.01).
Measurement of cellular GPx levels in primary CGN cultures revealed no significant differences in activity between WT and PrP−/− neurons under basal culture conditions at day 4 in vitro (Figure 3 ▶ , Control). Similarly, a 6- and 24-hour H2O2 exposure (60 μmol/L) caused no significant change in GPx activity in PrP−/− or WT cultures (Figure 3) ▶ . The activity of GPx was also measured in tissue extracts obtained from 2-week-old mouse cerebellum and was the same in both WT and PrP−/− mice (Figure 3) ▶ . These data indicate that PrPc expression is probably not an important modulator of neuronal GPx and is consistent with a recent report. 36
Figure 3.

GPx activity in WT and PrP−/− neurons. Lysates from CGN cultures or 2-week-old cerebellum (cer) from WT or PrP−/− mice were used to determine GPx enzyme activity. There was no significant difference in GPx activity between WT and PrP−/− neurons in control cultures or in cultures exposed to H2O2 (60 μmol/L) for 6 or 24 hours. No difference in GPx activity was observed between WT and PrP−/− cerebellum.
Inhibition of GR Activity Increases H2O2 Toxicity in CGN Cultures
To determine whether the reduced GR activity in PrP−/− neurons could account for the differences in cell viability in response to H2O2 toxicity, the GR-specific inhibitor BCNU was used. 37 WT and PrP−/− CGN were treated with different concentrations of BCNU and measured after 6 hours for GR activity. BCNU induced a dose-dependent reduction in enzyme activity in both PrP−/− and WT cultures (*P < 0.05, **P < 0.01, Figure 4A ▶ ). The susceptibility of the BCNU-treated cultures to H2O2 was tested by cotreating with 60 μmol/L H2O2. There was a significant decrease in cell viability with increasing BCNU concentration in both PrP−/− and WT cultures (*P < 0.01, Figure 4B ▶ ). BCNU alone had no effect on cell viability at concentrations up to 100 μmol/L. To examine whether a direct correlation exists between GR activity and H2O2 toxicity, we compared WT neurons exposed to 0.1 μmol/L BCNU with untreated PrP−/− neurons. Both cultures revealed a GR activity of approximately 27 mU/mg protein (Figure 4, A and C) ▶ and after exposure to 60 μmol/L H2O2 for 6 hours there was a similar decrease in cell viability (Figure 4C) ▶ . These data demonstrate a direct link between GR activity and peroxide toxicity and confirm that the lower GR activity in PrP−/− neurons can result in increased sensitivity to H2O2 toxicity.
Figure 4.
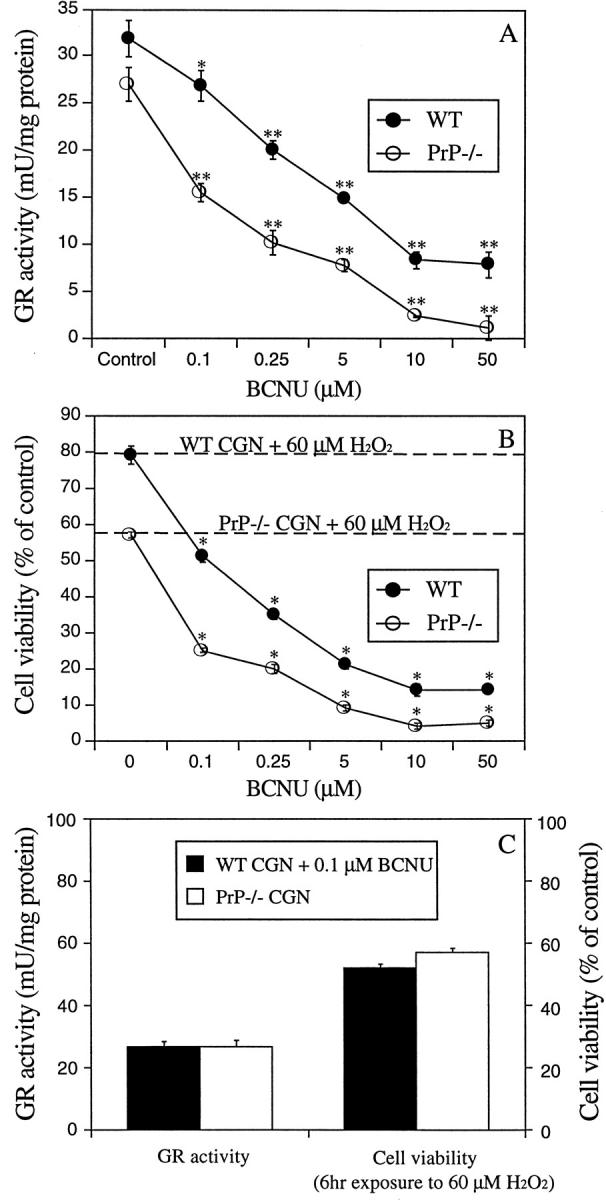
Effect of BCNU on H2O2 toxicity in CGN cultures. A: WT and PrP−/− CGN were treated with the GR inhibitor, BCNU (0.1 to 50 μmol/L) for 6 hours and cell lysates were used for measurement of GR activity. BCNU significantly inhibited GR activity at all concentrations used (*P < 0.05, **P < 0.01). B: WT and PrP−/− CGN were exposed to 60 μmol/L H2O2 for 6 hours alone (0 μmol/L BCNU and broken lines) or with 0.1 to 50 μmol/L BCNU (circles). Cell viability was determined with the MTT assay. Combined exposure to BCNU and H2O2 significantly decreased neuronal viability compared to H2O2 alone (*P < 0.01). BCNU alone was not toxic. C: Comparison of H2O2 toxicity in WT cultures treated with BCNU and untreated PrP−/− cultures. 0.1 μmol/L BCNU reduced WT neuronal GR activity to the same level as untreated PrP−/− neurons. After exposure to 60 μmol/L H2O2 for 6 hours there was no significant difference between cell viability in the WT and PrP−/− cultures. This demonstrates a close correlation between GR activity and H2O2 toxicity.
PrP106–126 Significantly Inhibits GR Activity and Increases H2O2 Toxicity in CGN
It has been demonstrated that the toxic PrP fragment PrP106–126 can reduce SOD activity and GSH levels in primary neurons. 20,38 We therefore tested the effect of PrP106–126 on neuronal GR activity. Exposure of WT and PrP−/− CGN to a nontoxic concentration of PrP106–126 (80 μmol/L) for 24 hours resulted in significantly lower GR activity compared to untreated controls (approximately 50 and 44% decrease in GR activity, respectively, *P < 0.01, Figure 5A ▶ ). Exposure of CGN to a nonfibrillogenic scrambled version of PrP106–126 failed to alter GR activity. Exposure of PrP106–126-treated cultures to 60 μmol/L H2O2 for 6 hours resulted in significantly increased toxicity compared to H2O2 alone (*P < 0.01, Figure 5B ▶ ). These results provide evidence that decreased GR activity may be relevent to the mechanism of PrP toxicity in vivo.
Figure 5.
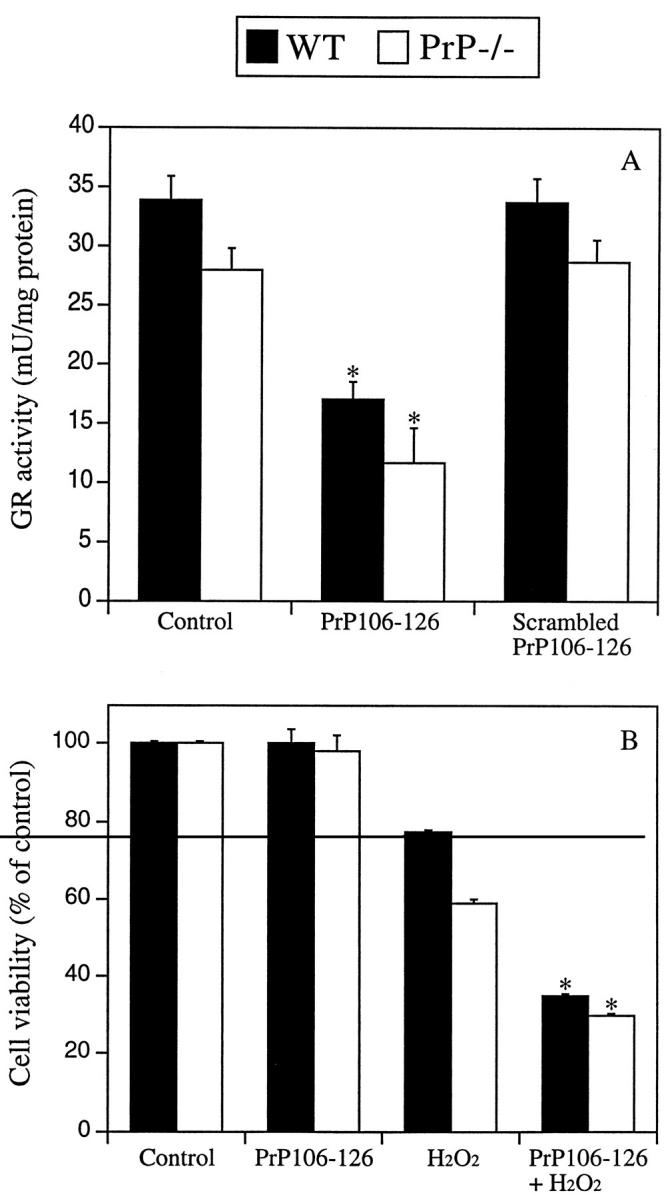
Effect of PrP106–126 on GR activity and H2O2 toxicity in CGN neurons. A: Neuronal cultures were exposed to 80 μmol/L PrP106–126 or scrambled PrP106–126 for 24 hours. No loss of cell viability was observed with either peptide after 24 hours’ exposure at these concentrations. Cell lysates were used for measurement of GR activity. PrP106–126-treated cultures revealed significantly lower GR activity than untreated cultures (*P < 0.01). Scrambled PrP106–126 had no effect on GR activity. B: PrP−/− and WT cultures were exposed to PrP106–126 for 24 hours followed by a 6-hour treatment with 60 μmol/L H2O2. Compared to H2O2 alone, PrP106–126 significantly decreased cell viability in both WT and PrP−/− cultures (*P < 0.01).
Discussion
Our studies indicate that PrPc expression appears to modulate neuronal GR activity levels. They also show that PrP−/− neurons are more susceptible to peroxide toxicity in the range of 40 to 100 μmol/L H2O2, which is within the reported range of H2O2 levels measured in rat brain during oxidative stress (25–160 μmol/L). 39 This is congruent with previous findings showing that a lack of PrPc expression increases susceptibility to O2− toxicity and lowers Cu/ZnSOD activity. 20 We found that PrP−/− CGN were up to 60% more susceptible to H2O2 toxicity than WT neurons compared to 20 to 30% lower viability of PrP−/− CGN exposed to the superoxide generator, xanthine/xanthine oxidase. 20 PrPc expression, therefore, appears to have an important role in modulating neuronal antioxidant homeostasis. These data also demonstrate important differences in the response of PrP−/− and amyloid precursor protein knockout (APP−/−) neurons to oxidative stress, because APP−/− and WT neurons revealed no differences in susceptibility to H2O2 toxicity. 28 As APP and PrPc are the respective parental protein molecules for generation of toxic amyloids in AD and CJD, our data highlight important divergences in the oxidative stress pathways in these neurodegenerative illnesses.
We believe the increased vulnerability of PrP−/− neurons to H2O2 is relevant to the pathophysiology of prion disease, because H2O2 toxicity is an important mediator of oxidative stress in neurons. 24 Peroxides play an important role in the pathology of several neurodegenerative disorders, as indicated by perturbations to GPx and GR activities and increased lipid peroxidation in Alzheimer’s disease, CJD, and amyotrophic lateral sclerosis. 25 Peroxides may also be involved in Aβ and PrP106–126 toxicity in vitro 14,21 and scrapie in vivo. 40 H2O2 is generated from many intracellular metabolic processes or through excitotoxicity 41 and is released by activated microglia. 42 H2O2 can also interact with copper to form the hydroxyl radical (OH·), one of the most destructive biological molecules generated in cells. 43 Because PrPc is involved in neuronal copper binding 44 and modulation of copper neurotoxicity, 19 interactions between H2O2 and copper via PrPc could promote neuronal degeneration in prion disease.
To counter H2O2 toxicity, neurons contain the GSH pathway and catalase. 41 The finding that PrP−/− neurons had lower GR, but not GPx, activity may be related to the increased sensitivity of PrP−/− neurons to H2O2. This finding is consistent with the lower SOD activity observed in PrP−/− neurons, which resulted in increased sensitivity to O2− toxicity. 20 The lack of significant differences in GPx levels between PrP−/− and WT neurons under basal conditions or after H2O2 exposure is also consistent with recent studies in PrP−/− and WT myoblasts, myotubes, and mouse brain. 36,45 This indicates that increased peroxide toxicity in PrP−/− neurons is not due to impaired GPx activity. However, the analogously lower GR activity in PrP−/− neurons in vitro and PrP−/− mouse cerebellum in vivo indicates that lower GR levels are unlikely to be irrelevant culture artifacts. Moreover, inhibition of GR activity in WT neurons with BCNU, a GR-specific inhibitor, 46-48 increased susceptibility to peroxide toxicity and correlated well with the GR level and toxicity in PrP−/− neurons (Figure 4C) ▶ . Furthermore, the PrP106–126 peptide reduced GR levels (this study) and SOD activity 20 in neurons and increased susceptibility to H2O2 toxicity, supporting the relevance of these findings to prion diseases.
A consequence of lower GR activity may be increased copper toxicity. Copper is a highly reactive metal and Cu(I) can combine with H2O2 to form OH·. 49 GSH is an important chelator of intracellular copper 49 and may contribute to transport of copper to Cu/ZnSOD and metalloproteins. 50 Lower GR activity would reduce the cellular capacity to regenerate glutathione, resulting in the destabilization of Cu homeostasis and increased OH· formation. In support of this, we have shown that GSH depletion dramatically increases Cu toxicity with no effect on Fe toxicity in cultured neurons. 35 Future studies will need to establish that the level of reduced GSH is lower in PrP−/− neurons due to the impaired GR activity. Perovic et al 38 have shown that PrP106–126 depletes neuronal GSH levels in vitro, a phenomenon that may be related to the reduced GR activity we observed in neurons treated with PrP106–126. Significantly, treatment of cultures with flupirtine to prevent GSH depletion also inhibited PrP106–126 neurotoxicity. 38 These studies strongly suggest that perturbations to GSH metabolism could be involved in neurodegeneration in prion disease. PrPres may induce a similar loss of GR and GSH function in neurons and therefore increase susceptibility to both O2− and H2O2. The fact that PrP106–126 inhibits Cu/ZnSOD 20 and GR activity in both WT and PrP−/− neurons indicates that the peptide can induce oxidative stress without direct interaction with PrPc. Only WT neurons are susceptible to PrP106–126-induced cell death, 12 suggesting that resistance to PrP106–126 toxicity by PrP−/− neurons may result in part from lower ROS generation in these cultures. This is consistent with the findings that microglia (which can release O2−, H2O2, and glutamate) are required for PrP106–126 toxicity in vitro and that WT microglia are more toxic than PrP−/− microglia when incubated with PrP106–126. 12
The evidence presented here and in previous studies 44,45 indicates that PrPc expression modulates neuronal antioxidant activity. This is supported by the reduced antioxidant activity (GR and Cu/ZnSOD) in neurons devoid of PrPc in vitro and in vivo and by the correlation between PrPc expression levels and Cu/ZnSOD activity in neurons overexpressing PrPc. 45,51 Whether PrPc directly interacts with GR and Cu/ZnSOD or acts upstream of antioxidant enzyme activation is still to be determined. The ability of PrP106–126 to inhibit GR and Cu/ZnSOD activity in PrP−/− neurons indicates that these antioxidant enzymes are affected by regulatory molecules downstream from PrPc (Figure 6) ▶ . Moreover, lower SOD and/or GR activity could also result from oxidative damage to the antioxidant enzymes. Interestingly, Barker et al 52 demonstrated that oxidative stress in rat brain significantly and specifically inhibited GR activity, an enzyme particularly susceptible to oxidative damage from peroxynitrite (ONOO−), a by-product of O2− and NO. Other studies have similarly shown that GR is particularly susceptible to oxidative damage from excessive free radical generation in vitro and in vivo. 53,54 However, the pathways linking PrPc expression to Cu/ZnSOD and GR activity are not understood and their study will be important in understanding how PrP modulates these key antioxidant enzymes.
Figure 6.

Schematic representation of Cu/ZnSOD and glutathione antioxidant pathways and putative interactions with PrP and copper. GR and Cu/ZnSOD activity are both reduced in PrP−/− neurons. ONOO− and OH· can damage GR and hence lower its activity (as indicated by dashed arrows). Pathways involving possible interactions with copper have been bolded. Cu/ZnSOD, copper/zinc superoxide dismutase; GR, glutathione reductase; GPx, glutathione peroxidase; H2O2, hydrogen peroxide; OH·, hydroxyl radical; ONOO−, peroxynitrite; O2−, superoxide radical.
The present study provides further evidence that PrPc modulates susceptibility to oxidative insults. Studies on animal models and cell lines infected with prions will help to determine whether PrPres accumulation results in perturbation of antioxidant activity and increases in oxidative stress leading to neuronal degeneration.
Acknowledgments
We thank Dr C. Weissmann (Institut für Molekularbiologie I, Zurich, Switzerland) for the PrP knockout mice, Denise Galatis for reading the manuscript, and Tricia Murphy for assistance with the mouse breeding.
Footnotes
Address reprint requests to Dr Roberto Cappai, Department of Pathology, The University of Melbourne, Parkville, Victoria 3052, Australia. E-mail: r.cappai@pathology.unimelb.edu.au.
Supported by grants from the National Health and Medical Research Council of Australia (to R. C. and C. L. M.) and The National Pituitary Hormones Advisory Council (to R. C. and S. J. C.).
References
- 1.Collinge J, Sidle KCL, Meads J, Ironside J, Hill AF: Molecular analysis of prion strain variation and the aetiology of a new variant of CJD. Nature 1996, 383:685-690 [DOI] [PubMed] [Google Scholar]
- 2.Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A, McCardle L, Chree A, Hope J, Birkett C, Cousens S, Fraser H, Bostock CJ: Transmissions to mice indicate that ’new variant’ CJD is caused by the BSE agent. Nature 1997, 389:498-501 [DOI] [PubMed] [Google Scholar]
- 3.Hill AF, Desbruslais M, Joiner S, Sidle KCL, Gowland I, Doey LJ, Lantos P, Collinge J: The same prion strain causes vCJD and BSE. Nature 1997, 389:448-450 [DOI] [PubMed] [Google Scholar]
- 4.DeArmond SJ, Prusiner SB: Etiology and pathogenesis of prion diseases. Am J Pathol 1995, 146:785-811 [PMC free article] [PubMed] [Google Scholar]
- 5.Gambetti P, Parchi P, Petersen RB, Chen SG, Lugaresi E: Fatal familial insomnia and familial Creutzfeldt-Jakob disease: clinical, pathological and molecular features. Brain Pathol 1995, 5:43-51 [DOI] [PubMed] [Google Scholar]
- 6.Hainfellner JA, Brantner-Inthaler S, Cervenakova L, Brown P, Kitamoto T, Tateishi J, Diringer H, Liberski PP, Regele H, Feucht M: The original Gerstmann-Straussler-Scheinker family of Austria: divergent clinicopathological phenotypes but constant PrP genotype. Brain Pathol 1995, 5:201-211 [DOI] [PubMed] [Google Scholar]
- 7.Stahl N, Borchelt DR, Hsiao K, Prusiner SB: Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 1987, 51:229-240 [DOI] [PubMed] [Google Scholar]
- 8.Hegde RS, Mastrianni JA, Scott MR, Defea KA, Tremblay P, Torchia M, DeArmond SJ, Prusiner SB, Lingappa VR: A transmembrane form of the prion protein in neurodegenerative disease. Science 1998, 279:827-834 [DOI] [PubMed] [Google Scholar]
- 9.Huang Z, Prusiner SB, Cohen FE: Scrapie prions: a three-dimensional model of an infectious fragment. Fold Des 1996, 1:13-19 [DOI] [PubMed] [Google Scholar]
- 10.Prusiner SB: Prions. Proc Natl Acad Sci USA 1998, 95:13363-13383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sailer A, Büeler H, Fischer M, Aguzzi A, Weissmann C: No propagation of prions in mice devoid of PrP. Cell 1994, 77:967-968 [DOI] [PubMed] [Google Scholar]
- 12.Brown DR, Herms J, Kretzschmar HA: Mouse cortical cells lacking cellular PrP survive in culture with a neurotoxic PrP fragment. Neuroreport 1994, 5:2057-2060 [DOI] [PubMed] [Google Scholar]
- 13.Forloni G, Bugiani O, Tagliavini F, Salmona M: Apoptosis-mediated neurotoxicity induced by β-amyloid and PrP fragments. Mol Chem Neuropathol 1996, 28:163-171 [DOI] [PubMed] [Google Scholar]
- 14.Brown DR, Schmidt B, Kretzschmar HA: Role of microglia and host prion protein in neurotoxicity of a prion protein fragment. Nature 1996, 380:345-347 [DOI] [PubMed] [Google Scholar]
- 15.Müller WE, Ushijima H, Schroder HC, Forrest JM, Schatton WF, Rytik PG, Heffner LM: Cytoprotective effect of NMDA receptor antagonists on prion protein (PrionSc)-induced toxicity in rat cortical cell cultures. Eur J Pharmacol 1993, 246:261-267 [DOI] [PubMed] [Google Scholar]
- 16.Whatley SA, Powell JF, Politopoulou G, Campbell IC, Brammer MJ, Percy NS: Regulation of intracellular free calcium levels by the cellular prion protein. Neuroreport 1995, 6:2333-2337 [DOI] [PubMed] [Google Scholar]
- 17.Florio T, Grimaldi M, Scorziello A, Salmona M, Bugiani O, Tagliavini F, Forloni G, Schettini G: Intracellular calcium rise through L-type calcium channels, as molecular mechanism for prion protein fragment 106–126-induced astroglial proliferation. Biochem Biophys Res Commun 1996, 228:397-405 [DOI] [PubMed] [Google Scholar]
- 18.Brown DR, Herms JW, Schmidt B, Kretzschmar HA: PrP, and β-amyloid fragments activate different neurotoxic mechanisms in cultured mouse cells. Eur J Neurosci 1997, 9:1162-1169 [DOI] [PubMed] [Google Scholar]
- 19.Brown DR, Schmidt B, Kretzschmar HA: Effects of copper on survival of prion protein knockout neurons and glia. J Neurochem 1998, 70:1686-1693 [DOI] [PubMed] [Google Scholar]
- 20.Brown DR, Schulz-Schaeffer WJ, Schmidt B, Kretzschmar HA: Prion protein-deficient cells show altered response to oxidative stress due to decreased SOD-1 activity. Exp Neurol 1997, 146:104-112 [DOI] [PubMed] [Google Scholar]
- 21.Mark RJ, Lovell MA, Markeberry WR, Uchida K, Mattson MP: A role for 4-hydoxynonenol, an aldehydic product of lipid peroxidation, in ion homeostasis and neuronal death by amyloid β-peptide. J Neurochem 1997, 68:255-264 [DOI] [PubMed] [Google Scholar]
- 22.Freedman JH, Ciriolo MR, Peisach J: The role of glutathione in copper metabolism and toxicity. J Biol Chem 1989, 264:5598-5605 [PubMed] [Google Scholar]
- 23.Vulpe CD, Packman S: Cellular copper transport. Ann Rev Nutr 1995, 15:293-322 [DOI] [PubMed] [Google Scholar]
- 24.Evans PH: Free radicals in brain metabolism and pathology. Br Med Bull 1993, 49:577-587 [DOI] [PubMed] [Google Scholar]
- 25.Bains JS, Shaw CA: Neurodegenerative disorders in humans: the role of glutathione in oxidative stress-mediated neuronal death. Brain Res Rev 1997, 25:335-358 [DOI] [PubMed] [Google Scholar]
- 26.Büeler H, Fischer M, Lang Y, Bluethmann H, Lipp H, DeArmond SJ, Prusiner SB, Aguet M, Weissmann C: Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 1992, 356:577-582 [DOI] [PubMed] [Google Scholar]
- 27.Tobler I, Gaus SE, Deboer T, Achermann P, Fischer M, Rülicke T, Moser M, Oesch B, McBride PA, Manson JC: Altered circadian activity rhythms and sleep in mice devoid of prion protein. Nature 1996, 380:639-642 [DOI] [PubMed] [Google Scholar]
- 28.White AR, Zheng H, Galatis D, Maher F, Hesse L, Multhaup G, Beyreuther K, Masters CL, Cappai R: Survival of cultured neurons from APP knockout mice against Alzheimer’s amyloid Aβ toxicity and oxidative stress. J Neurosci 1998, 18:6207-6217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu YB, Schubert D: Cytotoxic amyloid peptides inhibit cellular 3-(4,4-dimethylthiazoyl-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction by enhancing MTT formazan exocytosis. J Neurochem 1997, 69:2285-2293 [DOI] [PubMed] [Google Scholar]
- 30.Jobling MF, Barrow CJ, White AR, Masters CL, Collins SJ, Cappai R: The synthesis and spectroscopic analysis of the neurotoxic prion peptide 106–126: comparative use of manual Boc and Fmoc chemistry. Lett Pept Sci 1999, 6:129-134 [Google Scholar]
- 31.Puttfarcken PS, Getz RL, Coyle JT: Kainic acid-induced lipid peroxidation: protection with butylated hydroxytoluene and U78517F in primary cultures of cerebellar granule cells. Brain Res 1993, 624:223-232 [DOI] [PubMed] [Google Scholar]
- 32.Kim WK, Pae YS: Involvement of N-methyl-D-aspartate receptor and free radicals in homocysteine-mediated toxicity on rat cerebellar granule cells in culture. Neurosci Lett 1996, 216:117-120 [DOI] [PubMed] [Google Scholar]
- 33.Menniti F, Chenard B, Collins M, Ducat M, Shalaby I, White F: CP-101,606, a potent neuroprotectant selective for forebrain neurons. Eur J Pharmacol 1997, 331:117-126 [DOI] [PubMed] [Google Scholar]
- 34.Mischel RE, Kim YS, Sheldon RA, Ferriero DM: Hydrogen peroxide is selectively toxic to immature murine neurons in vitro. Neurosci Lett 1997, 231:17-20 [DOI] [PubMed] [Google Scholar]
- 35.White AR, Bush AI, Beyreuther K, Masters CL, Cappai R: Exacerbation of copper toxicity in primary neuronal cultures depleted of cellular glutathione. J Neurochem 1999, 72:2092-2098 [DOI] [PubMed] [Google Scholar]
- 36.Brown DR, Schmidt B, Groschup MH, Kretzschmar HA: Prion protein expression in muscle cells and toxicity of a prion protein fragment. Eur J Cell Biol 1998, 75:29-37 [DOI] [PubMed] [Google Scholar]
- 37.Reddan JR, Giblin FJ, Dziedzic DC, Wirebaugh BM, Peters JL: Hydrogen peroxide affects specific epithelial subpopulations in cultured rabbit lenses. Invest Ophthalmol Vis Sci 1995, 36:289-299 [PubMed] [Google Scholar]
- 38.Perovic S, Schroder HC, Pergande G, Ushijima H, Müller WEG: Effect of flupirtine on bcl-2 and glutathione level in neuronal cells treated in vitro with the prion protein fragment (PrP106–126). Exp Neurol 1997, 147:518-524 [DOI] [PubMed] [Google Scholar]
- 39.Hyslop PA, Zhang Z, Pearson DV, Phebus LA: Measurement of striatal H2O2 by microdialysis following global forebrain ischemia and reperfusion in the rat: correlation with the cytotoxic potential of H2O2 in vitro. Brain Res 1995, 671:181-186 [DOI] [PubMed] [Google Scholar]
- 40.Choi SI, Ju WK, Choi EK, Kim J, Lea HZ, Carp RI, Wisniewski HM, Kim YS: Mitochondrial dysfunction induced by oxidative stress in brains of hamsters infected with the 263K scrapie agent. Acta Neuropathol 1998, 96:279-286 [DOI] [PubMed] [Google Scholar]
- 41.Mattson MP, Lovell MA, Furukawa K, Markesbery WR: Neurotrophic factors attenuate glutamate-induced accumulation of peroxides, elevation of intracellular Ca2+ concentration, and neurotoxicity and increase antioxidant enzyme activities in hippocampal neurons. J Neurochem 1995, 65:1740-1751 [DOI] [PubMed] [Google Scholar]
- 42.Piani D, Spranger M, Frei K, Schaffner A, Fontana A: Macrophage-induced cytotoxicity of N-methyl-D-aspartate receptor positive neurons involves excitatory amino acids rather than reactive oxygen intermediates and cytokines. Eur J Immunol 1992, 22:2429-2436 [DOI] [PubMed] [Google Scholar]
- 43.Reiter R, Tang L, Garcia JJ, Munoz-Hoyos A: Pharmacological actions of melatonin in oxygen radical pathophysiology. Life Sci 1997, 60:2255-2271 [DOI] [PubMed] [Google Scholar]
- 44.Brown DR, Qin K, Herms JW, Madlung A, Manson J, Strome R, Fraser PE, Kruck T, Bohlens A, Schulz-Schaeffer W, Giese A, Westaway D, Kretzschmar HA: The cellular prion protein binds copper in vivo. Nature 1997, 390:684-687 [DOI] [PubMed] [Google Scholar]
- 45.Brown DR, Besinger A: Prion protein expression and superoxide dismutase activity. Biochem J 1998, 334:423-429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adamson GM, Harman AW: A role for the glutathione peroxidase/reductase enzyme system in the protection from paracetamol toxicity in isolated mouse hepatocytes. Biochem Pharmacol 1989, 38:3323-3330 [DOI] [PubMed] [Google Scholar]
- 47.Buckman TD, Sutphin MS, Mitrovic B: Oxidative stress in a clonal cell line of neuronal origin: effects of anti-oxidant enzyme modulation. J Neurochem 1993, 60:2046-2058 [DOI] [PubMed] [Google Scholar]
- 48.Spector A, Yang Y, Ho YS, Magnenat JL, Wang RR, Ma W, Li WC: Variation in cellular glutathione peroxidase activity in lens epithelial cells, transgenics and knockouts does not significantly change the response to H2O2 stress. Exp Eye Res 1996, 62:521-540 [DOI] [PubMed] [Google Scholar]
- 49.Da Costa Ferreira AM, Ciriola MR, Marcocci L, Rotilio G: Copper(I) transfer into metallothionein by glutathione. Biochem J 1993, 292:673-676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ascone I, Longo A, Dexpert H, Ciriolo MR, Rotilio G, Desideri A: An X-ray absorption study of the reconstitution process of bovine Cu: Zn superoxide dismutase by Cu(I)-glutathione complex. FEBS Lett 1993, 322:165-167 [DOI] [PubMed] [Google Scholar]
- 51.Brown DR, Schmidt B, Kretzschmar HA: Effects of oxidative stress on prion protein expression in PC12 cells. Int J Dev Neurosci 1997, 15:961-972 [DOI] [PubMed] [Google Scholar]
- 52.Barker JE, Heales SJR, Cassidy A, Bolanos JP, Land JM, Clark JB: Depletion of brain glutathione results in a decrease of glutathione reductase activity, an enzyme susceptible to oxidative damage. Brain Res 1996, 716:118-122 [DOI] [PubMed] [Google Scholar]
- 53.Nagendra SN, Shetty KT, Rao KM, Rao BSSR: Effect of disulfiram administration on rat-brain glutathione metabolism. Alcohol 1994, 11:7-10 [DOI] [PubMed] [Google Scholar]
- 54.Huang J, Philbert MA: Cellular responses of cultured cerebellar astrocytes to ethacrynic acid-induced perturbation of subcellular glutathione homeostasis. Brain Res 1996, 711:184-192 [DOI] [PubMed] [Google Scholar]