

Abstract
To elucidate the role of p53 and apoptosis in the pathogenesis of lung injury, we examined histological changes, expressions of p53 and p21waf1/cip1 (p21), apoptosis, DNA double strand breaks, cell kinetics, and DNA synthesis in C57/BL6 mice (p53+/+) and mice deficient for p53 (p53−/−) at 2 hours to 7 days after a single intravenous administration of bleomycin. We also compared these parameters between the lung cells and small intestinal epithelial cells to explore potential differences in their response to DNA damage. Bleomycin induced p21 expression in a p53-dependent manner in p53+/+ mice but neither p53 nor p21 expression in p53−/− mice. In the lung of both groups of mice, focal inflammation followed by fibrosis was observed, but there was no evidence of apoptosis. Cells with DNA breaks and those undergoing DNA synthesis were unequivocally increased, but the cycling cell fraction remained unchanged, suggesting that the DNA synthesis detected in the lung reflected unscheduled DNA synthesis for repair of damaged DNA. DNA breaks and unscheduled DNA synthesis were prolonged in p53−/− mice compared to p53+/+ mice. By contrast, in the small intestine, marked cell cycle arrest and extensive apoptosis were evoked in the cycling crypt cells of both groups of mice, but these changes were milder and DNA breaks remained detectable for a longer time in p53−/− mice than in p53+/+ mice. Among the resting enterocytes in the villi, apoptosis was observed almost equally in both groups, but repair of DNA breaks was significantly delayed in the p53−/− mice. These observations imply that apoptosis is mediated largely by the p53-dependent pathway in the crypts but exclusively by the p53-independent pathway in the villi, that this pathway is particularly important in DNA repair in the villi, and that despite this difference in the significance of apoptosis, p53 plays an important role in DNA repair in both the crypts and villi. Our results suggest that the lung cells and small intestinal cells respond to the bleomycin treatment in different ways in terms of the induction of apoptosis and that p53 carries out an essential role in the early response to and repair of DNA damage by a non-apoptotic mechanism which appears to be crucial in the noncycling lung cells and enterocytes. Importantly, the p53-p21 pathway and apoptosis are unlikely to be essential for bleomycin-induced tissue injury in the lung.
When cells are exposed to DNA-damaging agents, such as γ and UV irradiation, anticancer drugs, and so on, they undergo cell cycle arrest to allow repair of damaged DNA and/or apoptotic cell death to remove unrepairable DNA. 1-3 In these processes, p53 has been demonstrated to play key roles through its transcriptional activity and other unclarified mechanisms. 1,3,4 A cyclin-dependent kinase inhibitor, p21waf1/cip1, is a crucial downstream factor of p53, which mediates cell cycle arrest by binding to and inhibiting the functions of cyclin-dependent kinases and proliferating cell nuclear antigen. 5,6 Bax, another downstream factor of p53, is shown to mediate apoptotic cell death in vitro, 7 though its in vivo role in p53-mediated apoptotic cell death has been disputed. 8-10 Whether cells bearing DNA damage undergo cell cycle arrest or apoptotic cell death appears to depend on the type of cell (organ specificity) and/or conditions surrounding the cells, whereas the determinant of these two disparate cellular outcomes has not yet been clarified. 11
The lung is subject to damage caused by a variety of exogenous and endogenous insults. One line of evidence suggests that the p53-p21waf1/cip1 pathway, as well as apoptotic cell death, contributes to lung injuries induced by various agents. 12-15 An anticancer drug, bleomycin, generates superoxide radicals to induce DNA double strand breaks in vitro 16 and is well known to cause severe progressive pulmonary fibrosis. 16-23 Recent studies demonstrated that excessive apoptotic cell death was responsible for acute lung injury leading to pulmonary fibrosis induced by bleomycin. 12
To elucidate the role of the p53-p21waf1/cip1 pathway in the pathogenesis of chemical-induced lung injury, we examined the effects of bleomycin on the lung cells in p53 knockout mice in comparison with wild-type mice. To the best of our knowledge, a study of bleomycin-induced lung injury using the p53 knockout mice has not previously been conducted. We evaluated alterations of cell kinetics, expression of p53 and p21waf1/cip1, apoptotic cell death, DNA double strand breaks, and DNA synthesis at various time points after the bleomycin treatment. These parameters in the bronchial (epithelial) and alveolar cells were also compared with those in the small intestinal epithelial cells, because these two tissues are different in terms of cell kinetics under both physiological and pathological conditions. The small intestinal epithelium belongs to the renewal system consisting of clearly separated cycling and resting cell compartments, and its response to DNA damage has been examined extensively. 2,24-26 The respiratory epithelium belongs to the conditional renewal system, the study of which has been rather limited. 12-14 In this paper, we discuss the significance of the p53-p21waf1/cip1 pathway and refer to the difference between these two epithelial systems in response to bleomycin-induced DNA damage.
Materials and Methods
Animals and Chemicals
Specific-pathogen-free male C57/BL6 mice (wild-type mice), 10 weeks of age, were obtained from Japan SLC (Sizuoka, Japan). Age- and sex-matched C57/BL6 mice homozygously deficient for p53 (p53 knockout mice) 27 were obtained from Immuno-Biological Laboratories (Gunma, Japan). Experimental animals were treated with 100 mg/kg body weight of bleomycin hydrogen chloride (Nippon Kayaku Co. Ltd., Tokyo) by a single intravenous injection through the tail vein as previously described. 20-21 This dose corresponded to one-third of the Lethal dose 50% (LD50) described in the instructions (315 mg/kg body weight). In our preliminary study, 2 of 10 mice died within 7 days after the treatment and this dose was sufficient for the treated animals to develop pulmonary fibrosis. Control animals were treated with sterile saline alone. Wild-type and p53 knockout mice were divided into eight groups (controls and 2, 4, 6, and 12 hours and 1, 3, and 7 days after bleomycin injection) with 3 animals in each group. Mice were sacrificed under a pentobarbital anesthesia, and the lungs and small intestines were obtained. One hour before the sacrifice, mice were given 2 mg/kg body weight of bromodeoxyuridine (BrdU, Sigma Chemical, St. Louis, MO) by intraperitoneal injection.
Tissue Preparation
Tissues from the lower lobe of the right lung and from the small intestine were fixed in a buffered 4% paraformaldehyde solution for 3 days and embedded in paraffin. Other pieces of the right lung and small intestine were embedded in O.C.T. compound (Sakura, Tokyo) and snap-frozen in liquid nitrogen. Tissue sections were prepared from paraffin-embedded or frozen tissues for routine hematoxylin-eosin staining, immunohistochemistry, and in situ nick end labeling.
Immunohistochemistry
For p21waf1/cip1, Ki-67, and BrdU staining, 5-μm sections made from paraffin-embedded tissues and placed on silane-coated slides were dewaxed and rehydrated. The sections were immersed in 0.01 mol/L citrate buffer, pH 6.0, for p21waf1/cip1 or 0.05 mol/L Tris-HCl buffer containing 5% urea, pH 9.5, for Ki-67 28 and were heated in a microwave oven at 97°C for 30 minutes to retrieve the antigenic activities. For BrdU staining, the sections were immersed in a 4-N HCl solution for 20 minutes to denature double-strand DNA. The sections were incubated with 3% hydrogen peroxide/methanol at room temperature for 10 minutes to inactivate the endogenous peroxidase, and nonimmunospecific protein bindings were blocked with 5% normal goat serum and avidin-biotin blocking reagent (Vector Laboratories Inc., Burlingame, CA). The sections were incubated with one of the following primary antibodies at room temperature for 90 minutes, each diluted at 1:100: anti-p21waf1/cip1 rabbit polyclonal (sc-397, Santa Cruz Biotechnology, Santa Cruz, CA), anti-Ki-67 mouse monoclonal (MIB5, Immunotech, Marseille, France), and anti-BrdU mouse monoclonal antibody (Br3, Caltag, San Francisco, CA). Subsequently, they were incubated with biotinylated animal-matched secondary antibodies at room temperature for 60 minutes. The protein expressions were visualized by the avidin-biotin complex immunoperoxidase method using the SLAB kit (DAKO Japan Co., Ltd., Kyoto, Japan) with diaminobenzidine as substrate. Nuclear counterstaining was performed lightly with hematoxylin.
For p53 staining, 6-μm frozen sections were made and fixed with a buffered 4% paraformaldehyde solution for 5 minutes, then washed with distilled water. After treatment with 5% normal goat serum, the sections were incubated with 1:500 diluted rabbit polyclonal antibody against p53 (CM5, Novocastra, Newcastle, UK) at room temperature for 90 minutes. Then, tissue sections were incubated with the fluorescein isothiocyanate-conjugated goat antibody against rabbit immunoglobulin (DAKO) at room temperature for 60 minutes. The protein expression was observed with a fluorescence microscope (HB10101, Nikon, Tokyo).
In Situ Nick End Labeling (ISNEL)
DNA double strand breaks and/or apoptotic nucleosomal degradation were detected on paraffin tissue sections by the ISNEL method using the Trevigen apoptotic cell system (Trevigen, Gaithersburg, MD). Six-micron sections were dewaxed, rehydrated, and incubated first with a proteinase K solution at room temperature for 15 minutes, then with a 2% hydrogen peroxide solution for 5 minutes. After being immersed in a labeling buffer for 3 minutes, the sections were incubated with a reaction mixture containing Klenow fragment and dNTP in labeling buffer at 37°C for 60 minutes. After a brief wash with PBS, the streptavidin-biotin peroxidase method was applied with diaminobenzidine as a substrate. Light nuclear counterstaining was performed with hematoxylin.
Western Blotting
Fresh tissues from the left lung were homogenized in 1 ml of a homogenizing buffer solution containing 5 mmol/L Tris-HCl, pH 7.5, 0.25 mol/L sucrose, 2 mmol/L EDTA, 2 mmol/L EGTA, 0.5 mmol/L DDT, 0.5 mmol/L phenylmethylsulfonyl fluoride (PMSF), and 5 μg leupeptin with a politron homogenizer at 1000 rpm for 1 minute, and then filtrated through a nylon mesh (100 μm in pore size). The filtrates were centrifuged at 600 × g for 10 minutes. The pellets were resuspended in 100 μl of buffer A (50 mmol/L Tris-HCl, pH 7.5, 0.25 mol/L sucrose, 25 mmol/L KCl, 25 mmol/L MgCl2, 2 mmol/L EDTA, 2 mmol/L EGTA, 0.5 mmol/L DDT, 0.5 mmol/L PMSF, 5 μg leupeptin) and were incubated for 10 minutes on ice. Then, 200 μl of buffer B (50 mmol/L Tris, 2.3 mol/L sucrose, 25 mmol/L KCl, 25 mmol/L MgCl2, 2 mmol/L EDTA, 2 mmol/L EGTA, 0.5 mmol/L DDT, 0.5 mmol/L PMSF, 5 μg leupeptin) were added and mixed well. Each suspension was layered over 200 μl of buffer B and centrifuged at 12,000 × g for 30 minutes. The pellets were resuspended in hypotonic lysis buffer containing 25 mmol/L HEPES, pH 7.5, 0.4 mmol/L KCl, 5 mmol/L EDTA, 10 mmol/L NaF, 5 mmol/L DDT, 5 mmol/L EDTA, and 1% NP-40 and incubated for 60 minutes on ice. After centrifugation at 12,000 × g for 15 minutes, the supernatants were recovered as protein lysates. Equal volumes of protein lysate and 2× sodium dodecyl sulfate (SDS) sample buffer (0.1 mol/L Tris-HCl, pH 6.8, 4% SDS, 12% 2-mercapteethanol, 10% glycerol, and 0.0025% bromophenol blue) were mixed and boiled for 5 minutes. The protein concentration of the samples was determined by Bradford’s method.
Nuclear protein (30 μg) was subjected to SDS-polyacrylamide gel electrophoresis on 12% acrylamide gel and then transferred onto nitrocellulose membranes (Schleicher & Schuell, Dassel, Germany). Nonspecific protein binding was blocked with 5% nonfat milk in 0.01 mol/L PBS containing 0.1% Tween 20 (Tween-PBS) at room temperature for 60 minutes. The membranes were incubated with 1:500 diluted rabbit polyclonal antibody against p53 (CM5) or p21waf1/cip1 (sc-397) at room temperature for 90 minutes. After a brief washing with Tween-PBS, the membranes were incubated with a horseradish peroxidase-conjugated goat antibody against rabbit immunoglobulin (Amersham Life Science, Buckinghamshire, UK) at room temperature for 60 minutes. The membrane was briefly washed again with Tween-PBS. Immunoblotted proteins were visualized with the enhanced chemiluminescence system (Amersham).
Electrophoresis of DNA
Fresh tissues from the middle lobe of the right lung and those from the small intestine were minced with a razor blade and were suspended in a 500 μl DNA extraction buffer containing 50 mmol/L Tris-HCl, pH 7.7, 100 mmol/L NaCl, 100 mmol/L EDTA, 1% SDS, and 0.1 mg/ml proteinase K (Sigma). The samples were incubated at 55°C overnight and then subjected to phenol/chloroform extraction. After isopropanol precipitation, pellets of DNA were washed 3× with 70% ethanol. DNA samples (10 μg) were electrophoresed on a 1% agarose gel containing 1 μg/ml of ethidium bromide and visualized on an ultraviolet transilluminator.
Labeling Index and Apoptotic Index
More than 1000 cell nuclei of the bronchial epithelia, lung alveoli including epithelial cells, fibroblasts, endothelial cells, and macrophages, small intestinal crypts, and villi epithelia were counted. Percentages of cells positive for Ki-67, BrdU, and ISNEL were determined and used as the labeling index. The percentage of cells showing the morphological characteristics of apoptotic cell death, such as cell shrinkage and chromatin condensation and fragmentation, was determined on hematoxylin-eosin sections and used as the apoptotic index.
Statistical Analysis
The difference in mean values was analyzed by Student’s t-test. A P value <0.05 was considered significant.
Results
Histology
From 2 hours to 1 day after bleomycin treatment, no significant pathological changes were observed in the lungs of either the wild-type or p53 knockout mice. At 3 days, infiltration by small numbers of inflammatory cells, such as neutrophils, lymphocytes, and monocytes, was observed focally around the bronchioles and adjacent small blood vessels, as well as within the alveolar spaces (Figure 1, A and B) ▶ . The inflammatory infiltrates increased with time, and these changes were almost equivalent in the wild-type and p53 knockout mice.
Figure 1.
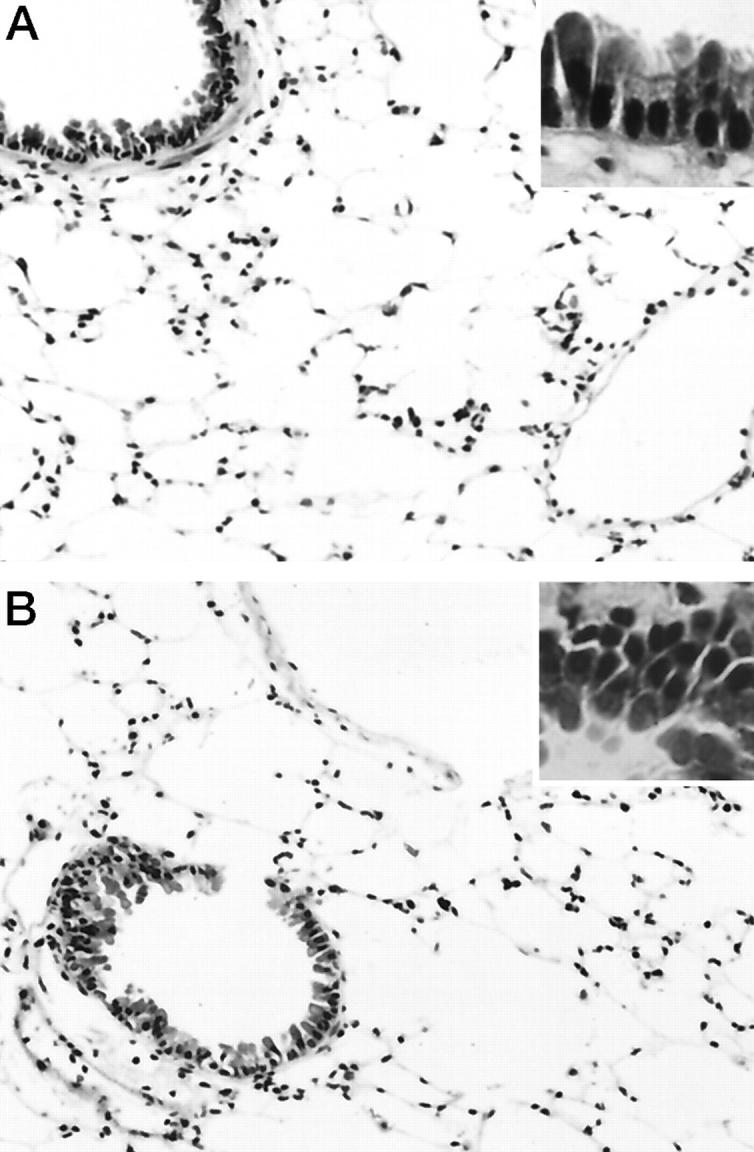
Histological appearance of the lung from a wild-type (A) and p53 knockout (B) mouse at 3 days after the treatment. In both the wild-type and p53 knockout mice, a few inflammatory cell infiltrates composed of neutrophils and mononuclear cells were observed around bronchi and small blood vessels and within alveolar spaces. No cells exhibiting apoptotic characteristics were seen in either bronchi or alveoli of either group of mice (insets). Hematoxylin and eosin; original magnification, ×200 (insets, ×400).
The small intestinal mucosa showed severe epithelial cell desquamation in the crypts of wild-type mice from 12 hours to 1 day after the treatment (data not shown). In the p53 knockout mice, epithelial desquamation also occurred in the crypts, but the changes were milder than in the wild-type mice. From day 1 to day 3, regeneration of epithelia with numerous mitotic figures was observed. These changes had almost completely disappeared by day 7 in both groups (data not shown).
Apoptotic Cell Death
In neither the wild-type nor the p53 knockout mice did bronchial and alveolar cells show signs of apoptotic cell death throughout the experimental period histologically (Figure 1, A and B ▶ , and Figure 2B ▶ , top, second panel). Agarose gel electrophoresis did not reveal evidence of DNA ladders at any time point examined (Figure 2A) ▶ .
Figure 2.

A: DNA electrophoresis of the lung and small intestine from wild-type mice. Lane 1, marker; Lanes 2–9, lung (control and at 2 hours to 7 days); Lane 10 , small intestine at 12 hours. DNA laddering was observed in the small intestine at 12 hours after the treatment, but not in the lung at any time point. B: Apoptotic index in the bronchi (top), alveoli (second panel), small intestinal crypts (third panel), and villi (bottom) of the wild-type (solid line) and p53 knockout (dashed line) mice. C and D: Histological appearance of the small intestine of a wild-type (C) and p53 knockout (D) mouse; left, crypts at 12 hours; right, villi at 3 days after the treatment. Cells with typical apoptotic characteristics were observed not only in the crypts but also in the villi of both groups (arrows). Hematoxylin and eosin; original magnification, ×400.
In contrast to the lung, apoptotic cell death was observed in the small intestine in both the crypts and villi, not only in the wild-type but also in the p53 knockout mice after treatment. In the crypts, the frequency of apoptotic cells increased rapidly after treatment in both groups (Figure 2B ▶ , third panel). The peak of apoptotic index was seen at 4 hours in the wild-type mice and at 12 hours in the p53 knockout mice, with the maximal value in the former being 1.7-fold higher than in the latter (P < 0.05). On the other hand, in the villi, the frequency of apoptotic cells gradually increased and peaked at 3 days in both groups (Figure 2B ▶ , bottom). The maximal value was slightly higher in the wild-type mice than in the p53 knockout mice, but the difference was not significant. In both groups, the apoptotic index in the crypts and villi had returned to the basal level 7 days after the treatment. Figure 2, C and D ▶ , shows typical features of apoptotic cell death observed in the crypts (left) and villi (right) of the wild-type and p53 knockout mice after the treatment, respectively. At this time point, a DNA ladder was observed on agarose gel electrophoresis (Figure 2A) ▶ .
ISNEL
In both the wild-type and p53 knockout mice, ISNEL-positive cells were observed among the bronchial and alveolar cells after the bleomycin treatment (Figure 3, A and B) ▶ . The ISNEL index in the bronchial cells of the wild-type mice was elevated soon after the treatment and peaked at 6 hours, whereas in the p53 knockout mice the peak was observed at 3 days (Figure 3C ▶ , top, and Table 1 ▶ ). The ISNEL index then gradually decreased in both groups, but in the p53 knockout mice it remained at higher levels than in the wild-type mice (P < 0.05). The ISNEL index in the alveolar cells of the wild-type mice was rapidly elevated soon after the treatment and peaked at 6 hours (Figure 3C ▶ , second panel, and Table 1 ▶ ). In the p53 knockout mice, the peak was somewhat delayed and seen at 12 hours. Similarly, sustained higher levels in the ISNEL index were observed in the p53 knockout mice compared to the wild-type mice (P < 0.05).
Figure 3.

A and B: In situ nick end labeling (ISNEL) of the lung from a wild-type (A) and p53 knockout (B) mouse at 24 hours after the treatment. Many ISNEL-positive cells were observed among the bronchial epithelial and alveolar cells in both groups. Original magnification, ×400. C: ISNEL index in the bronchi (top), alveoli (second panel), small intestinal crypts (third panel), and villi (bottom). Solid line, wild-type mice; dashed line, p53 knockout mice.
Table 1.
ISNEL Index
| Tissue | p53 status | Time after bleomycin treatment (hours) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 0 | 2 | 4 | 6 | 12 | 24 | 72 | 168 | ||
| Bronchi | Knockout | 0.9 ± 0.5 | 8.9 ± 3.3 | 8.7 ± 1.3 | 13.2 ± 1.1 | 16.4 ± 5.7 | 22.4 ± 10.1 | 46.9 ± 20.0 | 37.2 ± 20.6 |
| Wild-type | 0.1 ± 0.1 | 4.2 ± 2.8 | 4.7 ± 3.2 | 11.0 ± 1.2 | 8.5 ± 1.8 | 6.2 ± 1.9 | 5.0 ± 3.4 | 7.2 ± 6.7 | |
| Alveoli | Knockout | 1.3 ± 1.1 | 15.6 ± 14.9 | 14.5 ± 2.6 | 49.9 ± 3.2 | 51.3 ± 3.2 | 48.6 ± 1.9 | 52.1 ± 3.9 | 30.2 ± 3.8 |
| Wild-type | 0.1 ± 0.2 | 16.4 ± 7.6 | 14.6 ± 10.9 | 20.2 ± 5.7 | 11.0 ± 2.5 | 13.6 ± 3.6 | 7.9 ± 1.9 | 5.4 ± 4.7 | |
| Crypts | Knockout | 1.9 ± 1.6 | 7.0 ± 4.6 | 15.5 ± 0.7 | 17.5 ± 0.9 | 16.5 ± 1.4 | 17.8 ± 2.0 | 12.5 ± 3.7 | 19.8 ± 6.73 |
| Wild-type | 0.0 ± 0.0 | 26.3 ± 25.4 | 12.5 ± 10.8 | 17.6 ± 5.0 | 12.0 ± 3.8 | 8.7 ± 7.6 | 0.1 ± 0.1 | 0.1 ± 0.1 | |
| Villi | Knockout | 0.4 ± 0.2 | 25.0 ± 11.6 | 8.8 ± 1.8 | 8.6 ± 5.7 | 17.1 ± 8.0 | 23.9 ± 7.1 | 28.7 ± 8.7 | 10.7 ± 9.4 |
| Wild-type | 0.1 ± 0.2 | 18.9 ± 7.2 | 4.2 ± 4.7 | 16. ± 1.6 | 2.6 ± 3.2 | 3.4 ± 2.1 | 10.0 ± 5.7 | 0.8 ± 1.2 |
Values are presented as the mean ± SD (%).
In the small intestine, the ISNEL index in the crypts of the wild-type mice was rapidly elevated, peaking at 2 hours, then returning to the basal level 3 days after treatment (Figure 3C ▶ , third panel, and Table 1 ▶ ). By contrast, in the p53 knockout mice the ISNEL index of the crypts showed a gradual elevation and peaked at 12 hours, with a delay of 10 hours compared to the wild-type mice. It did not return to the basal level throughout the experimental period. On the other hand, in the villi, the ISNEL index in both groups rapidly increased, peaking at 2 hours, then decreasing. It gradually increased from 6 hours and peaked again at 3 days, but had returned to the basal level by 7 days in the wild-type mice (Figure 3C ▶ , bottom, and Table 1 ▶ ). As was seen in the crypts, the ISNEL index in the villi of the p53 knockout mice also did not return to the basal level. Virtually all of the apoptotic cell nuclei were ISNEL-positive, but many non-apoptotic cell nuclei were also stained positively with ISNEL (data not shown).
DNA Synthesis Determined by BrdU Labeling
The pattern of change in BrdU labeling after the treatment was similar between the bronchial and alveolar cells in both the wild-type and p53 knockout mice. There was no significant difference in the basal level of BrdU labeling between the two groups. After the treatment, the uptake of BrdU was observed in the bronchial and alveolar cells from both groups of mice (Figure 4, A and B) ▶ . The BrdU labeling in the wild-type mice rapidly increased, peaking at 12 hours, whereas in the p53 knockout mice it continued to increase for up to 3 days and showed significantly higher maximal values than in the wild-type mice (P < 0.05) (Figure 4C ▶ , top, second panel, and Table 2 ▶ ). Within 7 days, the BrdU labeling returned to the basal level in both the wild-type and p53 knockout mice.
Figure 4.

A and B: BrdU immunostain of the lung from a wild-type (A) and p53 knockout (B) mouse at 24 hours after the treatment. BrdU uptake was observed among the bronchial epithelial and alveolar cells in both groups. Original magnification, ×400. C: BrdU labeling index in the bronchi (top), alveoli (second panel), small intestinal crypts (third panel), and villi (bottom). Solid line, wild-type mice; dashed line, p53 knockout mice.
Table 2.
BrdU Labeling Index
| Tissue | p53 status | Time after bleomycin treatment (hours) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 0 | 2 | 4 | 6 | 12 | 24 | 72 | 168 | ||
| Bronchi | Knockout | 0.7 ± 0.2 | 5.3 ± 1.7 | 1.3 ± 0.5 | 1.9 ± 0.3 | 38.8 ± 2.5 | 36.4 ± 17.9 | 47.3 ± 13.3 | 1.3 ± 0.7 |
| Wild-type | 0.9 ± 0.7 | 3.5 ± 3.5 | 9.9 ± 5.8 | 4.8 ± 3.3 | 22.9 ± 3.9 | 10.9 ± 4.0 | 3.5 ± 1.7 | 3.7 ± 2.4 | |
| Alveoli | Knockout | 3.1 ± 0.3 | 9.1 ± 1.0 | 5.2 ± 0.9 | 12 ± 3.3 | 28.6 ± 4.1 | 31.5 ± 17.6 | 47.5 ± 4.0 | 8.5 ± 7.3 |
| Wild-type | 2.9 ± 1.2 | 7.7 ± 3.5 | 24.6 ± 5.0 | 12.5 ± 3.2 | 34.0 ± 8.9 | 16.9 ± 7.3 | 12.5 ± 5.2 | 7.4 ± 2.6 | |
| Crypts | Knockout | 34.6 ± 4.4 | 51.9 ± 0.8 | 51.7 ± 1.2 | 59.5 ± 2.8 | 33.2 ± 5.1 | 21.2 ± 5.0 | 59.8 ± 3.1 | 30.2 ± 1.0 |
| Wild-type | 39.3 ± 1.7 | 21 ± 2.5 | 16.7 ± 1.5 | 13.2 ± 1.1 | 72.2 ± 6.4 | 53.7 ± 3.7 | 49.8 ± 10.5 | 39.4 ± 6.6 | |
| Villi | Knockout | 0.1 ± 0.1 | 0.1 ± 0.1 | 0.0 ± 0.0 | 2.9 ± 3.0 | 37.6 ± 12.7 | 47.2 ± 13.3 | 2.0 ± 3.1 | 0.1 ± 0.1 |
| Wild-type | 0.1 ± 0.1 | 0.4 ± 0.2 | 0.1 ± 0.1 | 0.0 ± 0.0 | 51.3 ± 11.7 | 32.7 ± 7.2 | 0.6 ± 0.8 | 0.0 ± 0.0 |
Values are presented as the mean ± SD (%).
In the small intestine, the crypts and villi showed different changes in the BrdU uptake. In the crypts, the BrdU labeling ranged from 30 to 40% in the untreated wild-type and p53 knockout mice without any significant difference between the two (Figure 4C ▶ , third panel, and Table 2 ▶ ). In the wild-type mice, BrdU labeling in the crypts rapidly decreased after treatment, with the lowest level reached at 6 hours. Then it increased, peaking at 12 hours before returning to the basal level 7 days later. By contrast, in the p53 knockout mice BrdU labeling in the crypts transiently increased from 2 to 6 hours, then decreased to a low at 24 hours. Thus, in the crypts the initial decrease in BrdU labeling was delayed in the p53 knockout mice by 6 hours compared to the wild-type mice. It had also returned to the basal level by day 7 in the p53 knockout mice. In the villi, no BrdU uptake was observed in the untreated control, and its labeling was unchanged 6 hours after the treatment in both groups (Figure 4C ▶ , bottom, and Table 2 ▶ ). From 6 to 12 hours, it started to increase rapidly, peaking at 12 hours in the wild-type mice and at 24 hours in the p53 knockout mice. Thereafter, it decreased and had returned to the basal level by day 3 in both groups. Thus, the time at which the BrdU labeling peaked was delayed in the p53 knockout mice by 12 hours compared to the wild-type mice, but the maximal values were not significantly different between the two.
Expression of p53 and p21waf1/cip1
Western blot analysis showed that the expression of p53 in the lung of wild-type mice was first detected at 2 hours after bleomycin treatment and continued for 7 days (Figure 5A) ▶ . The expression of p21waf1/cip1 coincided with that of p53 (Figure 5A) ▶ . The expression of these proteins was also detected immunohistochemically in the bronchial epithelial and alveolar cell nuclei from 2 hours to 7 days after the treatment (Figure 5, B and C) ▶ . By contrast, in the p53 knockout mice the expression of p53 and p21waf1/cip1 was not detected by either immunohistochemistry (data not shown) or Western blotting at any time point examined (Figure 5A) ▶ . In the small intestine, the concomitant expression of these proteins was immunohistochemically detected exclusively in the crypts, but not in the villi, only in the treated wild-type mice (data not shown). In both groups of mice, only a weak expression of p21waf1/cip1 was seen in a few enterocytes of the villi, where p53 expression was not detected.
Figure 5.

Expression of p53 and p21waf1/cip1 in the lung. A: Western blot analysis for p53 and p21waf1/cip1 in the wild-type and p53 knockout mice. Expression of p53 and p21waf1/cip1 was observed in the wild-type mice after the treatment, whereas the expression of these proteins was not detected in the p53 knockout mice throughout the experimental period. B and C: Immunohistochemistry for p53 (B) and p21waf1/cip1 (C) in a wild-type mouse at 12 hours after the treatment. Numerous positive nuclei were observed in the bronchial epithelia and alveoli. B, immunofluorescent staining; original magnification, ×400. C, immunoperoxidase staining; original magnification, ×400.
Changes in Cell Kinetics Determined by Ki-67 Labeling
The labeling index of Ki-67 in the bronchial and alveolar cells showed no significant change at any time point examined in either the wild-type or the p53 knockout mice, although some fluctuation was observed soon after the treatment (Figure 6 ▶ , top and second panels).
Figure 6.

Ki-67 labeling index in the bronchi (top), alveoli (second panel), small intestinal crypts (third panel), and villi (bottom). Solid line, wild-type mice; dashed line, p53 knockout mice.
In the small intestinal crypts, by contrast, marked changes in Ki-67 labeling were seen (Figure 6 ▶ , third panel). In the wild-type mice, the Ki-67 labeling rapidly decreased after the treatment, bottoming out at 12 hours. It rapidly recovered and peaked at 24 hours, then decreased again before returning to the basal level on day 7. In the p53 knockout mice, the Ki-67 labeling transiently increased from 2 to 6 hours, then decreased to a low at 24 hours. Thereafter, it increased again, peaking at 3 days, and returned to the basal level by day 7. Thus, the lowest level of Ki-67 labeling in the p53 knockout mice was obtained some 12 hours later and the value was significantly higher than in the wild-type mice (P < 0.05). In the villi, Ki-67 labeling was very low, ranging from 0 to 0.2%, and was unchanged throughout the experimental period (Figure 6 ▶ , bottom).
Discussion
The purpose of the present study was to elucidate the in vivo role of the p53-p21waf1/cip1 pathway in the response to DNA damage of the lung cells. We assumed that bleomycin induces DNA double strand breaks, which in turn activate the p53-p21waf1/cip1 pathway in the wild-type mice, leading to cell cycle arrest and/or apoptotic cell death, but this pathway would not work in the p53 knockout mice. To monitor these potential cellular changes, we used three different methods; ie, analysis of cytomorphological changes, ISNEL, and the electrophoretic pattern of DNA. The DNA laddering on electrophoresis reflects nucleosomal degradation and is widely accepted as evidence of apoptotic cell death, but this method requires significant quantities of degraded DNA in the sample to be detected. On the other hand, ISNEL is suitable for visualizing apoptotic cell death at the single-cell level, but it detects not only cells with apoptotic nucleosomal degradation but also those with DNA double strand breaks. A single intravenous administration of bleomycin unequivocally evoked DNA double strand breaks in both the wild-type and p53 knockout mice, as demonstrated by many ISNEL-positive cell nuclei in the bronchial epithelia and alveoli in the absence of cells exhibiting apoptotic morphologies and of DNA laddering. Western blot analysis and immunohistochemistry confirmed that p21waf1/cip1 was expressed in a p53-dependent manner in response to the DNA damage induced by bleomycin in the bronchial and alveolar cells, because its expression coincided with that of p53 in the wild-type mice, but the expression of these proteins was not detected in the p53 knockout mice. Focal inflammation and fibrosis (data not shown) were observed almost equally in both groups. However, we did not obtain any evidence of apoptotic cell death preceding the inflammatory and fibrotic responses in either the p53 knockout mice or the wild-type mice. Inconsistent with our results, a previous study showed extensive apoptotic cell death in the bronchial and alveolar epithelia after intratracheal bleomycin administration. 12 In the present study, bleomycin was given intravenously given through the tail vein, because this route of bleomycin administration is also used in human patients. To our knowledge, there has been no report showing that the intravenous administration of bleomycin caused apoptotic cell death in the lung. 20,21 Bleomycin may exert its effects more directly on and cause more severe damage to the airway cells on intratracheal instillation than on intravenous administration.
Several recent studies emphasized the importance of the p53-p21waf1/cip1 pathway and apoptotic cell death in acute alveolar damage induced by bleomycin and other agents. 12-14 Our results were inconsistent with these and suggested it is unlikely that the p53-p21waf1/cip1 pathway is obligatory for bleomycin-induced acute alveolar damage and pulmonary fibrosis, as apoptotic cell death is. There appear to be other essential factors initiating the early inflammatory reactions. 18-23 For example, a transcriptional factor, NF-κβ, which is ubiquitously expressed and activated in response to a variety of stimuli including DNA damage and transactivates proinflammatory cytokines, is presumed to be involved in the pathway mediating bleomycin-induced acute alveolar damage. 29-31 It would be of interest to examine the role of p53 in transactivation of proinflammatory cytokines using this experimental system with the aid of quantitative assessment of bleomycin-induced pulmonary lesions.
In the small intestine, in contrast to the lung, a number of cells undergoing apoptotic cell death were observed in both the treated wild-type and p53 knockout mice. This result appears to support the conclusion of previous studies that this type of cell death was mediated not only in a p53-dependent, but also in a p53-independent manner. 26 Consistent with previous studies, 2,26 we confirmed here that p53-dependent apoptotic cell death occurred earlier and predominantly in the crypts, where p53 was expressed immediately after the bleomycin treatment, whereas p53-independent apoptotic cell death occurred a little later in the crypts and much later in the villi, where no p53 expression was detected. The differences in response to bleomycin treatment observed between the lung and small intestine, and between the crypts and villi, may be related either to the organ specificity of bleomycin toxicity 16,32 or to the difference in cell types and particularly in cell kinetics, as discussed below.
As described above, no apoptotic cell death was seen in the bronchial and alveolar cells, but the ISNEL index was elevated soon after the bleomycin treatment. In the small intestine, the ISNEL index was also elevated in the crypts and villi; its change coincided with that in the apoptotic index in the wild-type mice, but it continued to be much higher in the p53 knockout mice. The ISNEL method can detect not only those DNA double strand breaks induced by endogenous apoptotic mechanisms, but also those directly caused by exogenous DNA-damaging agents. 33,34 Thus, the ISNEL index in the small intestine is thought to reflect preferentially the apoptotic cell death in the wild-type mice, whereas in the p53 knockout mice it probably reflected both the apoptotic cell death and the DNA damage directly caused by bleomycin. However, it most likely reflected only the latter in the bronchial and alveolar cells of both groups of mice. The extremely high value of and delayed reduction in the ISNEL index observed in the lung of p53 knockout mice could be attributed exclusively to a delay of the repair of damaged DNA, implying that p53 is an important factor in the early response to and repair of DNA damage in vivo. The sustained high ISNEL index observed in the villi of the p53 knockout mice appears strongly supportive of the importance of this p53-dependent DNA repair in noncycling cells.
To evaluate the effect of the p53-p21waf1/cip1 pathway on cell kinetics, we determined the fraction of cycling cells as measured by Ki-67 antigen expression and the frequency of cells undergoing DNA synthesis as measured by BrdU uptake, 28,35 and compared the values between the wild-type and p53 knockout mice. The incidence of cycling cells among the bronchial and alveolar cells as well as among the enterocytes of small intestinal villi was not changed significantly by the bleomycin treatment; it remained quite low in both the wild-type and p53 knockout mice. The BrdU uptake, in contrast, was rapidly elevated in these cell compartments in both groups of mice. Previous studies by others showed that the BrdU uptake in noncycling cells correlates well with unscheduled DNA synthesis in the repair of damaged DNA. 36,37 Thus, the elevated BrdU uptakes observed here in the bronchial and alveolar cells and in the enterocytes of the villi most likely reflect the repair of damaged DNA. The delayed reduction of BrdU uptake as well as of the ISNEL index in the p53 knockout mice reconfirmed the significance of p53 in the early response to and repair of DNA damage in the lung. In the crypts of the small intestine, where Ki-67 expression and BrdU uptake showed almost coincidental changes, the BrdU uptake represents mostly DNA replication in the S phase and, to a small extent, unscheduled DNA synthesis. It is suggested that the p53-p21waf1/cip1 pathway is essential for induction of cell cycle arrest in response to bleomycin-induced DNA damage in the small intestinal crypt cells, because both the Ki-67 expression and BrdU uptake rapidly decreased in the wild-type mice, whereas these parameters increased, transiently but significantly, in the p53 knockout mice. This transient increase of cycling cells may be associated with the effects of early responsible genes, such as jun and fos, which are reported to be immediately expressed after DNA damage and to promote various cellular mechanisms including cell cycle progression. 38,39
Taken together, our results confirmed that p53 plays important roles in the early response to and repair of the bleomycin-induced DNA damage in both the lung and small intestine in vivo. However, as observed here and in other studies, 16,32 cellular responses to bleomycin treatment are different between these two tissue systems, and even in the small intestine they are distinctly different between the crypts and villi. This difference may be attributable mostly to cell type, because DNA damage was unequivocally evoked by the bleomycin treatment in all of the tissues as determined by the ISNEL index. The lung cells belong to the conditional renewal system, which is quiescent under normal physiological conditions, and the enterocytes of the villi are nonreplicating resting cells. On the other hand, the crypt epithelial cells belong to the renewal system, which constantly proliferates and turns over. The mode of response to DNA damage and the main pathway mediated by p53 appear to differ among these epithelial systems. That is, in the renewal system, either p53-dependent or p53-independent apoptotic cell death and cell cycle arrest play a major role, but in the conditional renewal system and in the resting cells as well, other mechanism(s) mediated by p53 may carry out an essential role, particularly under conditions where cell turnover and proliferation are not enhanced. In this context, it would be of interest to elucidate whether the cycling cells in the lung undergo either p53-dependent or p53-independent apoptotic cell death in response to bleomycin treatment, as do the crypt cells of the small intestine. The frequency of lung cells expressing p53 in the bleomycin-treated wild-type mice was much higher than that of Ki-67-labeled cells. Identification of the type of cells that expressed p53 was not easy, but both replicating cells (Clara cells, basal cells, and type 2 alveolar cells) and nonreplicating cells (ciliated cells) appeared to be labeled by p53 immunostaining. Although the possibility cannot be ruled out that the cycling cells, comprising a very small fraction of the lung cells, undergo apoptotic cell death in response to bleomycin treatment, we were unable to obtain any evidence of apoptotic cell death in the lung by the methods used in the current study.
p53 is known to have a variety of functions in the response to DNA damage and other stimuli. 1,3 Several recent studies demonstrated that p53 directly binds to and repairs damaged DNA by catalyzing DNA renaturation and strand transfer. 4,41,42 Such a function of p53 is presumed to be essential to the response to DNA damage in the conditional renewal system, including the lung cells, and in the resting cell populations, such as the enterocytes of the small intestinal villi, although further study on the response to DNA damage and the roles of p53 and other factors in noncycling cells is needed to confirm this possibility.
Acknowledgments
We thank Messrs. T. Suzuki and M. Ikeda for their technical assistance.
Footnotes
Address reprint requests to Dr. Koji Okudela, Department of Pathology, School of Medicine,Yokohama City University, 3–9 Fukuura, Kanazawa-ku, Yokohama 236-0004, Japan. E-mail: kojixok@med.yokohama-cu.ac.jp.
Supported in part by a grant from the Smoking Research Foundation, Japan.
References
- 1.Cox LS: Multiple pathways control cell growth and transformation: overlapping and independent activities of p53 and p21waf1/cip1/sdi1. J Pathol 1997, 183:134-140 [DOI] [PubMed] [Google Scholar]
- 2.Willson JW, Prichard DM, Hickman JA, Potten CS: Radiation induced p53 and p21WAF1/CIP1 expression in the murine intestinal epithelium. Apoptosis and cell cycle arrest. Am J Pathol 1998, 153:899-909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weinert T: DNA damage and check point pathways: molecular anatomy and interaction with repair. Cell 1998, 94:555-558 [DOI] [PubMed] [Google Scholar]
- 4.Bakalkin G, Yakovleva T, Selivanova G, Magnusson PK, Szekely L, Kiseleva E, Klein G, Terenius L, Wiman KG: p53 binds single-stranded DNA ends and catalyzes DNA renaturation and strand transfer. Proc Natl Acad Sci USA 1994, 91:413-417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.El-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B: WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75:817-825 [DOI] [PubMed] [Google Scholar]
- 6.Cayrol C, Kinbiehler M, Ducommun B: p21 binding to PCNA cause G1 and G2 cell cycle arrest in p53-deficient cells. Oncogene 1998, 16:311-320 [DOI] [PubMed] [Google Scholar]
- 7.Miyashita T, Reed JC: Tumor suppresser p53 is direct transcriptional activator of the human bax gene. Cell 1995, 80:293-299 [DOI] [PubMed] [Google Scholar]
- 8.Miyashita T, Krajewski S, Krajewska M, Wang HG, Lin HK, Leibermann DA, Hoffman B, Reed JC: Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene 1994, 9:1799-1805 [PubMed] [Google Scholar]
- 9.Guinee D, Jr, Brambilla E, Fleming M, Hayashi T, Rahn M, Koss M, Ferrans V, Travis W: The potential role of bax and bcl-2 expression in diffuse alveolar damage. Am J Pathol 1997, 151:999-1007 [PMC free article] [PubMed] [Google Scholar]
- 10.Strbel T, Swanson L, Korsmeyer S, Cannistra SA: Radiation-induced apoptosis is not enhanced by either p53 or Bax in SW626 cancer cells. Oncogene 1997, 14:2753-2758 [DOI] [PubMed] [Google Scholar]
- 11.Canman CE, Gilmer TM, Coutts SB, Kastan MB: Growth factor modulation of p53-mediated growth arrest versus apoptosis. Genes Dev 1995, 9:600-611 [DOI] [PubMed] [Google Scholar]
- 12.Hagimoto N, Kuwano K, Nomoto Y, Kunikake R, Hara N: Apoptosis and expression of Fas/Fas ligand mRNA in bleomycin-induced pulmonary fibrosis in mice. Am J Respir Cell Mol Biol 1997, 16:91-101 [DOI] [PubMed] [Google Scholar]
- 13.Kuwano K, Hagimoto N, Nomoto Y, Kawasaki M, Kunikake R, Fujita M, Miyazaki H, Hara N: p53 and p21 (Waf1/Cip1) mRNA expression associated with DNA damage and repair in acute immune complex alveolitis in mice. Lab Invest 1997, 76:161-169 [PubMed] [Google Scholar]
- 14.Barazzone C, Horowitz S, Donati YR, Rodriguez I, Piguet PF: Oxygen toxicity in mouse lung: pathways to cell death. Am J Respir Cell Mol Biol 1998, 19:573-581 [DOI] [PubMed] [Google Scholar]
- 15.Hayashi H, Miyamoto H, Ito T, Kameda Y, Nakamura N, Kubota Y, Kitamura H: Analysis of p21waf1/cip1 expression in normal, premalignant, and malignant cells during the development of human lung adenocarcinoma. Am J Pathol 1997, 151:461-470 [PMC free article] [PubMed] [Google Scholar]
- 16.Harrison JH, Jr, Hoyt DG, Lazo JS: Acute pulmonary toxicity of bleomycin: DNA scission and matrix protein mRNA level in bleomycin-sensitive and -resistant strains of mice. Mol Pharmacol 1989, 36:231-238 [PubMed] [Google Scholar]
- 17.Thrall RS, McCormick JR, Jack RM, McReynold RA, Ward PA: Bleomycin-induced pulmonary fibrosis in the rats. Am J Pathol 1979, 95:117-130 [PMC free article] [PubMed] [Google Scholar]
- 18.Smith RE, Strieter RM, Phan SH, Kunkel S: C-C chemokines: novel mediator of the profibritic inflammatory response to bleomycin challenge. Am J Respir Cell Mol Biol 1996, 15:693-702 [DOI] [PubMed] [Google Scholar]
- 19.Gharaee-Kermani M, Phan SH: Lung interleukin-5 expression in murine bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol 1997, 16:438-447 [DOI] [PubMed] [Google Scholar]
- 20.Hoyt DG, Lazo JS: Murine strain difference in acute lung injury and activation of poly (ADP-ribose) polymerase by in vitro lung slices to bleomycin. Am J Respir Cell Mol Biol 1992, 7:645-651 [DOI] [PubMed] [Google Scholar]
- 21.Piguet PF, Vesin C: Pulmonary platelet trapping induced by bleomycin: correlation with fibrosis and involvement of the β integrin. Int J Exp Pathol 1994, 75:321-328 [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang K, Flanders KC, Phan SH: Cellular localization of transforming growth factor-β expression in bleomycin-induced pulmonary fibrosis. Am J Pathol 1995, 147:352-361 [PMC free article] [PubMed] [Google Scholar]
- 23.Santana A, Saxena B, Noble NA, Gold LI, Marshall BC: Increased expression of transforming growth factor β isoforms (β1, β2, β3) in bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol 1995, 13:34-44 [DOI] [PubMed] [Google Scholar]
- 24.Hall PA, Mckee PH, Menage HD, Dover R, Lane DP: High level of p53 protein in UV-irradiated normal human skin. Oncogene 1993, 8:203-207 [PubMed] [Google Scholar]
- 25.Clarke AR, Howard LA, Harrison D, Winton DJ: p53, mutation frequency and apoptosis in the murine small intestine. Oncogene 1997, 14:2015-2018 [DOI] [PubMed] [Google Scholar]
- 26.Merritte AJ, Allen TD, Potten CS, Hickman JA: Apoptosis in small intestinal epithelia from p53-null mice: evidence for a delayed, p53-independent G1/M-associated cell death after a γ-irradiation. Oncogene 1997, 14:2759-2766 [DOI] [PubMed] [Google Scholar]
- 27.Donehower LA, Harvey M, Slagle BA, McArthur MJ, Montogomery CA, Jr, Butel JS, Bradley A: Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumor. Nature 1992, 356:215-221 [DOI] [PubMed] [Google Scholar]
- 28.Ito T, Udaka N, Hayashi H, Okudela K, Kanisawa M, Kitamura H: Ki-67 (MIB5) immunostaining of mouse lung tumors induced by 4-nitroquinoline 1-oxide. Histochem Cell Biol 1998, 110:589-593 [DOI] [PubMed] [Google Scholar]
- 29.Baeuerle PA, Baltimore D: NF-κβ B: ten years after. Cell 1996, 87:13-20 [DOI] [PubMed] [Google Scholar]
- 30.Fiedler MA, Wernke-Dollies K, Stark JM: Inhibition of TNFα-induced NF-κβ activation and IL-8 release in A549 cells with the proteasome inhibitor MG132. Am J Respir Cell Mol Biol 1998, 19:259-268 [DOI] [PubMed] [Google Scholar]
- 31.Lee S, Dimtchev A, Lavin MF, Dritschila A, Jung M: A novel ionizing radiation-induced signaling pathway that activates the transcriptional factor NF-κβ. Oncogene 1998, 17:1821-1826 [DOI] [PubMed] [Google Scholar]
- 32.Cohen AM, Philips FS, Sternberg SS: Studies on the cytotoxicity of bleomycin in the small intestine of the mouse. Cancer Res 1972, 32:1293-1300 [PubMed] [Google Scholar]
- 33.Ansari B, Coates PJ, Greenstein BD, Hall PA: In situ end-labeling detects DNA strand brakes in apoptosis, and other physiological, and pathological states. J Pathol 1993, 170:1-8 [DOI] [PubMed] [Google Scholar]
- 34.Coates PJ, Save V, Ansari B, Hall PA: Demonstration of DNA repair in individual cells using in situ end labeling. J Pathol 1995, 176:19-26 [DOI] [PubMed] [Google Scholar]
- 35.Gerdes J, Lemke H, Baisch H, Wacher H-H, Schwab U, Stein H: Cell cycle analysis of a cell proliferation associated human nuclear antigen defined by monoclonal antibody Ki-67. J Immunol 1984, 133:1710-1715 [PubMed] [Google Scholar]
- 36.Beisker W, Hittelman WN: Measurement of the kinetics of DNA repair synthesis after UV irradiation using immunohistochemical staining of incorporated 5-bromo-2-deoxyuridine and flow cytometry. Exp Cell Res 1988, 174:156-167 [DOI] [PubMed] [Google Scholar]
- 37.Lehtinen M, Kulomaa P, Kallioniemi OP, Paavonen J, Leinikki P: Analysis of DNA synthesis in herpes simplex-infected cells by the incorporation of 125IdU and BrdU. Arch Virol 1989, 107:215-223 [DOI] [PubMed] [Google Scholar]
- 38.Nagler RM, Nagler A: Effects of ionizing irradiation and β-adrenergic stimulation on gene expression pattern in rat submandibular glands. Anticancer Res 1996, 16:2749-2756 [PubMed] [Google Scholar]
- 39.Liu ZG, Baskaran R, Lea-Chou ET, Wood LD, Chen Y, Karin M, Wang JY: Three distinct signalling responses by murine fibroblasts to genotoxic stress. Nature 1996, 384:273-276 [DOI] [PubMed] [Google Scholar]
- 40.MacCallum DE, Hupp T, Midgley CA, Stuart D, Campbell S, Harper A, Walsh FS, Wright EC, Balmain A, Lane DP, Hall PA: The p53 response to ionizing radiation in adult and developing murine tissue. Oncogene 1996, 13:2575-2587 [PubMed] [Google Scholar]
- 41.Reed M, Woelker B, Wang P, Anderson ME, Tegtmeyer P: The C-terminal domain of p53 recognizes DNA damage by ionizing radiation. Proc Natl Acad Sci USA 1995, 92:9455-9459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee S, Elenbaas B, Levine AJ, Griffith J: p53 and its 14 kd C-terminal domain recognize primary DNA damage in the form of insertion/deletion mismatches. Cell 1995, 81:1016-1030 [DOI] [PubMed] [Google Scholar]