

Abstract
Coxsackieviruses are important human pathogens, frequently causing myocarditis, pancreatitis, and a variety of less severe diseases. B lymphocytes appear central to the interaction between these viruses and their mammalian hosts, because agammaglobulinemic humans, genetically incapable of antibody production, are susceptible to chronic infections by coxsackieviruses and related enteroviruses, such as poliovirus and echovirus. However, recent studies show that Type B coxsackievirus (CVB) infects B lymphocytes soon after infection, suggesting the possibility that these cells may play some role in virus dissemination and/or that the virus may be able to modulate the host immune response. We analyzed the role of B lymphocytes in CVB infection and confirmed that CVB infects B lymphocytes, and extended these findings to show that this is a productive infection involving approximately 1 to 10% of the cells; however, infectious center assays show that other splenocytes are infected at approximately the same frequency. Virus is readily detectable by in situ hybridization in the spleen of immunocompetent mice but is difficult to detect in mice deficient in B cells (BcKO mice), consistent with much of the splenic signal being the result of B cell infection. Surprisingly, given the extent of their infection, B cells express barely detectable levels of the murine coxsackievirus-adenovirus receptor (mCAR), suggesting that another means of cell entry may be used. We found no evidence of B cell depletion following CVB infection, indicating that this is not the explanation for the transient immunosuppression previously reported. Virus replication and dissemination are slightly delayed in BcKO mice, consistent with B cells’ playing a role as an important early target of infection and/or a means to distribute the virus to many tissues. In addition, we show that BcKO mice recapitulate a central feature of human agammaglobulinemia: CVB establishes chronic infection in a variety of organs (heart, liver, brain, kidney, lung, pancreas, spleen). In most of these tissues the viral titers remain high (105-108 plaque forming units (pfu) per gram of tissue) for the life of the mouse, and in several there is severe pathology, particularly severe myocardial fibrosis with ventricular dilation, reminiscent of the dilated cardiomyopathy seen in humans with chronic enteroviral myocarditis. Transfer of B and/or T cells from non-immune mice had no discernible effect, whereas equivalent transfers from immune mice often resulted in transient or permanent disappearance of detectable CVB.
Coxsackieviruses are members of the family Picornaviridae and lie in the enterovirus genus along with polioviruses, echoviruses, and unclassified enteroviruses. There are two types of coxsackievirus, A and B, classified by their pathogenicity in newborn mice, 1 and within each type are several strains. Type B coxsackieviruses (CVB) are common causes of human disease. As the genus name suggests, they replicate in the gastro-intestinal tract, but they are not restricted to this site; they also infect, for example, skeletal muscle (causing intercostal myalgia (Bornholm disease) and polymyositis), cardiac muscle (with resulting myocarditis), and acinar cells in the exocrine pancreas (causing fulminant pancreatitis), as well as various cells of the immune system. Foci of infection can also be found in the liver, kidney, central nervous system, spleen, and elsewhere. Enteroviruses infect humans with epidemic frequency, reflected by the remarkably high prevalence of myocarditis; necropsy of victims of accidental death revealed that approximately 1% had ongoing myocarditis. 2
Several avenues of research have implicated B cells as important to the interaction between the host and these viruses. First, antibodies seem to be critical to the control of these viral infections. Individuals genetically deficient in antibody production can resist most viruses but are susceptible to chronic enterovirus infections; although many of these are echoviral, 3,4 CVB encephalitis also has been noted. 5,6 Adoptive transfer of immune serum sometimes, but not always, ameliorates disease. Second, recent work suggests that B cells may be an important site of early CVB replication in vivo, 7,8 raising the possibility that these cells may contribute to the virus’ ability to establish or disseminate infection, and it is tempting to suggest that infection of these cells might contribute to the immunosuppression reported more than 20 years ago. 9,10
We therefore sought to clarify the role of B cells in CVB pathogenesis and investigated (i) whether CVB3 infection of B lymphocytes is productive, (ii) the frequency of productively infected B and non-B splenocytes, (iii) the splenic distribution of CVB in mice lacking B lymphocytes (BcKO mice), (iv) the role of the murine coxsackievirus-adenovirus receptor (mCAR) in B cell infection, (v) the contribution of B cells to early replication and dissemination of CVB3, (vi) the susceptibility of BcKO mice to long-term enteroviral infection as seen in human agammaglobulinemia, and (vii) the efficacy of B and T cell transfer in control of long-term infection.
Materials and Methods
Virus
A myocarditic strain of CVB3 (Nancy) was cloned and sequenced 11 (GenBank accession number U57056), and plasmid pH3, encoding this genome, was kindly provided by Dr. Kirk Knowlton of the University of California (San Diego, CA). Transfection of HeLa cells with this plasmid yields infectious virus, used for the studies reported below.
Mice
C57BL/6 mice (H-2b MHC haplotype) were acquired from The Scripps Research Institute (TSRI) breeding facility, and μMt/μMt (−/−) mice were purchased from Jackson Laboratories and bred at TSRI. The μMt mice (also H-2b) are B-cell-knockout (BcKO); a membrane exon of the gene encoding the μ chain was deleted, and B cell development in the resulting homozygous mice is arrested at or before the pre-B cell stage. 12 All mice used were 8 weeks of age or older.
Organ Preparation for Titrations and Histology
Immediately before removal of organs, mice were injected with 500 μl of 3.5% chloral hydrate. Mice were then perfused with normal saline to clear blood from organs, thus preventing overestimation of organ viral titers resulting from blood-borne CVB3. Heart, pancreas, liver, kidney, lung, spleen, and brain were removed from mice; half of each organ was placed in a cryotube and fast-frozen in liquid nitrogen for titrations. These materials were weighed and subsequently homogenized in 1 ml of serum-free Dulbecco’s modified Eagle’s medium (DMEM). The other half of the organ was fixed (Bouin’s or 10% normal buffered formalin) and embedded in paraffin for sectioning before appropriate histological analysis. Immunohistochemistry was carried out using 6-μm frozen sections.
Plaque Assays
Six-well plates were plated with 7.5 × 10 5 HeLa cells/well 24 hours before infection and grown overnight in a 37°C incubator with 5% CO2. Cells were 90 to 100% confluent at the time of titration. Organs to be titered were serially diluted (10-fold) in serum-free DMEM. Media was aspirated, and 400 μl of serially diluted homogenized organ was added to each well. Infected plates were placed back in the incubator for 1 hour and rocked every 10 minutes. After 1 hour, the inoculum was removed by aspiration, and cells were overlaid with 3 ml of 1× DMEM in 0.5% agar (1:1 mixture of 2× DMEM (Gibco-BRL, Gaithersburg, MD) at 37°C and 1% agar at 55°C). Between 40 and 50 hours after infection, cells were fixed by adding 2 ml of methanol:acetic acid (3:1 v/v) to each well and letting the plate sit for 10 minutes. The fixative was poured off, and agarose plugs were removed. Cells were stained with 1 ml 0.5% crystal violet in 20% ethanol per well. Plates were rinsed in tap water and plaques were counted.
Cell Sorting before Infectious Center Assay
Splenocytes from infected C57BL/6 mice were harvested 1, 2, 3, or 4 days after infection, and a single-cell suspension of splenocytes was prepared by homogenization, followed by lysis of red blood cells (5 minutes at room temperature, 0.83% NH4Cl). Polystyrene petri dishes (100 × 15mm) were coated with anti-Ig antibodies by incubation with 5 ml of buffer (0.05 mol/L Tris-HCl, 0.15 mol/L NaCl, pH 9.5) containing 16 μg of anti-Ig antibody and 350 μg of bovine serum albumin at room temperature for 1 hour or longer, followed by washes with phosphate-buffered saline (PBS) and PBS + 5% serum. Approximately 3 × 10 7 splenocytes, resuspended in 3 ml of PBS + 5% serum, were incubated on the coated dishes for 1 hour at 4°C. The B-cell-depleted supernatant was incubated on a second plate with 4 times more antibody to ensure complete depletion of B cells. The cells attached to the dish were washed with ice-cold PBS + 5% serum and then recovered in 5 ml of PBS + 5% serum (37°C) by vigorous pipetting. The efficiency of the depletion/enrichment was assessed by staining with fluorescein isothiocyanate (FITC)-labeled anti-B220 antibody followed by fluorescence-activated cell sorter (FACS) analysis.
Infectious Center Assay
Total splenocytes, as well as the B-cell-enriched and the B-cell-depleted fractions, were counted using Trypan blue, and serial 10-fold dilutions were made. One hundred microliters of each dilution were added to HeLa monolayers, allowed to settle, and overlaid with agar-containing medium. After 36 hours’ incubation, cells were fixed and stained and plaques were counted. Each plaque indicates a single productively infected cell.
Preparation of RNA and Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
Total liver RNA was obtained using TRIZOL reagent (Gibco-BRL) and the conditions recommended by the manufacturer. Purified B cell mRNA was obtained as follows: 5 × 10 6 splenocytes were mixed with 1.2 × 10 8 magnetic beads (PerSeptive Biosystems, Framingham, MA) coupled to anti-B220 antibody. After incubation, B220+ cells were selected using a magnetic field (MPC-2 magnet, Dynal, Lake Success, NY). Poly(A)+ RNA from the enriched B cells was obtained using Dynal’s mRNA Direct Kit, and the conditions recommended by the supplier. Briefly, B cells were lysed, and the lysate was incubated with oligo(dT)-coupled magnetic beads. The Dynal MPC-2 magnet was used to select the poly(A)+ RNA which was washed and eluted in low salt buffer at 65°C, and used as template for the synthesis of first strand cDNA using oligo(dT) as primer and RT Superscript II (Gibco). The first strand cDNA was used in a PCR reaction (35 cycles) with primers specific for the mCAR gene (mCAR1, 5′-GGCGCGCCTACTGTGCTTCG-3′; mCAR2, 5′-CTGCCAGCCATGGCGTAGGC-3′) or for the housekeeping gene glyceraldehyde phosphate dehydrogenase (GAPDH1, 5′-CCATCACCATCTTCCAGGAG-3′; GAPDH2, 5′-CCTGCTTCACCACCTTCTTG-3′).
In Situ Hybridization (ISH)
CVB3-specific, P33-labeled, RNA probes were transcribed in vitro from a linearized plasmid containing a 421-base fragment from the capsid region of CVB using RNA polymerase T7 to generate negative sense probe or T3 to generate positive sense probe. A previously published protocol 13 was followed using 5-μm paraffin sections from tissues previously fixed in 10% neutral buffered formalin. After preparation of the slides and prehybridization at 42°C, 2 × 10 6 to 5 × 10 6 cpm of the P33-labeled RNA probe was applied and allowed to hybridize overnight at 45°C. After washing, slides were dipped in photographic emulsion and held at 4°C for 1 to 7 days, when they were developed and fixed. Slides were then stained with hematoxylin (3–5 minutes) and eosin (2–3 minutes) and mounted.
Adoptive Transfers
C57BL/6 mice, previously infected with 500 plaque forming units (pfu) of CVB3, were killed at >4 weeks after infection and their spleens were removed and placed immediately in complete 1× DMEM (DMEM supplemented with 10% fetal bovine serum, 2 mmol/L L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin). Spleens were disrupted into a single-cell suspension and red blood cells were lysed using 0.83% ammonium chloride lysing buffer. Pan-B cells and pan-T cells were isolated from splenocytes using VarioMACS magnetic cell isolation kit (Miltenyi Biotec, Bergisch Gladbach, Germany). Samples of both B cell and T cell populations were FITC-labeled for FACS analysis to evaluate the efficiency of sorting. Recipient mice were uninfected BcKO mice that received one of six sets of inocula by the i.p. route: (i) no cells, (ii) B cells from an immune mouse, (iii) T cells from an immune mouse, (iv) a combination of B cells and T cells from an immune mouse, (v) B cells from a non-immune mouse, or (vi) T cells from a non-immune mouse. In all cases where cells were given, each recipient mouse received 0.5 × 10 7 to 4 × 10 7 lymphocytes (precise numbers varied between experiments, but all gave similar results). Immediately after adoptive transfer injections, all mice were challenged with 5000 pfu CVB3 i.p. All mice were monitored for survival, and serum CVB3 titers were done at days 4, 7, 14, 21, and 28. In a separate experiment, two chronically infected BcKO females were identified by the presence of high titers of virus at 21 days after infection; at 26 days postinfection both females were injected i.p. with 5.4 × 10 7 B cells from an immune C57BL/6 mouse. Virus titers were measured at the indicated times, and both mice were sacrificed at day 21 posttransfer for organ viral titers and histology.
Results
Identification of Productively Infected B and Non-B Cells
Others have reported that, in immunocompetent mice, CVB can be detected in splenic lymphocytes, particularly B cells, during the acute stage of infection. 7,8 However, the mere identification of virus RNA or protein within cells is not proof of productive infection because, at least in tissue culture, CVB can establish persistent infection in several cell types, 14,15 including lymphoid cells. 16 Although one study showed that splenocytes were productively infected, 7 the precise source of nascent virus was not identified, because cell sorting was not carried out before assay, nor were individual cells evaluated for virus production. To determine whether B cells were productively infected by CVB, we sorted the splenocytes from infected C57BL/6 mice at 1, 2, 3, and 4 days after infection (4 mice per time point), separating them into B cell and non-B cell populations, which were then evaluated by infectious center assay. For each cell population, dilutions of cells were plated on a HeLa cell monolayer, then immobilized by an agarose overlay. The development of plaques was observed by microscopy, the monolayers were fixed and stained, and the plaques were counted. As shown in Figure 1 ▶ , no infected cells were detected on day 1, but on days 2 and 3, approximately 1% of both cell populations scored positive for productive CVB infection. By day 4 after infection the number had decreased slightly, to approximately 0.5%. These data show that B cells are indeed productively infected; however, the frequency of B cell infection appears very similar to that of non-B cells. Note that positive controls were carried out in parallel to determine the sensitivity of the infectious center assay. HeLa cells were infected with a known number of pfu, and these infected cells were diluted and plated on an uninfected HeLa monolayer. However, the number of cells scoring positive usually were ∼10% of the number expected from the input pfu. Therefore, the absolute values in these experiments may be ∼10-fold underestimates of the percentages of B and non-B cells productively infected.
Figure 1.

Infectious center assay of B and non-B cells. C57BL/6 mice were infected with CVB3 (5000 pfu i.p.) and, at each of the indicated time points postinfection, 4 mice were sacrificed. The splenocytes were separated by panning into B cells and non-B cells, and these cells were analyzed by infectious center assay (see Materials and Methods). Each bar represents the mean number of infectious centers detected per 10 6 cells for each cell population. SE bars are included.
No Significant Depletion of B Cells during Acute CVB3 Infection
The productive infection of immune cells by CVB3 might have serious biological consequences for the host. Perhaps the most obvious is immunosuppression; transient global immunosuppression has been reported in this infection, 9 and it was suggested that this resulted from defects in splenic function, perhaps antigen presentation. 10 In these early studies the infection of lymphocytes, although sought, went undetected. Our studies, together with those of other labs, 7,8 suggest that at any given time in the acute infection, 1 to 10% of B cells are productively infected by this lytic virus. To exclude the possibility that large-scale depletion of B cells was effected by the virus, we carried out FACS analyses of the spleen at up to day 21 after infection; as shown in Figure 2 ▶ , the proportion of B cells increased transiently at 3 days after infection, most likely the result of virus-specific B cell proliferation in germinal centers, but remained quite stable thereafter, demonstrating that acute CVB infection does not dramatically reduce splenic B cell levels.
Figure 2.

CVB3 infection does not deplete splenic B cells. Mice were infected with CVB3 (500 pfu i.p.) and at the times indicated were sacrificed and their spleens harvested and disrupted into a single-cell suspension. The percentage of B cells was analyzed by FACS.
Delayed Replication and Dissemination of CVB3 in Mice Lacking B Cells
Thus, it appears that B cells are productively infected early in CVB infection, but that their numbers remain relatively stable as infection progresses. Do these cells act as an important primary target for CVB replication and/or might they be implicated in virus dissemination to other host tissues? To investigate this, we compared C57BL/6 mice with BcKO mice, which lack mature B lymphocytes. C57BL/6 or BcKO mice were infected, and CVB titers were measured at 12 hours and 1, 2, and 3 days postinfection. As shown in Figure 3 ▶ , virus replication/dissemination appeared delayed in BcKO mice. By 24 hours after infection a difference was readily detected; for example, virus was presented in the hearts of C57BL/6 mice, but not of BcKO mice. Of 10 tissue samples analyzed in each mouse strain at this time point, only 3 were positive in BcKO compared to 9 in C57BL/6; titers were much higher in the latter, reaching a maximum of ∼4 × 10 9 pfu/gram (pancreas) compared to a maximum almost 50,000-fold lower in the pancreas of one BcKO mouse. Marked differences were also noted in the serum and spleen. At day 2 after infection virus titers in serum, heart, and spleen of BcKO mice were 10-fold to 1000-fold lower than in the same tissues from C57BL/6 mice. By 3 days after infection, viral titers in BcKO mice were similar to those in immunocompetent mice, suggesting that viral replication cannot be constrained in the absence of antibody. These data show definitively that mature B cells are not required for CVB3 infection, but suggest strongly that these cells play a role early in infection, in providing the virus with a susceptible cell population and/or in disseminating the virus to distant tissues such as heart, spleen, and pancreas. Similar results were seen at early times in a separate experiment (see Figure 6 ▶ ).
Figure 3.

Delayed dissemination of CVB in BcKO mice. BcKO or C57BL/6 mice (8 mice/strain) were infected with CVB3 (5000 pfu i.p.), and tissues were harvested at 12 hours or 1, 2, or 3 days after infection (2 mice per strain per time point). Viral titers were determined, and corrected for tissue weight. Titers shown are per gram of tissue (or per milliliter, for serum). Gray bars, BcKO mice; black bars, C57BL/6 mice.
Figure 6.

Chronic high-titer coxsackievirus infection in B-cell-deficient mice. C57BL/6 and BcKO mice were infected with CVB3. At the indicated time points, 2 mice were bled, sacrificed, and perfused with saline. Serum and tissues were titered; mean titers are shown for 2 mice per strain at each time point. All titers are shown as pfu/gram, except for serum (pfu/ml). Open symbols, C57BL/6 mice; solid symbols, BcKO mice.
Marked Difference in CVB Distribution in Spleens of BcKO and Immunocompetent Mice
If B cells are an important focus of primary infection, one might anticipate differences in the distribution of CVB3 in the spleen when comparing BcKO mice with their immunocompetent counterparts. We used ISH to compare virus-infected cells in the spleens of immunocompetent and BcKO mice at days 2 and 3 postinfection. As shown in Figure 4, A and D ▶ , virus was difficult to detect by ISH in either mouse strain at 2 days after infection, although in C57BL/6 mice, which had a splenic titer of ∼5 × 10 8 pfu/gram (Figure 3) ▶ , a weak signal was detected in the marginal zone surrounding the white pulp (Figure 4B) ▶ . In contrast, the next day virus was readily detectable in the spleen of C57BL/6 mice (Figure 4E) ▶ , mainly in the outer areas of the white pulp (Figure 4F) ▶ , an area comprising predominantly B cells (Figure 4G) ▶ . Despite the much stronger ISH signal on day 3 after infection, the splenic virus titers in C57BL/6 mice are similar on both days, suggesting altered distribution of the infectious materials in the spleen; this has previously been described. 7,17 The strong day 3 signal was absent from the spleen of BcKO mice (Figure 4H) ▶ , consistent with the signal being predominantly in B cells. At this point it is impossible to determine whether the infected B cells seen in C57BL/6 mice in Figure 4 ▶ were infected in the spleen or whether they were infected in an extra-splenic area and later congregated in the spleen.
Figure 4.
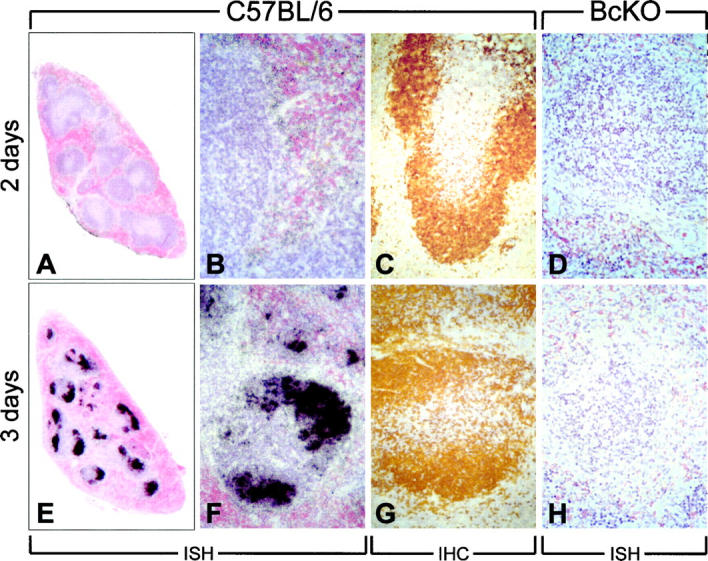
Distribution kinetics of CVB3 in spleens of normal and BcKO mice. C57BL/6 (A-C, E-G) or BcKO mice (D and H) were infected with CVB3, and spleens were harvested 2 or 3 days after infection. In situ hybridization was carried out using CVB-specific negative-sense RNA probe (ISH, A, B, D-F, and H), or sections were analyzed by immunohistochemistry (IHC, C and G) using B-cell-specific antibody.
Expression of mCAR on Purified B and Non-B Cells
To continue our investigation of CVB infection of splenic cells, we sought evidence of CVB receptor expression on the surfaces of B cells. Many enteroviruses, 18 including coxsackieviruses, 19,20 bind to the cell surface molecule decay accelerating factor (DAF), but DAF binding is insufficient for internalization; the latter requires another protein, 21 also important for internalization of adenovirus, and thus termed the coxsackievirus-adenovirus receptor (CAR). 22,23 The murine homologue, mCAR, has recently been cloned, 24 and the gene was generously provided to us by Dr. J. Bergelson. An RNA probe complementary to the ORF was used to evaluate mCAR mRNA levels in the spleens of C57BL/6 and BcKO mice by ISH, but no convincing signal was seen (not shown), leading us to attempt to identify mRNA expression in purified B cells using RT-PCR. As a positive control template we used total RNA from liver, a tissue known to be strongly positive for mCAR. 24 After 35 PCR cycles, mCAR mRNA signal from total liver RNA was easily visualized on an ethidium-stained gel (Figure 5A) ▶ and was overwhelmingly positive on Southern blot (Figure 5B) ▶ . In contrast, despite using oligo-dT-selected mRNA as template, the mCAR signal from purified B cells was invisible on the ethidium-stained gel (Figure 5A) ▶ and only very weakly positive on probing of the Southern blot (Figure 5B) ▶ . The quality of the oligo-dT-selected B cell mRNA was confirmed using control primers for the mRNA encoding the housekeeping enzyme GAPDH (Figure 5A) ▶ . Given the extraordinary sensitivity of RT-PCR followed by Southern blotting, it is difficult to conclude with certainty that this weak mCAR signal is B-cell-derived; it is possible that the signal comes from a low level of contamination by non-B cells. Identical RT-PCR analysis of total splenocytes also gave only a weak signal (not shown).
Figure 5.
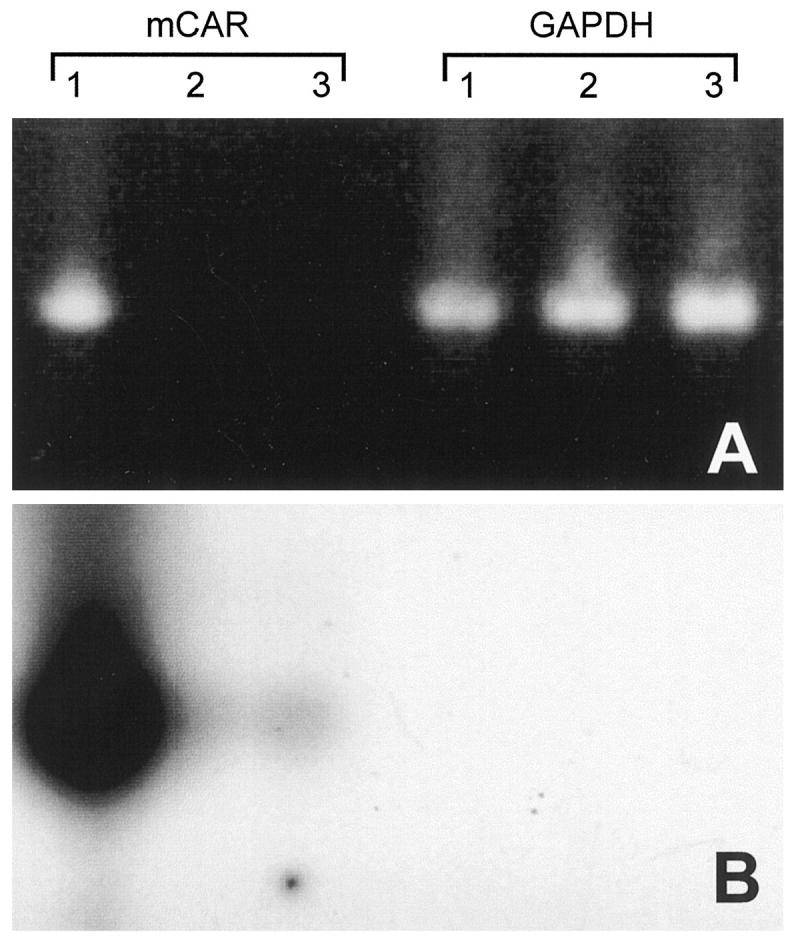
mCAR mRNA is barely detectable in B lymphocytes. Total RNA was prepared from liver (Lane 1), or mRNA was isolated from purified B cells of 2 C57BL/6 mice (Lanes 2 and 3), and equal amounts of RNA were used as templates in RT-PCR, using primers specific either for mCAR or for glyceraldehyde phosphate dehydrogenase (GAPDH), as indicated. Equal portions of the PCR reactions (35 cycles) were run on an agarose gel and visualized with ethidium bromide staining (A). A Southern blot transfer was carried out and probed with mCAR-specific 32P-labeled DNA. The resulting autoradiograph (2-hour exposure) is shown in B.
Lifelong Chronic Virus Infection of All Tested Tissues of BcKO Mice
The preceding studies compare immunocompetent and BcKO mice during the acute phase of infection and show differences in early virus titers (Figure 3) ▶ . This result is confirmed in Figure 6 ▶ , which also demonstrates the profound differences seen later in infection in BcKO mice. By day 14 after infection CVB3 had, as expected, been cleared from most organs in the C57BL/6 mice, whereas viral titers were high in BcKO mice. At 45 days after infection virus was undetectable in C57BL/6 mice but remained at high titers in all BcKO organs titered: heart, pancreas, brain, liver, kidney, lung, spleen, and serum. The chronically infected BcKO mice exhibited visible steatorrhea and profound wasting (perhaps the result of the malabsorption) and rarely survived beyond 60 days.
Severe Myocardial Damage in Chronically Infected BcKO Mice
The capacity of CVB to cause acute myocarditis and subsequent myocardial fibrosis in mice is well recognized; we have shown that both processes are exacerbated by CD8+ T cells 25 and, more specifically, require perforin. 26 BcKO mice can mount apparently normal CD8+ T cell responses 27 and one might expect that these mice would develop myocardial lesions. This is indeed the case. Acute myocarditis develops by 7 days after infection (not shown) and is maintained as ongoing myocarditis with concurrent fibrosis. By 45 days after infection severe scarring is visible, along with ventricular dilation (Figure 7A) ▶ . The fibrosis extends widely throughout the myocardium, and in some places spans at least 50% of the ventricular wall (Figure 7B) ▶ . ISH using a negative sense (genome-detecting) probe shows a focal pattern of virus distribution (Figure 7, C and D) ▶ similar to that seen in the acute phase in immunocompetent mice. 25 Despite the high virus titers and easily detected genomic-sense viral RNA, use of a positive sense probe yields a barely detectable signal (Figure 7, E and F) ▶ , indicating that there is a great excess of genome-sense RNA compared to antigenomic material. Others have shown that, during acute CVB infection when high levels of infectious virus are produced, the ratio of CVB genome to antigenome is approximately 100:1, but when persistence is established in immunocompetent mice, infectious virus is undetectable and the genome:antigenome ratio drops to around 1:1. 28 The virus RNA present in BcKO mice at 45 days after infection is predominantly genomic (compare Figure 7, C and D ▶ to Figure 7, E and F ▶ ), suggesting that the virus replication strategy remains as in acute infection, consistent with the high titers of virus shed.
Figure 7.
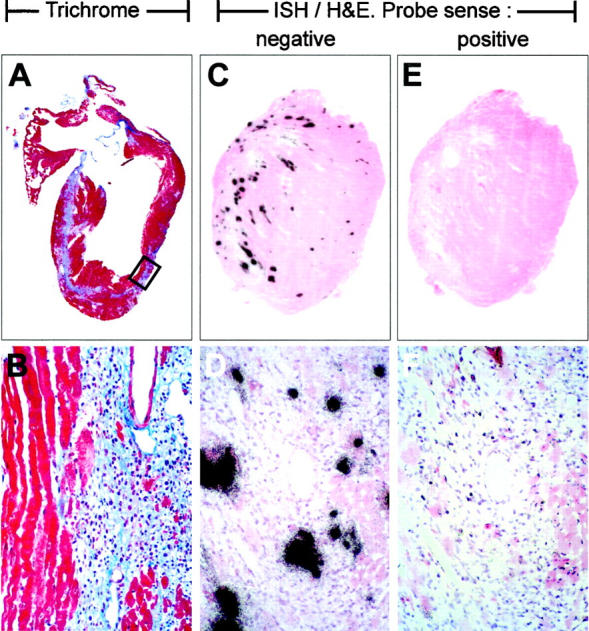
Histopathology and ISH at 45 days after infection in a BcKO mouse. Heart of a BcKO mouse taken 45 days postinfection and analyzed by histology. A and B: Trichrome stain, in which fibrous tissue appears light blue. The boxed area in A is enlarged in B; original magnification, ×80. C-F: ISH, counterstained with H&E. C and E are adjacent sections, and higher magnifications of identical areas (centered over a vessel) are shown in D and F, respectively. A negative-sense probe to detect CVB genomic-sense RNA resulted in the signal seen in C and D, whereas a positive-sense probe to detect genome-complementary RNA failed to generate a strong signal (E and F). Probes were of comparable specific activity.
Beneficial Effects of Adoptive Transfer of Cells from a CVB-Immune Mouse Immediately before Virus Challenge
Uninfected BcKO mice received one of six sets of cells by adoptive transfer; no cells, B cells, or B and T cells from non-immune C57BL/6 mice; or B cells, T cells, or B and T cells from CVB-immune C57BL/6 mice. The following day the BcKO mice were challenged with CVB and virus titers in serum were evaluated. As shown in Figure 8A ▶ , all mice developed viremia by 4 days after infection, and several died within a few days of infection. Of the surviving mice, none of those which received cells from a non-immune host cleared the virus, while 8 of 13 mice primed with immune cells showed transient (5 mice) or permanent (3 mice) clearance of detectable virus in the serum. Of these 8 mice, 7 received immune B cells (5 received B and T cells, 2 received only B cells); the eighth mouse received T cells alone. That B cells appear more effective than T cells in this assay is reflected by the fact that 100% (7 of 7 mice) of B cell recipients surviving beyond day 6 cleared virus at least transiently, whereas this occurred in only 55% of surviving T cell recipients (6 of 11 mice; 5 of these 6 mice had also received B cells).
Figure 8.

The effects of adoptive cell transfer on CVB infection in BcKO mice. A: B cells and/or T cells were transferred from CVB-immune or non-immune C57BL/6 mice into naïve BcKO mice, and 1 day later the recipient mice were infected with CVB3 (500 pfu i.p.). Sera were drawn at the indicated time points and were titered for virus. In each panel, different symbols represent individual recipient mice. Day of death is shown where relevant. B: At day 0 (arrow), B cells from immune C57BL/6 mice were transferred into BcKO mice chronically infected with CVB3. Sera were drawn at 4, 7, 14, and 21 days after transfer and were titered for virus. Mice were sacrificed on day 21 and their organs were titered (see text).
Adoptive Transfer of B Cells from a CVB-Immune Mouse Clears Chronic Infection from All Tissues of a BcKO Mouse
The above demonstrates that immune cells can limit acute viral replication in BcKO mice, sometimes effecting complete viral clearance. To confirm the therapeutic benefit of immune B cells and to extend the finding into a different clinical situation, 2 BcKO mice chronically infected with CVB3 were adoptively transferred with immune B-cells. We have shown previously that such chronically infected mice do not spontaneously clear CVB. As shown in Figure 8B ▶ , within 4 days of B cell transfer, virus was undetectable in the serum of both mice, and this clearance was accompanied by an improvement in the appearance of the mice (increased locomotor activity, normal posture, no ruffled fur). The serum clearance was maintained for at least 21 days, when the mice were killed and their organs titered. No virus was detected in any of the titered tissues (liver, heart, lung, spleen, brain, pancreas, kidney).
Discussion
We show here that B cells are infected with CVB, confirming the findings of other labs; however, we extend the analysis in several ways. First, we show that the B cell infection is productive, and that a significant number of B cells are infected (Figure 1) ▶ . Furthermore, virus appears to replicate in splenic B cells. B cells enter the spleen through the splenic artery and migrate through the marginal zone into the white pulp; CVB3 infection is first detected in the spleen at 2 days after infection, around the edge of the white pulp, in the marginal zone (Figure 4B) ▶ , and it appears likely that replication within these cells leads to the stronger splenic ISH signal at day 3 after infection, seen only in B-cell-containing mice (Figure 4, E and F) ▶ . The change in location of the ISH signal between day 2 and day 3 is consistent with migration of the infected B cells into the white pulp over this 24-hour period. In addition to being an early site of infection and replication, B cells play a role in virus dissemination, as demonstrated by the delayed spread at early times in BcKO mice (Figures 3 and 6) ▶ ▶ . Particularly striking is the finding that virus titers in solid tissues (eg, heart and pancreas) at 1 day postinfection are 103-fold to 105-fold lower in mice deficient in B cells. The potential benefits to the virus of replicating inside mobile host cells such as lymphocytes are clear; it can replicate while in transit to distant tissues, and at the same time sequester itself from neutralization by antibodies. This “Trojan horse” hypothesis is consistent with the recent observation that B cells containing CVB RNA are detectable within the myocardium. 29 However, although B cells play host to CVB3 in normal mice, virus dissemination does not require their presence, as shown by our demonstration of successful infection by and dissemination of CVB in mice lacking B lymphocytes (Figures 3 and 6) ▶ ▶ . Presumably, any role of B cells in dissemination can be paralleled by other splenocytes, albeit with a slight delay. Our infectious center analyses show that, at several time points after infection, the frequencies of productive infection in non-B cells are comparable to those of B cells (Figure 1) ▶ ; these data are underlined by the observation that splenic titers are similar at days 2 and 3 in C57BL/6 mice, despite the marked difference in ISH B cell signal. Thus, we speculate that the major benefit to the virus of B cell infection might lie not in virus dissemination, but instead in evasion of the host response. All mammalian viruses so far studied attempt to escape the host immune response, in many cases by disarming the aspect of the immune response that most strongly opposes them. For example, antibodies play a critical role in immunity to influenza viruses, and in response to this host pressure, the viruses undergo antigenic drift and shift. Conversely, herpesviruses, adenoviruses, and lymphocytic choriomeningitis virus are controlled mainly by T cells, and these viruses make strenuous attempts to avoid the unwelcome attention of T lymphocytes. As described above, antibodies play a vital role in controlling enteroviral infections, and the strikingly high titers of CVB3, which are maintained for up to 60 days in BcKO mice, underline the critical role played by B cells in controlling this infection. It is therefore tempting to suggest that the infection by CVB of B cells (and, perhaps, of other immunologically important cells) represents an attempt by the virus to undercut this antagonistic immune response. Studies published more than two decades ago suggested that coxsackievirus infection results in suppression of humoral immune responses. 9 We show here that CVB infection does not effect a wholesale depletion of B cells, but other possible immunosuppressive consequences of CVB infection of immune cells are under investigation. It will be interesting to determine the antigen specificity of the B cells that become infected; it has been shown in the lymphocytic choriomeningitis virus (LCMV) model that B cells specific for the viral glycoprotein are specifically deleted, apparently because B cells displaying glycoprotein -specific antibodies are selectively infected, and subsequently destroyed by anti-LCMV cytotoxic T lymphocytes. 30
For many years it was considered likely that, for any given virus, a single cell surface protein would act as its receptor. However, it is now recognized that, for several viruses, cell binding and entry may require more than one protein, acting serially or in concert. Many enteroviruses, 21,31 including CVB types 1, 3, and 5, 20 bind to the cell surface protein CD55 (DAF, a member of the complement regulation system). However, DAF binding does not invariably result in infection 20 and, conversely, some CVB3 isolates fail to bind DAF, but nevertheless can infect and cause disease in mice. 19 Recently a second protein has been identified that appears to be critical to cell entry. 22,24 This protein, also used by adenovirus and hence termed the coxsackievirus-adenovirus receptor (CAR), is widely distributed (as judged by Northern blot analysis of RNA expression), having been found on liver, heart, lung, and kidney; however, it is barely detectable in human spleen, and was not detected in mouse spleen. 24,32 Because we found extensive productive B cell infection, we evaluated mCAR mRNA expression in B cells and found it to be extremely low, detectable only after Southern blot analysis of PCR products (Figure 5B) ▶ . These results suggest that mCAR may not be required for B cell infection. Because the tissue tropisms of coxsackievirus and adenovirus are markedly different, it seems likely that additional factors, rather than CAR, are the ultimate determinants of coxsackievirus and adenovirus tropism. How else might CVB enter B cells? DAF is one candidate and is expressed on B cells. A recent study implicated complement component 3 in localization of CVB3 antigen to splenic germinal centers, and the authors suggested that this component may act as a bridge through which DAF binds CVB. 17 Our findings are consistent with this hypothesis and suggest that such interactions may permit B cell infection, obviating the need for mCAR.
CVB infection in mouse and man can result in both acute and chronic myocarditis, the latter sometimes leading to dilated cardiomyopathy. In chronically infected BcKO mice the heart was severely scarred and dilated (Figure 7) ▶ , as is often seen in human dilated cardiomyopathy, which is one of the most serious sequelae of acute viral myocarditis and which can be treated effectively only by heart transplantation. 33,34 The mechanism underlying the human chronic inflammatory disease is controversial. Although most observers agree that there is a large immunopathological component, some groups believe it to be autoimmune; others advance virus-specific explanations. CD8+ T cells greatly exacerbate acute myocarditis 25 by perforin-mediated lysis of infected cells. 26 We are currently investigating whether the chronic myocarditis seen in BcKO mice is dependent on CD8+ T cells. Others have suggested that CVB-induced myocarditis may be mediated by anti-myosin antibodies induced by CVB infection, 35 although this is controversial. 36 The data in the present study demonstrate clearly that the ongoing myocarditis in BcKO mice cannot be antibody-mediated. If the chronic myocarditis is indeed driven by virus-specific immune responses, rather than by autoimmune phenomena, then at the very least some CVB antigen must be present in the target cells. Although CVB infection, in common with most picornaviruses, is often rapidly cytolytic, there is solid evidence that the virus can persist in tissue culture cell populations continually passaged for 1 year. 15,16 Here we show that long-term virus shedding takes place in B-cell-deficient mice. Others have previously demonstrated long term infection in T-cell-deficient mice. 37,38 Thus, chronic CVB infection can occur in vivo. However, is this limited to immunodeficient mice? Although there appears to be little doubt that viral RNA can be detected up to at least 30 days postinfection in several immunocompetent mouse strains, 28,39 there is little evidence in such mice of long-term productive infection. We have confirmed the presence of CVB RNA in various organs of C57BL/6 mice up to 45 days after infection, but we have not succeeded in isolating infectious virus (Mena, Fischer, and Whitton, unpublished). However, other picornaviruses (for example, Theiler’s virus), can persist for many months in immunocompetent mice, and it would be premature to exclude this possibility for coxsackieviruses. In humans there is little evidence that infectious virus persists, and although enteroviral RNA sequences have been reported in various tissues long after the apparent primary infection, 40 even this issue is controversial. 41 But many studies of human heart used myocardial biopsy material which is, for obvious reasons, limited in quantity. As we show in Figure 7 ▶ , CVB distribution is very focal even at a titer of 10 8 pfu/gram of heart tissue, so perhaps it is not surprising that CVB materials sometimes cannot be identified in biopsies of hearts in which infectious virus is undetectable.
Several of our findings in BcKO mice parallel those in human X-linked agammaglobulinemia, and thus may serve as a model with which to evaluate novel approaches to the management of this syndrome, in which susceptibility to microbial infection is a major concern. Many of these infections are established by pyogenic bacteria, but certain virus infections also are more common. For example, agammaglobulinemics are susceptible to chronic enteroviral infections 3,4 ; they may continue to shed virulent poliovirus for many years following live virus vaccination 42 ; and CVB can establish long-term productive infection with encephalitis. 5,6 In the present study we demonstrate long-term productive CVB infection of several tissues including the brain (Figure 6) ▶ . We have previously shown that CD8+ T cells reduce CVB load by up to 95%, 25 and BcKO appear to mount normal CD8+ T cell responses, 27 although they may have ancillary immune defects. 43 However, the chronic infection of CVB3 in BcKO mice shows that CD8+ T cells alone cannot control CVB infection in the absence of antibodies, underlining the enormous importance of humoral immunity in the control and eradication of this virus. During acute CVB infection the ratio of CVB genome to antigenome is approximately 100:1. Studies have suggested that, when persistence is established in immunocompetent mice, the genome:antigenome ratio changes to around 1:1. 28 The virus RNA present in BcKO mice at 45 days after infection is predominantly genomic (compare Figure 7, C and D ▶ to Figure 7, E and F ▶ ), consistent with the high titers of virus shed. Thus, in BcKO mice the virus maintains an acute replication scheme. We suggest that, in normal mice, the antibody response imposes on the virus the requirement that it alter its replication strategy, resulting in a 1:1 genome:antigenome ratio and undetectable infectious virus, whereas in BcKO mice (or in agammaglobulinemic humans), the lack of selective pressure from antibodies allows the virus to retain a 100:1 genomic ratio, along with the high levels of infectious virus. The BcKO mouse therefore appears to be a good model for enterovirus infection in human agammaglobulinemia. Further adoptive transfer studies may help us identify the mechanisms underpinning CVB persistence in an immunocompetent host.
Acknowledgments
We thank Annette Lord for excellent secretarial support. This is manuscript 11983-NP from The Scripps Research Institute.
Footnotes
Address reprint requests to J. Lindsay Whitton, Department of Neuropharmacology, CVN-9, The Scripps Research Institute, 10550 N. Torrey Pines Road, La Jolla, CA 92037. E-mail: lwhitton@scripps.edu.
Supported by National Institutes of Health grant AI-42314.
References
- 1.Hyypia T, Kallajoki M, Maaronen M, Stanway G, Kandolf R, Auvinen P, Kalimo H: Pathogenetic differences between coxsackie A and B virus infections in newborn mice. Virus Res 1993, 27:71-78 [DOI] [PubMed] [Google Scholar]
- 2.Gravanis MB, Sternby NH: Incidence of myocarditis: a 10-year autopsy study from Malmo, Sweden. Arch Pathol Lab Med 1991, 115:390-392 [PubMed] [Google Scholar]
- 3.Misbah SA, Spickett GP, Ryba PC, Hockaday JM, Kroll JS, Sherwood C, Kurtz JB, Moxon ER, Chapel HM: Chronic enteroviral meningoencephalitis in agammaglobulinemia: case report and literature review. J Clin Immunol 1992, 12:266-270 [DOI] [PubMed] [Google Scholar]
- 4.McKinney REJ, Katz SL, Wilfert CM: Chronic enteroviral meningoencephalitis in agammaglobulinemic patients. Rev Infect Dis 1987, 9:334-356 [DOI] [PubMed] [Google Scholar]
- 5.Geller TJ, Condie D: A case of protracted coxsackie virus meningoencephalitis in a marginally immunodeficient child treated successfully with intravenous immunoglobulin. J Neurol Sci 1995, 129:131-133 [DOI] [PubMed] [Google Scholar]
- 6.Hertel NT, Pedersen FK, Heilmann C: Coxsackie B3 virus encephalitis in a patient with agammaglobulinaemia. Eur J Pediatr 1989, 148:642-643 [DOI] [PubMed] [Google Scholar]
- 7.Anderson DR, Wilson JE, Carthy CM, Yang D, Kandolf R, McManus BM: Direct interactions of coxsackievirus B3 with immune cells in the splenic compartment of mice susceptible or resistant to myocarditis. J Virol 1996, 70:4632-4645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klingel K, Stephan S, Sauter M, Zell R, McManus BM, Bultmann B, Kandolf R: Pathogenesis of murine enterovirus myocarditis: virus dissemination and immune cell targets. J Virol 1996, 70:8888-8895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bendinelli M, Ruschi A, Campa M, Toniolo A: Depression of humoral and cell-mediated immune responses by coxsackieviruses in mice. Experientia 1975, 31:1227-1229 [DOI] [PubMed] [Google Scholar]
- 10.Bendinelli M, Matteucci D, Toniolo A, Patane AM, Pistillo MP: Impairment of immunocompetent mouse spleen cell functions by infection with coxsackievirus B3. J Infect Dis 1982, 146:797-805 [DOI] [PubMed] [Google Scholar]
- 11.Knowlton KU, Jeon ES, Berkley N, Wessely R, Huber SA: A mutation in the puff region of VP2 attenuates the myocarditic phenotype of an infectious cDNA of the Woodruff variant of coxsackievirus B3. J Virol 1996, 70:7811-7818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kitamura D, Roes J, Kuhn R, Rajewsky K: A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature 1991, 350:423-426 [DOI] [PubMed] [Google Scholar]
- 13.Lane TE, Paoletti AD, Buchmeier MJ: Disassociation between the in vitro and in vivo effects of nitric oxide on a neurotropic murine coronavirus. J Virol 1997, 71:2202-2210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cao Y, Schnurr DP: Persistent infection of YAC-1 cells by coxsackievirus B3. J Gen Virol 1988, 69:59-65 [DOI] [PubMed] [Google Scholar]
- 15.Schnurr DP, Schmidt NJ: Persistent infection of mouse fibroblasts with coxsackievirus. Arch Virol 1984, 81:91-101 [DOI] [PubMed] [Google Scholar]
- 16.Matteucci D, Paglianti M, Giangregorio AM, Capobianchi MR, Dianzani F, Bendinelli M: Group B coxsackieviruses readily establish persistent infections in human lymphoid cell lines. J Virol 1985, 56:651-654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anderson DR, Carthy CM, Wilson JE, Yang D, Devine DV, McManus BM: Complement component 3 interactions with coxsackievirus B3 capsid proteins: innate immunity and the rapid formation of splenic antiviral germinal centers. J Virol 1997, 71:8841-8845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bergelson JM, Chan M, Solomon KR, St. John NF, Lin H, Finberg RW: Decay-accelerating factor (CD55), a glycosylphosphatidylinositol-anchored complement regulatory protein, is a receptor for several echoviruses. Proc Natl Acad Sci USA 1994, 91:6245-6249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bergelson JM, Mohanty JG, Crowell RL, St. John NF, Lublin DM, Finberg RW: Coxsackievirus B3 adapted to growth in RD cells binds to decay-accelerating factor (CD55). J Virol 1995, 69:1903-1906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shafren DR, Bates RC, Agrez MV, Herd RL, Burns GF, Barry RD: Coxsackieviruses B1, B3, and B5 use decay accelerating factor as a receptor for cell attachment. J Virol 1995, 69:3873-3877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shafren DR, Dorahy DJ, Ingham RA, Burns GF, Barry RD: Coxsackievirus A21 binds to decay-accelerating factor but requires intercellular adhesion molecule 1 for cell entry. J Virol 1997, 71:4736-4743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bergelson JM, Cunningham JA, Droguett G, Kurt-Jones EA, Krithivas A, Hong JS, Horwitz MS, Crowell RL, Finberg RW: Isolation of a common receptor for coxsackie B viruses and adenoviruses 2 and 5. Science 1997, 275:1320-1323 [DOI] [PubMed] [Google Scholar]
- 23.Shafren DR, Williams DT, Barry RD: A decay-accelerating factor-binding strain of coxsackievirus B3 requires the coxsackievirus-adenovirus receptor protein to mediate lytic infection of rhabdomyosarcoma cells. J Virol 1997, 71:9844-9848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bergelson JM, Krithivas A, Celi L, Droguett G, Horwitz MS, Wickham T, Crowell RL, Finberg RW: The murine CAR homolog is a receptor for coxsackie B viruses and adenoviruses. J Virol 1998, 72:415-419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Henke A, Huber SA, Stelzner A, Whitton JL: The role of CD8+ T lymphocytes in coxsackie virus B3-induced myocarditis. J Virol 1995, 69:6720-6728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gebhard JR, Perry CM, Harkins S, Lane T, Mena I, Asensio VC, Campbell IL, Whitton JL: Coxsackievirus B3-induced myocarditis: perforin exacerbates disease, but plays no detectable role in virus clearance. Am J Pathol 1998, 153:417-428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Asano MS, Ahmed R: CD8 T cell memory in B cell-deficient mice. J Exp Med 1996, 183:2165-2174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klingel K, Hohenadl C, Canu A, Albrecht M, Seemann M, Mall G, Kandolf R: Ongoing enterovirus-induced myocarditis is associated with persistent heart muscle infection: quantitative analysis of virus replication, tissue damage, and inflammation. Proc Natl Acad Sci USA 1992, 89:314-318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klingel K, Rieger P, Mall G, Selinka HC, Huber M, Kandolf R: Visualization of enteroviral replication in myocardial tissue by ultrastructural in situ hybridization: identification of target cells and cytopathic effects. Lab Invest 1998, 78:1227-1237 [PubMed] [Google Scholar]
- 30.Planz O, Seiler P, Hengartner H, Zinkernagel RM: Specific cytotoxic T cells eliminate cells producing neutralizing antibodies. Nature 1996, 382:726-729 [DOI] [PubMed] [Google Scholar]
- 31.Karnauchow TM, Tolson DL, Harrison BA, Altman E, Lublin DM, Dimock K: The HeLa cell receptor for enterovirus 70 is decay-accelerating factor (CD55). J Virol 1996, 70:5143-5152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tomko RP, Xu R, Philipson L: HCAR and MCAR: the human and mouse cellular receptors for subgroup C adenoviruses and group B coxsackieviruses. Proc Natl Acad Sci USA 1997, 94:3352-3356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heck CF, Shumway SJ, Kaye MP: The Registry of the International Society for Heart Transplantation: sixth official report 1989. J Heart Transplant 1989, 8:271-276 [PubMed] [Google Scholar]
- 34.Editorial: dilated cardiomyopathy and enteroviruses. Lancet 1990, 336:971–973 [PubMed]
- 35.Wolfgram LJ, Beisel KW, Rose NR: Heart-specific autoantibodies following murine coxsackievirus B3 myocarditis. J Exp Med 1985, 161:1112-1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neu N, Ploier B, Ofner C: Cardiac myosin-induced myocarditis: heart autoantibodies are not involved in the induction of the disease. J Immunol 1990, 145:4094-4100 [PubMed] [Google Scholar]
- 37.Sato S, Tsutsumi R, Burke A, Carlson G, Porro V, Seko Y, Okumura K, Kawana R, Virmani R: Persistence of replicating coxsackievirus B3 in the athymic murine heart is associated with development of myocarditic lesions. J Gen Virol 1994, 75:2911-2924 [DOI] [PubMed] [Google Scholar]
- 38.Schnurr DP, Cao Y, Schmidt NJ: Coxsackievirus B3 persistence, and myocarditis in N: NIH(S) II nu/nu, and +/nu mice. J Gen Virol 1984, 65:1197-1201 [DOI] [PubMed] [Google Scholar]
- 39.Tam PE, Schmidt AM, Ytterberg SR, Messner RP: Duration of virus persistence and its relationship to inflammation in the chronic phase of coxsackievirus B1-induced murine polymyositis. J Lab Clin Med 1994, 123:346-356 [PubMed] [Google Scholar]
- 40.Muir P, Archard LC: There is evidence for persistent enterovirus infections in chronic medical conditions in humans. Rev Med Virol 1994, 4:245-250 [Google Scholar]
- 41.Melchers W, Zoll J, van Kuppeveld F, Swanink C, Galama J: There is no evidence for persistent enterovirus infections in chronic medical conditions in humans. Rev Med Virol 1994, 4:235-243 [Google Scholar]
- 42.Kew OM, Sutter RW, Nottay BK, McDonough MJ, Prevots DR, Quick L, Pallansch MA: Prolonged replication of a type 1 vaccine-derived poliovirus in an immunodeficient patient. J Clin Microbiol 1998, 36:2893-2899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Homann D, Tishon A, Berger DP, Weigle WO, von Herrath MG, Oldstone MBA: Evidence for an underlying CD4 helper and CD8 T-cell defect in B-cell-deficient mice: failure to clear persistent virus infection after adoptive immunotherapy with virus-specific memory cells from μm MT/μm MT mice. J Virol 1998, 72:9208-9216 [DOI] [PMC free article] [PubMed] [Google Scholar]