

Abstract
Inflammation of the intestine causes pain and altered motility, at least in part through effects on the enteric nervous system. While these changes may be reversed with healing, permanent damage may contribute to inflammatory bowel disease (IBD) and post-enteritis irritable bowel syndrome. Since little information exists, we induced colitis in male Sprague-Dawley rats with dinitrobenzene sulfonic acid and used immunocytochemistry to examine the number and distribution of enteric neurons at times up to 35 days later. Inflammation caused significant neuronal loss in the inflamed region by 24 hours, with only 49% of neurons remaining by days 4 to 6 and thereafter, when inflammation had subsided. Eosinophils were found within the myenteric plexus at only at the earliest time points, despite a general infiltration of neutrophils into the muscle wall. While the number of myenteric ganglia remained constant, there was significant decrease in the number of ganglia in the submucosal plexus. Despite reduced neuronal number and hyperplasia of smooth muscle, the density of axons among the smooth muscle cells remained unchanged during and after inflammation. Intracolonic application of the topical steroid budesonide caused a dose-dependent prevention of neuronal loss, suggesting that evaluation of anti-inflammatory therapy in inflammatory bowel disease should include quantitative assessment of neural components.
Inflammatory bowel disease (IBD) is a chronic idiopathic inflammation of the intestine that affects an increasing percentage of the population in Western society, with the highest incidence among the younger population, for whom there are no specific or effective treatments. The broad acting anti-inflammatory agents in wide use have serious side effects of immune suppression, loss of bone calcium, and growth retardation. Therefore, a strong need exists for increased information about the cellular basis of this disease, as well as new pharmacological tools and improved methods of use.
Studies of IBD have relied heavily on immunological approaches, relating activation of immune cells to the periodic exacerbation and remissions of disease. However, a particular challenge lies in understanding the long-term or permanent changes present in the intestine in IBD, which are evident even in the periods of remission between acute episodes. Recently, attention has been paid to the other cell types in the intestine that may acquire the ability to participate in inflammation or that are previously unexpected targets of inflammatory change. 1 This has shown that nonimmune cells can participate directly in inflammation, as well as pointing out the potential for long-term alterations in cell structure and function that may predispose to repeated episodes of inflammation. Thus, intestinal smooth muscle has been shown to have the potential to present antigen to activated T cells and may also be a source of cytokines that can directly affect neural function. 1,2
Most, if not all, aspects of normal gut function can be traced back to the correct functioning of the enteric nervous system, which resides within the intestinal wall as ganglionated plexuses of considerable structural complexity and integrative capacity. That transient or even permanent alterations to the intrinsic nervous system of the intestine (ENS) can occur in IBD is supported by an array of findings, such as evidence for altered sensory perception, shifts in amount and proportion of neurotransmitters, and the direct action of proinflammatory cytokines on neural function. 1 Cytokines present in the inflamed intestine such as interleukin (IL)-1 and IL-3 3 can alter release of neurotransmitters and may thus affect the ENS acutely. However, longer term challenges to neural function may come from either an adaptive response to an altered cellular environment, such as a requirement for innervation for newly arisen target cells in the mucosa or smooth muscle layers or from irreversible damage to the non-renewing population of neurons. Earlier, we had shown that intestinal inflammation causes extensive smooth muscle growth in rat models of jejunitis and colitis, 4 a process that contributes to the thickened intestinal wall seen in IBD and can ultimately lead to intestinal stricturing in Crohn’s disease. This is evidence of both an increase in cells which are normally innervated, as well as further evidence that inflammation can affect cells throughout the intestinal wall. Therefore, we used a model of chemically induced colitis in the rat 5,6 to study the effects of inflammation on neuronal number in the ENS, as well as axonal density within the smooth muscle. In addition, we have explored the potential for beneficial effects of topical application of the novel steroid budesonide of preserving intestinal innervation.
Materials and Methods
Animals
Male Sprague-Dawley rats (180–200 g) were obtained from Charles River Laboratories (Quebec, Canada) and were housed in pairs in microfilter isolated cages with free access to food and water. All experimental procedures were approved by the local Animal Research Ethics Board, in accordance with the guidelines of the Canadian Council on Animal Care.
All rats were housed for at least 7 days before experimental use, and food was removed for 24 hours before induction of colitis by intrarectal instillation of dinitrobenzene sulfonic acid (DNBS; ICN). 6 For this, 29 mg of DNBS was dissolved in 250 μl of 50% ethanol and instilled into the colon 8 cm proximal to the anus with a PE50 catheter while the rat was under light anesthesia. Control and vehicle control groups received either 0.9% saline or 50% ethanol alone. Some animals received the anti-inflammatory steroid budesonide (Sigma) in 10% ethanol at doses from 300 to 1000 μg/kg body weight, delivered by rectum as a 1-ml solution, as described by Jacobson et al. 7 Budesonide was given at 24 hours and 2 hours before the installation of DNBS or the control solutions and at daily intervals thereafter.
Histology
Animals were sacrificed by cervical dislocation under halothane anesthesia at various times after initiation of colitis. The descending colon was rapidly removed after noting the location of the inflamed region and fixed in 10% neutral buffered formalin for 24 hours before routine processing for paraffin sectioning. For longitudinal sections, the colon was cut open along the midline following fixation and embedded so that subsequent sectioning produced parallel sections through the mid-region. For cross-sections, 0.5-cm segments of colon were removed beginning distal to the middle of the inflamed region. For comparison, 0.5-cm segments of noninflamed colon were taken beginning 0.5 cm proximal to the margin of the inflamed region.
Histological sections (4 μm) were stained with hematoxylin and eosin for routine examination or with Congo red for detection of eosinophils. Immunocytochemistry with an antibody to the pan-neuronal marker PGP 9.5 (UltraClone, Isle of Wight, UK) was used to detect neurons and their extensions, with visualization with diaminobenzidine and counterstaining with hematoxylin.
Ganglia were identified as discrete aggregations of PGP-positive cells, located either between the smooth muscle layers (myenteric plexus) or between the mucosa and circular smooth muscle layer (submucosal plexus). Within the ganglia, neurons were considered as cells with PGP-positive cytoplasm and a hematoxylin-stained nucleus. The number and distribution of myenteric neurons and ganglia were determined in longitudinal sections by analysis of adjacent microscope fields throughout the length of the inflamed area, defined as showing mucosal damage and inflammatory infiltrate. This represented at least 35 adjacent microscope fields (approximately 7.5 mm) in each of the two parallel intestinal profiles for each tissue. The mean number of neurons per millimeter was calculated for each tissue, and then averaged among animals (n = 5 to 7 per time point). The mean number of ganglia/mm of colon was obtained similarly. To detect changes in the neuronal content of the ganglia, the average number of neurons appearing within the myenteric or submucosal ganglia was determined in these sections. This was reported as the arbitrary score of apparent neuronal content, a value that is proportional to the total number of neurons within the ganglia. These estimates of neuronal number and distribution require a constant nuclear diameter for comparison among different conditions. 8 This was verified by image analysis (data not shown).
The density of innervation was considered to be proportional to the number of axons (“axon density”) and this was determined by counting the number of PGP-positive axon profiles in 6 nonadjacent microscope fields taken within each of the longitudinal or circular smooth muscle layers, with exclusion of fields containing blood vessels or the myenteric plexus. At the same time, the number of smooth muscle nuclei within the fields was recorded, using nuclear appearance to exclude immune cells. The axon density was calculated and expressed as axons per 10 smooth muscle nuclei.
Myeloperoxidase
Samples of colon (approximately 0.5 cm) were removed from the inflamed region or from areas 1.5 to 2.0 cm proximal to the affected region, cleaned of mesentery and luminal contents, snap-frozen in liquid nitrogen and stored at −70°C until assay. The tissues were weighed, homogenized on ice in buffer, centrifuged for 2 minutes (13,000 × g) and 10-μl aliquots reacted with 100 μl of the peroxidase substrate (TNB peroxidase substrate system; K&P) for 10.0 minutes, when the reaction was stopped with 100 μl of sulfuric acid. The optical density (450 nm) of the reaction product was determined and expressed as ng/ml based on a standard curve using horseradish peroxidase (Sigma).
Statistical Analysis
Results are expressed as the mean ± SE of n observations, where n is the number of animals. Statistical significance was determined by analysis of variance, where P < 0.05 was considered significant.
Results
DNBS Colitis
Rats receiving DNBS developed bloody diarrhea by day 6, while control groups receiving either saline or ethanol solution appeared normal. DNBS-treated rats showed an initial weight loss over the first 2 days, which was then reversed to show an average weight gain by day 6 of 6.7 ± 0.8% 9 relative to starting values, while the saline control group increased in weight by 19.0 ± 0.3% 9 over the same period. By day 6, the mid-descending colon of DNBS-treated rats showed a prominently inflamed region, with a mean length of 2.7 ± 0.4 10 cm. Figure 1A ▶ shows the typical microscopic appearance of the colon on day 6, with severe mucosal damage, submucosal ulceration, and prominent inflammatory infiltrate. No abnormalities were detected in the histology of tissues from animals receiving saline or ethanol solutions. Examination of cohort rats on day 35 after DNBS treatment showed an overtly normal colon.
Figure 1.

Changes to the myenteric plexus during DNBS colitis. A: Typical appearance of the inflamed region of the colon on day 6, showing mucosal damage and extensive inflammatory infiltrate (asterisk) between the mucosa (MU) and smooth muscle (SM). B: Typical appearance of PGP-9.5 immunocytochemistry on day 6, showing positively stained neurons within a ganglion of the myenteric plexus (large arrow), and numerous darkly stained axons (small arrows) within the longitudinal (left) and circular (right) muscle layers. C: Quantitation of PGP-9.5-stained neuronal nuclei within the ganglia of the myenteric plexus of the rat colon during DNBS colitis. There was significant reduction in myenteric neurons per millimeter compared with control (saline) by day 1, within further decrease by day 2. Values were similar from day 2 to day 35. Inset: Examination of myenteric neurons per millimeter at early time points following DNBS treatment, showing a significant decrease by 24 hours, with nonsignificant decrease by 6 hours. No significant differences were seen between saline and ethanol controls at any time points. Bars, mean ± SEM of 5 to 9 rats per time point.
Assessment of Neuron Number
PGP immunocytochemistry of the control colon showed prominent neurons within the ganglia of the myenteric and submucosal plexuses, and darkly stained axons within the smooth muscle layers. Examination of the number of neurons in the myenteric plexus of the control colon showed a uniform distribution among regions spanning the area likely to be affected by DNBS: 1.5 cm segments taken at the splenic flexure, the mid-descending region and the distal colon had similar numbers of neurons at 12.2 ± 0.5, 11 11.9 ± 1.4, 12 and 11.3 ± 0.9 11 neurons/mm, respectively.
In DNBS-treated rats at day 6, myenteric plexus neurons in the inflamed region of the colon could still be clearly distinguished with unchanged intensity of staining (Figure 1B) ▶ . However, the number of myenteric neurons per millimeter was significantly decreased when compared with the equivalent location in control animals, decreasing by nearly 50% to 6.8 ± 0.4 12 neurons/mm compared with saline control (12.4 ± 0.9 3 neurons/mm; Figure 1C ▶ ). The decrease in neuron number was limited to the overtly inflamed region, since examination of 0.5-cm segments taken 0.5 cm from the proximal and distal margins of the inflamed area in day 6 rats showed that the neuron number was similar to control values (data not shown). There was no significant change in the number of myenteric neurons per millimeter in the colons of rats receiving either saline or ethanol control solutions when compared with untreated controls (data not shown).
Examination of the time course of DNBS treatment showed that neuron number per millimeter was significantly decreased by day 2, to 62.0% of control (Figure 1C) ▶ . Further decrease occurred by day 4, reaching a level of 48.8% of control. All values of myenteric neuron number/mm were similar from day 4 and thereafter, up to day 35 after DNBS, indicating an irreversible loss of myenteric neurons. Examination at very early time points (Figure 1C ▶ , inset) showed that a trend to decrease was detectable by even 6 hours after DNBS, with significant decrease by 24 hours.
Effect of Colitis on Myenteric and Submucosal Ganglia
Initially, we suspected that the decreased neuron number in DNBS-treated rats could be due to loss of entire ganglia from the myenteric plexus, due to the obvious mucosal damage and transmural inflammation. However, the number of myenteric plexus ganglia per millimeter did not decrease significantly through the time course of colitis (Figure 2A) ▶ . To estimate the neuronal content of myenteric ganglia, we determined the mean number of neurons per ganglion appearing in the histological sections by counting the number of PGP-positive cells containing a nucleus within each ganglion. This value (“apparent neuronal content”) was consistent among control tissues (3.4 ± 0.4 1 ), and was assumed to be representative of ganglionic neuronal content. A comparison among tissues from animals during DNBS-induced inflammation showed a significant decrease in neuronal content by day 1 (P < 0.05),which was then constant thereafter, through day 35 (Figure 2B) ▶ .
Figure 2.
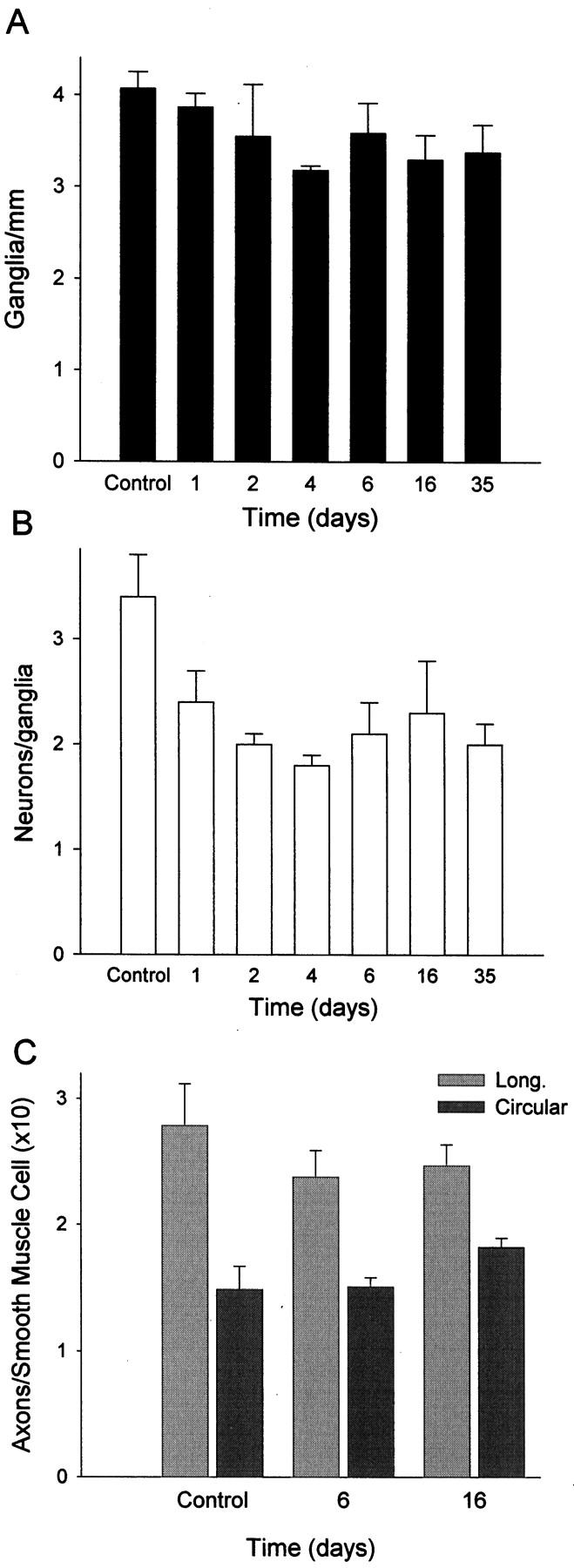
Reduction in neuronal content of myenteric ganglia in DNBS-induced colitis. A: Evaluation of the number of myenteric ganglia per millimeter showed no significant change during the course of inflammation. B: Estimation of the apparent neuronal content of the myenteric ganglia during DNBS colitis. The number of myenteric neurons (PGP-positive cells with nuclei present in the section) was determined in histological sections and showed significant reduction on day 1 after DNBS and thereafter. Bars, means ± SEM of values from 3 to 5 animals per time point. C: Determination of axon density within the longitudinal and circular smooth muscle layers of the colon during DNBS colitis. In areas within the smooth muscle layers, the ratio of PGP-positive axon profiles to the number of smooth muscle nuclei was determined, reflecting innervation by axons originating from both intrinsic and extrinsic sources. This value was not significantly different among day 0 (saline control), day 6, and day 16 tissues, despite decreased neuronal cell number and a thickened smooth muscle layer.
Analysis of neurons within the submucosal plexus, lying much closer to the region of overt damage, showed a significant decrease in neuron number during colitis, from 68.3 ± 4.8 12 to 41.7 ± 8.7 neurons per cross-section by day 6 of inflammation. This was correlated with a significant drop in the number of ganglia, from 36.6 ± 6.0 12 to 21.7 ± 3.3 11 ganglia/cross section by day 6. There was no significant change in the apparent neuronal content of the submucosal ganglia (1.94 ± 0.2 12 versus 2.04 ± 0.5 11 on day 6). Therefore, we concluded that colitis caused the loss of ganglia from the submucosal plexus, in contrast to the partial loss of neuronal content from the myenteric plexus.
Axon Density in Smooth Muscle
The decrease in neuronal number seen in colitis might lead to decreased innervation of the intestinal smooth muscle, where hyperplasia of the smooth muscle cells is also present and responsible for thickening of the smooth muscle layers. 4 Therefore, we tested whether colitis was associated with a decrease in axon density among the smooth muscle cells, using PGP immunoreactivity to detect axons in the smooth muscle layers, and comparing this with the number of smooth muscle nuclei. The axon density in the smooth muscle layers was unchanged during and after the DNBS-induced inflammation, measured separately in longitudinal and circular smooth muscle layers at days 6, 16, and 35 after DNBS treatment (Figure 2C) ▶ . This implies that an increase in axon number has occurred rapidly during inflammation.
Involvement of Immune Cells in Neural Damage
Measurement of myeloperoxidase (MPO) activity was used to reflect the presence of neutrophils in the tissue, and this showed the greatest increase in the colon of animals at early time points following DNBS administration. The MPO was significantly increased from the control level of 0.1 ± 0.1 11 by day 1 (11.5 ± 1.1 11 ng/ml), and remained elevated through day 6 (2.3 ± 0.4 11 ng/ml). No significant increase occurred in saline or ethanol-treated controls, and the MPO was similar to control by day 16 and thereafter. Due to the correlation between the rapid increase in MPO values and the early onset of damage to the myenteric plexus, we examined the histology of tissues at the early time points of 6 and 24 hours after DNBS. We found that eosinophils were characteristically detected within the myenteric plexus ganglia at these times (Figure 3, A and B) ▶ . While large numbers of neutrophils were already present in the surrounding smooth muscle, they were rarely detected within the ganglia until day 2 (Figure 3B) ▶ , suggesting that eosinophils may be responsible for neuronal damage and loss.
Figure 3.

Immune cell presence within myenteric ganglia of the inflamed rat colon and the effect of an anti-inflammatory treatment. A: Histological section with Congo red staining demonstrating the typical finding of eosinophils (black arrows) within and adjacent to the myenteric ganglia of the inflamed mid-descending colon on day 1 of DNBS-induced colitis. One neutrophil (open arrow) is present in this section, a rare finding at this time. N, neuronal nucleus; asterisk, glial cell nucleus. Magnification, ×500. B: Distribution of eosinophils and neutrophils within the ganglia of the myenteric plexus during DNBS-induced colitis. Eosinophils were detected within the ganglia at earlier time points (≥6 hours) than neutrophils (≥2 days), despite maximal myeloperoxidase levels on day 1 reflecting infiltration of the surrounding tissues. Bars, cumulative frequency of observations from 2 or 3 animals per time point. C: Effect of anti-inflammatory treatment during DNBS colitis on neuronal number within the myenteric plexus. Animals received 1 ml of saline or budesonide (300–1000 μg/kg) daily per rectum. There was a dose-dependent prevention of neuronal loss with budesonide versus saline, with complete prevention of damage at 1000 μg/kg. D: Myeloperoxidase levels within the intestinal wall of animals treated with budesonide or saline as in C. Bars, mean ± SEM of values from 3 to 5 animals per time point. *P < 0.05 compared with control.
Due to this evidence suggesting a direct involvement of immune cells with neuronal loss, we investigated whether anti-inflammatory therapy could influence the extent of damage to the enteric nervous system in this model of colitis. Delivered as a daily enema, budesonide caused the dose-dependent prevention of the loss of myenteric plexus neurons in DNBS-treated rats, with a dose of 1000 μg/kg being completely successful in preventing significant loss of myenteric plexus neurons, while lower doses also achieved significant improvement over untreated controls (Figure 3C) ▶ . Reduction in the MPO levels of DNBS-treated rats occurred in proportion to the dose of budesonide, while carrier alone was without effect (Figure 3D) ▶ . Budesonide treatment did not affect organ weight of thymus, spleen, or adrenals at any dose (n = 3 per dose; data not shown).
Discussion
The autonomous function of the intestine largely depends on appropriate regulation by the ENS. 13 We have used an animal model to show that the ENS undergoes major structural damage during inflammation and we present evidence suggestive of compensating axonal proliferation. This suggests that the loss of neurons within the ENS is an important consequence of intestinal inflammation and may be a significant cause of dysfunction seen in post-enteritis IBS in humans.
In our study of DNBS colitis, we found severe transmural inflammation from day 2 onward, which appeared to be resolved by day 35, with only a thickened muscle wall as a consequence. However, examination showed a significant and lasting decrease in neuron number, by nearly 50% on day 6 that remained through day 35. While an extensive inflammatory infiltrate developed in the muscle wall, and was correlated with significant increases in MPO levels, immune cells were observed within the myenteric ganglia only at the earliest time points of ≤24 hours after DNBS. This suggests that the early events that occur during the inflammatory episode are responsible for the loss of both submucosal and myenteric plexus neurons.
The observed decrease in the number of neurons was not due to transient loss of expression of the marker protein, since PGP-positive neurons remained detectable in inflamed tissue with an unaltered intensity of staining, and the number remained low after inflammation was resolved. Since the apparent number of neurons in each myenteric ganglion decreased during colitis and remained unchanged after healing, while the number of ganglia remained largely unchanged, it appears that inflammation caused loss of a proportion of the neurons within each ganglion. In contrast, a decrease in the number of submucosal neurons was associated with a significant decrease in the number of submucosal ganglia. This may occur due to the potentially greater severity of inflammatory damage closer to the mucosal surface.
It is not yet clear whether cell death occurs proportionally among all neural phenotypes in the inflamed intestine, which could have the additional consequences of altered control of diverse intestinal functions including contractility and mucosal transport. However, the effects of loss of enteric neurons will be superimposed on alterations in neural function that are already present in the inflamed intestine. This is most clearly seen in animal models, where altered neurotransmitter release occurs, 1 and long-lasting up-regulation in acetylcholine synthesis can be detected. 14
Further studies are required to identify the mechanism of death of enteric neurons. It is possible that increased production of nitric oxide during inflammation is involved, since NO production has been implicated in neuronal injury following ischemia, trauma, and in several neurodegenerative diseases. 15 Elsewhere, inhibition of nitric oxide synthase was found to prevent apoptotic death of neurons in vivo. 16 Earlier, we found that oral delivery of a NO synthase inhibitor prevented the appearance of damage to enteric neurons in colitis, 4 thus supporting a role for this molecule in neuronal cell death.
This study suggests that neuronal plasticity is an important homeostatic mechanism in the intestine, both during and following inflammation. Challenges to the normal patterns of innervation in this model include loss of neurons, an increase in smooth muscle cell number, and an unknown degree of damage to surviving axons. Nonetheless, the density of axons among the smooth muscle cells was maintained during and after inflammation, suggesting rapid axonal proliferation occurs in the inflamed colon. Since the methods used did not discriminate among axons of extrinsic versus intrinsic origins, it is possible that axonal proliferation by both populations is involved in the maintenance of innervation density. In a recent study, the distribution of axons containing substance P and VIP showed extensive changes during TNBS-induced colitis, with an initial decrease followed by increased expression and a return on day 7 after TNBS to control values. 17 This suggests that intrinsic neurons may undergo axonal proliferation to maintain the innervation of the smooth muscle cells. In the mucosa, neural plasticity may be an essential part of repair, as suggested by the early degeneration and later regeneration of mucosal axons in the jejunum of the rat during Nippostrongylus-induced inflammation. 18
The mechanisms that lead to axonal proliferation in the intestine are not known. Intestinal smooth muscle cells may produce neurotrophic factors that regulate their own innervation, which could be critical during inflammation-induced remodelling of the intestine. For example, nerve growth factor (NGF) and its receptor trkA are expressed on vascular smooth muscle in vivo 11,19 and in vitro, where promotion of NGF expression by platelet-derived growth factor, transforming growth factor-β and IL-1 9 supports the likelihood of its production in the inflamed intestine. Also, NT-3 is implicated due its ability to cause neural crest cells from fetal gut to differentiate into neurons and glia. 10
The effects of inflammation on intestinal innervation in IBD are less clear, where there is controversy as to the extent, nature, and diagnostic value of such alterations. 12 There are no definitive studies of neuron number in IBD, although older reports suggest that there could be either an increase 20 or a decrease in a limited study. 21 It seems clear that there is axonal necrosis 2,22,23 as well as axonal hyperplasia. 24,25 Alterations in neurotransmitter levels occur, although conflicting findings may arise from local variation in tissue characteristics as well as variable progression and disease among patients. 26
To mimic one form of clinical therapy relevant to treatment of colitis, we used the novel anti-inflammatory steroid budesonide delivered as a daily enema and investigated its ability to control the loss of enteric neurons described above. While the lower doses of budesonide achieved a significant reduction in neuronal loss, the highest dose administered completely prevented this decrease. Similarly, Palmen et al 27 recently described that intracolonic budesonide was more effective than dexamethasone in reducing macroscopic damage score in TNBS colitis. Previously, Jacobson et al 7 showed that inflammation caused a decrease in K+-evoked release of 3H-noradrenaline from extrinsic nerves of the rat colon, and that this could be attenuated by treatment with budesonide or an IL-1 receptor antagonist.
In these studies, budesonide was effective at doses that did not cause alterations in organ weights of thymus, spleen, or adrenals. These findings reflect the reduced systemic effects of this drug relative to other steroids due to the nearly complete first-pass metabolism after topical action, making it desirable as a therapeutic agent. 28 Taken in conjunction with the present study, this shows that anti-inflammatory therapy can both prevent permanent damage as well as ameliorate transient changes in functional properties of intestinal innervation. This may explain the part of the beneficial actions of budesonide and similar compounds in the therapy of IBD. 29 Overall, there is a strong need to evaluate neuronal damage in IBD and to determine the best way for current therapies to minimize permanent damage.
Footnotes
Address reprint requests to M.G. Blennerhassett, Ph.D., Gastrointestinal Diseases Research Unit, Hotel Dieu Hospital, 166 Brock Street, Kingston, Ontario K7L 5G2, Canada. E-mail: mblen@meds.queensu.ca.
Supported by a grant from the Medical Research Council of Canada.
D. P. Lamb’s present address: MDS Capital Corporation, 100 International Boulevard, Toronto, Ontario M9W 6J6, Canada.
References
- 1.Collins SM: The immunomodulation of enteric neuromuscular function: implications for motility and inflammatory disorders. Gastroenterology 1996, 111:1683-1699 [DOI] [PubMed] [Google Scholar]
- 2.Hogaboam CM, Snider DP, Collins SM: Neuromuscular regulation of T-cell activation. J Neuroimmunol 1997, 75:123-134 [DOI] [PubMed] [Google Scholar]
- 3.Collins SM, Hurst SM, Main C, Stanley E, Khan I, Blennerhassett P, Swain M: Effect of inflammation of enteric nerves: cytokine-induced changes in neurotransmitter content and release. Ann NY Acad Sci 1992, 664:415-424 [DOI] [PubMed] [Google Scholar]
- 4.Hogaboam CM, Jacobson K, Collins SM, Blennerhassett MG: The selective beneficial effects of nitric oxide inhibition in experimental colitis. Am J Physiol 1995, 268:G673-G684 [DOI] [PubMed] [Google Scholar]
- 5.Hawkins JV, Emmel EL, Feuer JJ, Nedelman MA, Harvey CJ, Klein HJ, Rozmiarek H, Kennedy AR, Lichtenstein GR, Billings PC: Protease activity in a hapten-induced model of ulcerative colitis in rats. Dig Dis Sci 1997, 42:1969-1980 [DOI] [PubMed] [Google Scholar]
- 6.Wallace JL, Le T, Carter L, Appleyard CB, Beck PL: Hapten-induced chronic colitis in the rat: alternatives to trinitrobenzene sulfonic acid. J Pharmacol Toxicol Methods 1995, 33:237-239 [DOI] [PubMed] [Google Scholar]
- 7.Jacobson K, McHugh K, Collins SM: The mechanism of altered neural function in a rat model of acute colitis. Gastroenterology 1997, 112:156-162 [DOI] [PubMed] [Google Scholar]
- 8.Coggeshall RE, Lekan HA: Methods for determining numbers of cells and synapses: a case for more uniform standards of review. J Comp Neurol 1996, 364:6-15 [DOI] [PubMed] [Google Scholar]
- 9.Creedon DJ, Tuttle JB: Synergistic increase in nerve growth factor secretion by cultured vascular smooth muscle cells treated with injury-related growth factors. J Neurosci Res 1997, 47:277-286 [PubMed] [Google Scholar]
- 10.Chalazonitis A, Rothman TP, Chen J, Lamballe F, Barbacid M, Gershon MD: Neurotrophin-3 induces neural crest-derived cells from fetal rat gut to develop in vitro as neurons or glia. J Neurosci 1994, 14:6571-6584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bono F, Lamarche I, Herbert JM: NGF exhibits a pro-apoptotic activity for human vascular smooth muscle cells that is inhibited by TGFbeta1. FEBS Lett 1997, 416:243-246 [DOI] [PubMed] [Google Scholar]
- 12.Brewer DB, Thompson H, Haynes IG, Alexander-Williams J: Axonal damage in Crohn’s disease is frequent, but non-specific. J Pathol 1990, 161:301-311 [DOI] [PubMed] [Google Scholar]
- 13.Gershon MD, Chalazonitis A, Rothman TP: From neural crest to bowel: development of the enteric nervous system. J Neurobiol 1993, 24:199-214 [DOI] [PubMed] [Google Scholar]
- 14.Davis KM, Masella J, Blennerhassett MG: Acetylcholine metabolism in the inflamed rat intestine. Exp Neurol 1998, 152:251-258 [DOI] [PubMed] [Google Scholar]
- 15.Beckman JS, Koppenol WH: Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol 1996, 271:C1424-C1437 [DOI] [PubMed] [Google Scholar]
- 16.Wu W, Li L: Inhibition of nitric oxide synthase reduces motoneuron death due to spinal root avulsion. Neurosci Lett 1993, 153:121-124 [DOI] [PubMed] [Google Scholar]
- 17.Miampamba M, Sharkey KA: Distribution of calcitonin gene-related peptide, somatostatin, substance P and vasoactive intestinal polypeptide in experimental colitis in rats. Neurogastroenterol Motil 1998, 10:315-329 [DOI] [PubMed] [Google Scholar]
- 18.Stead RH, Kosecka-Janiszewska U, Oestreicher AB, Dixon MF, Bienenstock J: Remodeling of B-50 (GAP-43)- and NSE-immunoreactive mucosal nerves in the intestines of rats infected with Nippostrongylus brasiliensis. J Neurosci 1991, 11:3809-3821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Donovan MJ, Miranda RC, Kraemer R, McCaffrey TA, Tessarollo L, Mahadeo D, Sharif S, Kaplan DR, Tsoulfas P, Parada L: Neurotrophin and neurotrophin receptors in vascular smooth muscle cells: regulation of expression in response to injury. Am J Pathol 1995, 147:309-324 [PMC free article] [PubMed] [Google Scholar]
- 20.Davies DR, Dockerty MB, Mayo CW: The myenteric plexus in regional enteritis: a study of the number of ganglion cells in the ileum in 24 cases. Surg Gynecol Obstet 1955, 101:208-216 [PubMed] [Google Scholar]
- 21.Oehmichen M, Reifferscheid P: Intramural ganglion cell degeneration in inflammatory bowel disease. Digestion 1977, 15:482-496 [DOI] [PubMed] [Google Scholar]
- 22.Dvorak AM, Silen W: Differentiation between Crohn’s disease and other inflammatory conditions by electron microscopy. Ann Surg 1985, 201:53-63 [PMC free article] [PubMed] [Google Scholar]
- 23.Steinhoff MM, Kodner IJ, DeSchryver-Kecskemeti K: Axonal degeneration/necrosis: a possible ultrastructural marker for Crohn’s disease. Mod Pathol 1988, 1:182-187 [PubMed] [Google Scholar]
- 24.Leonard N, Hourihane DO, Whelan A: Neuroproliferation in the mucosa is a feature of coeliac disease and Crohn’s disease. Gut 1995, 37:763-765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Strobach RS, Ross AH, Markin RS, Zetterman RK, Linder J: Neural patterns in inflammatory bowel disease: an immunohistochemical survey. Mod Pathol 1990, 3:488-493 [PubMed] [Google Scholar]
- 26.Belai A, Boulos PB, Robson T, Burnstock G: Neurochemical coding in the small intestine of patients with Crohn’s disease. Gut 1997, 40:767-774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Palmen MJ, Dieleman LA, Soesatyo M, Pena AS, Meuwissen SG, van Rees EP: Effects of local budesonide treatment on the cell-mediated immune response in acute and relapsing colitis in rats. Dig Dis Sci 1998, 43:2518-2525 [DOI] [PubMed] [Google Scholar]
- 28.Robinson M: Optimizing therapy for inflammatory bowel disease. Am J Gastroenterol 1997, 92:12S-17S [PubMed] [Google Scholar]
- 29.Borley NR, Mortensen NJ, Jewell DP: Preventing postoperative recurrence of Crohn’s disease. Br J Surg 1997, 84:1493-1502 [PubMed] [Google Scholar]