

Abstract
Using a murine breast cancer model, we earlier found a positive correlation between the expression of nitric oxide synthase (NOS) and tumor progression; treatment with inhibitors of NOS, NG-methyl-l-arginine (NMMA) and NG-nitro-l-arginine methyl ester (L-NAME), had antitumor and antimetastatic effects that were partly attributed to reduced tumor cell invasiveness. In the present study, we used a novel in vivo model of tumor angiogenesis using subcutaneous implants of tumor cells suspended in growth factor-reduced Matrigel to examine the angiogenic role of NO in a highly metastatic murine mammary adenocarcinoma cell line. This cell line, C3L5, expresses endothelial (e) NOS in vitro and in vivo, and inducible (i) NOS in vitro on stimulation with lipopolysaccharide and interferon-γ. Female C3H/HeJ mice received subcutaneous implants of growth factor-reduced Matrigel inclusive of C3L5 cells on one side, and on the contralateral side, Matrigel alone; L-NAME and D-NAME (inactive enantiomer) were subsequently administered for 14 days using osmotic minipumps. Immediately after sacrifice, implants were removed and processed for immunolocalization of eNOS and iNOS proteins, and measurement of angiogenesis. Neovascularization was quantified in sections stained with Masson’s trichrome or immunostained for the endothelial cell specific CD31 antigen. While most tumor cells and endothelial cells expressed immunoreactive eNOS protein, iNOS was localized in endothelial cells and some macrophages within the tumor-inclusive implants. Measurable angiogenesis occurred only in implants containing tumor cells. Irrespective of the method of quantification used, tumor-induced neovascularization was significantly reduced in L-NAME-treated mice relative to those treated with D-NAME. The quantity of stromal tissue was lower, but the quantity of necrotic tissue higher in L-NAME relative to D-NAME-treated animals. The total mass of viable tissue (ie, stroma and tumor cells) was lower in L-NAME relative to D-NAME-treated animals. These data suggest that NO is a key mediator of C3L5 tumor-induced angiogenesis, and that the antitumor effects of L-NAME are partly mediated by reduced tumor angiogenesis.
Nitric oxide, an inorganic free radical gas, is synthesized from the amino acid l-arginine by a group of enzymes, the NO synthases (NOS). At least three isoforms of NOS have been cloned, characterized, and localized: endothelial (e) and neuronal (n) NOS isoforms are Ca2+/calmodulin-dependent and are expressed constitutively in these and other cells. The inducible (i) isoform is Ca2+/calmodulin-independent and usually induced in the presence of inflammatory cytokines and bacterial products in macrophages, hepatocytes, and other cells. Under certain conditions, iNOS can also be expressed constitutively in some cells. When constitutively expressed, NO produced at low levels is an important mediator of physiological functions such as vasodilation, inhibition of platelet aggregation, and neurotransmission. Under inductive conditions, high levels of NO produced by macrophages and other effector cells can mediate antibacterial and antitumor functions. However, chronic induction of NOS may contribute to many pathological processes including inflammation and cancer. 1,2
Much scientific research has focused on the role of NO in tumor progression; although two apparently conflicting views exist, overall an overwhelming amount of clinical and experimental evidence supports a positive association between NO production and tumor progression. The level of NOS protein and/or activity in the tumor has been positively correlated with the degree of malignancy for tumors of the human reproductive tract, 3 breast, 4,5 and central nervous system. 6 In a majority of gastric carcinomas, iNOS was detected in stromal elements, and eNOS was detected in the tumor vasculature. 7 iNOS expression was higher in prostatic carcinomas relative to benign prostatic hyperplasia. 8 Similarly, relative to normal healthy control tissue, total NOS activity was higher in carcinomas of the larynx, oropharynx, oral cavity, 9 and adenocarcinomas of the lung. 10
Experimental tumor models have provided more direct evidence of a contributory role of NO in tumor progression. Using a rat adenocarcinoma model in which cells of the tumor vasculature expressed iNOS, treatment of the host with the NOS inhibitor NG-nitro-l-arginine methyl ester (L-NAME) reduced NO production and tumor growth. 11 Furthermore, despite in vitro cytostatic effects of NO induction with lipopolysaccharide (LPS) and interferon (IFN)-γ in EMT-6 murine mammary cells, this induction stimulated tumor growth and metastasis in vivo. 12 Finally, in our own studies using a murine mammary adenocarcinoma model (C3H/HeJ spontaneous mammary tumors and their clonal derivatives), NO-mediated stimulation of tumor progression was observed. 13 The spontaneously developing tumors showed heterogeneous expression of eNOS within primary tumors, whereas their metastatic counterparts were homogeneously eNOS positive, suggesting that eNOS expression promoted metastasis. A highly metastatic cell line, C3L5, clonally derived from a spontaneous mammary tumor showed strong eNOS expression in vitro and in vivo, and iNOS in vitro on stimulation with LPS and IFN-γ. 13 Treatment of C3L5 mammary tumor-bearing mice with inhibitors of NOS, L-NAME and NG-methyl-l-arginine (NMMA), had antitumor and antimetastatic effects. 14,15 Reduced tumor cell invasiveness was identified as one of the mechanisms mediating these effects. 13,16 We hypothesized that, in this tumor model, additional mechanisms likely played critical roles in mediating the therapeutic effects of NOS inhibition.
In contrast to the above, some studies reported an inverse association between NO and tumor progression. For example, the levels of NOS enzymes and NOS activity declined during the transition of human colonic mucosa to polyps, and then to carcinomas. 17 However, a later study revealed higher NOS activity in adenomatous polyps, which was believed to promote increased angiogenesis before the transition of adenomas into carcinomas. 18 In a murine melanoma cell line, NOS activity was inversely correlated with capacity for metastasis. 19 When genetically transduced to overexpress iNOS, the melanoma cells, 19 as well as renal carcinoma cells, 20 lost their tumorigenic and metastatic abilities as a result of NO-mediated tumor cell apoptosis. These opposing findings suggest a dual role for NO in tumor growth and metastasis; the susceptibility of tumor cells to NO-mediated injury may depend on levels of NO produced and the genetic makeup of the tumor cells. During clonal evolution of tumors, high NO-producing cells may self-delete by apoptosis, and those making lower levels of NO or capable of resisting NO-mediated injury may have an in vivo advantage, resulting from NO-mediated stimulation of tumor cell invasiveness, tumor blood flow or tumor angiogenesis. 13
A body of recent evidence suggests a stimulatory role of NO in angiogenesis. For example, NO donors were found to increase proliferation and migratory function of endothelial cells in vitro. 21,22 Using the in vivo rabbit cornea assay, angiogenesis induced by vasoactive molecules such as substance P and prostaglandin E1 was blocked with NOS inhibition. 22 Similarly, NOS inhibitors reduced neovascularization in acetic acid-induced gastric ulcers in rats, 23 and human squamous cell carcinoma xenografts in the rabbit cornea. 9 An angiogenesis-promoting role for tumor-derived NO was also suggested by the in vivo behavior of a human colon adenocarcinoma cell line engineered to continuously express iNOS. When transplanted into nude mice, the iNOS-transduced cells resulted in tumors with enhanced growth rate and vascularity relative to those derived from wild-type control cells. 24
In the present study, we have evaluated the contributory role of NO in C3L5 mammary tumor-induced angiogenesis. To achieve this, we devised a novel in vivo Matrigel implant model of tumor-induced angiogenesis and subjected the host animals to chronic treatment with the NOS inhibitor L-NAME, or as controls, its inactive enantiomer, D-NAME.
Materials and Methods
Animals
Female C3H/HeJ mice (6–8 weeks old) were obtained from Jackson Laboratories (Bar Harbor, ME). On arrival at the vivarium, animals were immediately randomized to treatment groups (ie, L-NAME and D-NAME); experimental procedures began after a one-week acclimatization period. Throughout the investigation, animals had free access to food (standard mouse chow) and water and were maintained on a 12-hour light/dark cycle. Animals were treated in accordance with guidelines set out by the Canadian Council on Animal Care.
Tumor Cell Line
A spontaneously occurring mammary tumor in a female retired breeder C3H/HeJ mouse was the source of a primary transplantable tumor T58 from which a metastatic C3 cell line was derived. Since the metastatic potential of the C3 line declined over a number of years following repeated in vitro passages, a highly metastatic C3L5 line was derived by five cycles of repeated in vivo selections for spontaneous lung micrometastases following subcutaneous transplantation of C3 cells into C3H/HeJ mice. 25 The C3L5 cells used in the present study were grown from frozen stock and maintained in RPMI 1640 medium (GIBCO; Burlington, ON) supplemented with 5% fetal calf serum (GIBCO) and 1% penicillin-streptomycin (Mediatech; Washington, DC) in a humidified incubator, 5% CO2.
In Vivo Assay for Tumor-Induced Angiogenesis
We devised a novel in vivo model of tumor angiogenesis based on the protocol of Kibbey et al. 26 These authors used conventional Matrigel, a reconstituted basement membrane, which is liquid at 4°C and forms a solid gelatinous mass at body temperature. Measurable angiogenesis occurred within these implants, possibly due to angiogenic growth factors present in the conventional Matrigel. In our application of the assay we used growth factor-reduced Matrigel, which, unlike conventional Matrigel, did not stimulate angiogenesis on its own. However, when tumor cells were suspended in growth factor-reduced Matrigel as a component of the subcutaneous implant, a strong angiogenic response was observed, which was easily and objectively quantifiable. Based on several pilot experiments in which the implant volume, tumor cell number, and implant duration were varied, we standardized the assay (the detailed kinetics of tumor-induced angiogenesis in these implants are not presented here). This assay was used to examine the effects of NO on the angiogenic response by administering L-NAME or its inactive enantiomer, D-NAME, to mice using osmotic minipumps (pilot experiments established an equivalent angiogenic response in animals receiving D-NAME and those receiving no treatment). The angiogenic response was evaluated by examining the gross morphology of the Matrigel implants and quantifying neovascularization in sections stained with Masson’s trichrome or immunostained for the endothelial-specific CD31 antigen (PECAM). In addition, we documented the mass (weight in mg) of the implants on retrieval and systematically analyzed sections of tumor cell-inclusive implants for area quantification of three histologically distinct regions: peripheral tumor-free stromal tissue feeding blood vessels into the more deeply located tumor tissue; viable tumor tissue; and necrotic regions, to determine the effects of therapy on the various components of the implants.
In the inguinal region, mice received subcutaneous implants of 5 × 10 4 C3L5 cells suspended in growth factor reduced Matrigel (Collaborative Research, Bedford, MA) (3.5 mg of Matrigel in 0.5 ml of RPMI 1640), and on the contralateral side as controls, the equivalent amount of Matrigel alone. Immediately thereafter, osmotic minipumps (ALZA Corporation, Palo Alto, CA) were implanted subcutaneously, providing a constant systemic supply (0.5 ml/hour; 25 mg/200 μl 0.9% NaCl) of L-NAME to one group (n = 15), or D-NAME to the other group (n = 15) (both drugs purchased from Sigma Chemical Co., St. Louis, MO) for the experiment duration (14 days). This experiment was performed on two separate occasions; both experiments were conducted using the same protocol and sample size (ie, n = 15 animals/group).
Mice were sacrificed using an overdose of pentobarbital and the Matrigel implants were removed and divided in half; therefore paraffin and frozen sections were obtained from the same sample. Samples fixed in 4% paraformaldehyde, processed for paraffin embedding, and sectioned were stained with Masson’s trichrome or immunostained for eNOS and iNOS proteins. Alternatively, samples frozen in OCT were sectioned and analyzed immunohistochemically for CD31. Both types of sections (ie, Masson’s trichrome stained or CD31 immunostained) were scanned at low power for areas containing new blood vessels (researcher blind to experimental condition); these areas were systematically imaged at 160× magnification using Northern Exposure (Empix Imaging Inc.), and individual vessel counts for each field were documented using Mocha Image Analysis Software (Jandel Scientific) to identify fields of maximum blood vessel density (ie, “hot spots”). Subsequently, “hot spots” were statistically analyzed for between-group differences using two different approaches: the average (n = 15 animals/group) of the maximal number of blood in one field per animal, and the average (n = 15 animals/group) of the average of three fields of maximal blood vessel density (taken in descending order) per animal. Masson’s trichrome-stained sections were also used to quantify histologically distinct regions within implants (ie, peripheral stromal, healthy tumor, and necrotic regions); entire cross sections of the implants were digitally imaged; areas were then quantified, and data expressed as the number of pixels, using Mocha Image Analysis Software (researcher blind to experimental condition).
Immunohistochemical Localization of CD31 Antigen (PECAM-1)
OCT-fixed samples were stored at −80°C until sectioned; samples were sectioned at 5 μm thickness and stored at −20°C before immunostaining (sections stored for maximum of 2 days at −20°C). Frozen sections were fixed in ice-cold methanol (5 minutes, −20°C). Endogenous peroxidase activity was blocked with methanol containing 3% H2O2 (30 minutes, room temperature) before application of blocking serum: normal mouse serum (Cedarlane Laboratories Limited, Hornby, ON) diluted in 1% bovine serum albumin (1:10; 1 hour at room temperature in humidified chamber). Sections were then incubated with primary antibody: purified rat anti-mouse CD31 monoclonal antibody (1:50; overnight at 4°C in humidified chamber; Cedarlane Laboratories) followed by secondary antibody: biotinylated mouse anti-rat IgG-2a monoclonal antibody (1:100; 1 hour at room temperature in humidified chamber; Caltag Laboratories, San Francisco, CA). Avidin-biotin complex (ABC) (Vector Laboratories, Inc., Burlingame, CA) was then applied (1 hour at room temperature), followed by diaminobenzidine chromogen (Sigma); sections were then lightly counterstained with Mayer’s hemalum. Negative controls were incubated with the equivalent concentration of rat IgG-2a (Caltag Laboratories) in place of primary antibody.
Immunohistochemical Localization of eNOS and iNOS Antigens
Paraffin-embedded implants were sectioned at 7 μm thickness. Following deparaffinization and rehydration of sections, endogenous peroxidase activity was blocked using methanol containing 3% H2O2 before application of blocking serum (normal horse serum, 1:10; 1 hour at room temperature). Sections were then incubated with primary antibody: mouse monoclonal anti-eNOS or mouse monoclonal anti-macrophage iNOS (1:80; overnight at 4°C, or 1:50; overnight at 4°C; Transduction Laboratories, Lexington, KY) for eNOS and iNOS localization, respectively. Secondary antibody (biotinylated horse anti-mouse, 1:200; 1 hour at room temperature) was then applied, followed by ABC (1 hour at room temperature) and diaminobenzidine chromogen. Sections were lightly counterstained with Mayer’s hemalum.
Data Analysis
Data were analyzed using SAS v6.12 on a Unix mainframe computer, and treatment groups (ie, L-NAME and D-NAME) compared using one-way analysis of variance. In quantifying neovascular response for each treatment group (n = 15 mice/group), results were expressed as the mean of the maximum number of microvessels in a single field (160× magnification) and the mean number of microvessels in three fields of maximum blood vessel density (160× magnification). Data from the duplicate experiment (n = 15 mice/group) were analyzed in the same manner. A probability of 0.05 was used in determining statistical significance.
Results
Gross Morphology of Implants
Figure 1 ▶ shows the gross morphology of tumor-exclusive implants (Figure 1A) ▶ and tumor-inclusive implants obtained from L-NAME and D-NAME-treated (Figure 1, B and C ▶ , respectively) animals. Tumor-exclusive implants from L-NAME and D-NAME-treated animals were small, translucent, and avascular. Tumor-inclusive implants were larger, and implants obtained from L-NAME-treated animals were less vascular than those obtained from D-NAME-treated animals.
Figure 1.

Gross morphology is shown for tumor-exclusive Matrigel implants (A) and tumor-inclusive Matrigel implants obtained from L-NAME- and D-NAME-treated animals (B and C, respectively). Tumor-exclusive implants were translucent, avascular, and unaffected by L-NAME or D-NAME treatment. Tumor-inclusive implants were larger, and those obtained from L-NAME-treated animals were less vascular relative to those obtained from D-NAME-treated animals. Photomicrographs of Masson’s trichrome staining are shown for tumor-exclusive (D) and tumor-inclusive Matrigel implants obtained from L-NAME-treated (E) and D-NAME-treated (F) animals. Tumor-exclusive implants obtained from L-NAME- and D-NAME-treated animals were avascular and contained a few fibroblasts. The sections of tumor-inclusive implants in both treatment groups showed three histologically distinct areas, shown in G (from L-NAME-treated animal): peripheral stroma (S), adjacent tumor-dominant area (T), and central zone of necrosis (N). The stromal components in L-NAME-treated animals appeared thinner and less vascular relative to those in D-NAME-treated animals. Scale bars: A–C, 1 mm; D–G, 30 μm.
Histological Evaluation of Vascularity of Implants—Masson’s Trichrome Staining
Figure 1 ▶ shows Masson’s trichrome staining of tumor-exclusive Matrigel implant (Figure 1D) ▶ (sections of tumor cell-exclusive implants were identical for L-NAME and D-NAME-treated animals) and tumor-inclusive implants obtained from L-NAME (Figure 1E) ▶ and D-NAME-treated animals (Figure 1F) ▶ . This method stains fibrous tissue and stroma bluish-green. Blood vessels containing red blood cells stand out because of bright red staining of red blood cells. Other cells (including tumor cells) show pink staining of cytoplasm and dark magenta colored nuclei. Tumor-exclusive Matrigel implants obtained from L-NAME and D-NAME-treated animals were avascular and contained a few fibroblasts. Tumor-inclusive implants obtained from both treatment groups consisted of three histologically distinct regions, shown in Figure 1G ▶ (implant obtained from L-NAME-treated animal): a peripheral zone of stroma (S) containing feeder blood vessels; healthy tumor areas (T); and more centrally located necrotic areas (N) infiltrated with leukocytes. Areas of highest microvascular count were best identified in the stroma of tumor-inclusive implants in Masson’s trichrome-stained sections. The stroma of tumor-inclusive implants obtained from L-NAME-treated animals appeared thinner and less vascular relative to those obtained from D-NAME-treated animals.
Histological Evaluation of Vascularity of Implants—CD31 Immunostaining
Figure 2 ▶ shows immunohistochemical localization of CD31 antigen in implants obtained from D-NAME and L-NAME-treated animals (Figure 2, A and B ▶ , respectively). This method stains endothelial cells brown and correctly identifies cells lining the microvasculature within the tumor component of the implants (in contrast to Masson’s trichrome-stained sections, which predominantly identifies blood vessels within stromal areas). Nuclei are lightly counterstained with Mayer’s hemalum. Neovascularization was reduced in tumor-inclusive implants obtained from L-NAME-treated animals relative to those obtained from D-NAME-treated animals.
Figure 2.
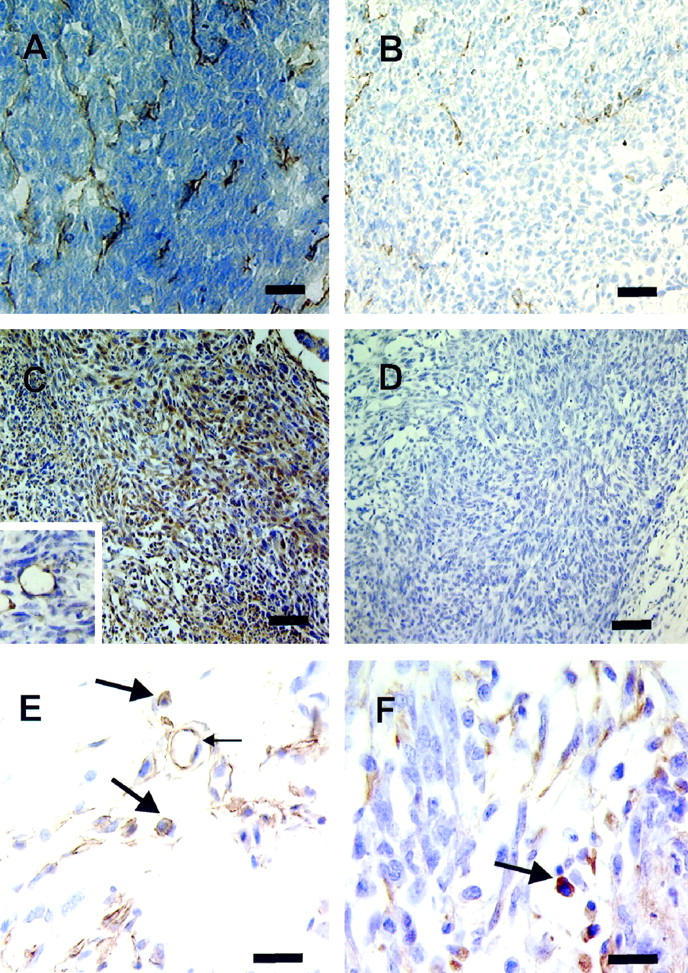
Immunohistochemical localization of CD31 antigen is shown at top in tumor-inclusive implants from D-NAME- and L-NAME-treated animals (A and B, respectively), identifying endothelial cells lining the microvasculature within the tumor component of the implants. Neovascularization was reduced in L-NAME-treated animals relative to those treated with D-NAME. Immunohistochemical localization of eNOS antigen is shown at center in tumor-inclusive implants; positive immunostaining and negative control are shown (C and D, respectively). A high proportion of tumor cells and endothelial cells lining the tumor vasculature (inset in C) within implants expressed eNOS, regardless of treatment group. The bottom shows immunohistochemical localization of iNOS antigen in peripheral stroma (E) and healthy tumor (F) bordering necrotic area. Expression of iNOS did not differ between treatment groups. Positive immunoreactivity for iNOS protein was evident in macrophages (large arrows) located in stromal (E) and tumor (F), as well as within central necrotic (not shown) areas of the implants in both treatment groups. Endothelial cells (small arrow) stained positively for iNOS, and some nonspecific staining of stromal tissue was observed in both treatment groups. Scale bars: A–D, 30 μm; E–F, 50 μm; C, inset, 60 μm.
Histological Evaluation of Implants—eNOS and iNOS Immunostaining
Immunohistochemical localization of eNOS antigen in tumor-inclusive implant and a negative control are shown in Figure 2, C and D ▶ , respectively. eNOS expression was observed in endothelial cells of the tumor vasculature (Figure 2C ▶ , inset), and a high proportion of tumor cells within implants obtained from animals treated with either L-NAME or D-NAME. Immunohistochemical localization of iNOS antigen is shown in Figure 2, E and F ▶ . Regardless of treatment group, tumor cells within the implants did not stain positively for iNOS. However, positive immunoreactivity for iNOS protein was observed in a significant proportion of macrophages located in peripheral stroma (Figure 2E) ▶ , in the healthy tumor bordering the necrotic area (Figure 2F) ▶ , and within the central necrotic area (not shown in Figure 2 ▶ ). Endothelial cells also stained positively for iNOS (Figure 2E ▶ , small arrow).
Quantification of Tumor-Induced Neovascularization—Masson’s Trichrome Staining and CD31 Immunostaining
Figure 3 ▶ shows the number of blood vessels per unit area for Masson’s trichrome-stained and CD31 immunostained sections; data are expressed as the maximum number of blood vessels per field and as the average number of blood vessels in three fields of maximal density. Irrespective of staining protocol or method of quantification used, the neovascular response was reduced in L-NAME-treated mice relative to those treated with D-NAME. Tumor-induced neovascularization, measured by Masson’s trichrome, was reduced in implants obtained from L-NAME-treated animals when data were expressed as 1) the maximum number of blood vessels per field (L-NAME, 52.3 ± 5.9; D-NAME, 92.3 ± 11.3, P < 0.003) and 2) as the average number of blood vessels in three fields (L-NAME, 42.1 ± 5.4; D-NAME, 83.2 ± 10.2, P < 0.001) (Figure 3, A and B ▶ , respectively). L-NAME treatment reduced tumor-induced neovascularization, as measured by CD31 immunostaining, when data were expressed 1) as the maximum number of blood vessels per field (L-NAME, 50.8 ± 7.6; D-NAME, 79.9 ± 6.5, P < 0.0099) and 2) as the average number of blood vessels in three fields (L-NAME, 40.1 ± 5.6; D-NAME, 69.6 ± 4.8, P < 0.0009) (Figure 3, C and D ▶ , respectively). Results obtained from the duplicate experiment were similar and are not presented.
Figure 3.

Quantification of tumor-induced neovascularization in sections stained with Masson’s trichrome (A and B) and immunostained for CD31 (C and D). The data were expressed as the maximum number of blood vessels per field (mean ± SE; n = 15 animals/group) and as the average number of blood vessels in 3 fields of maximal density (mean ± SE; n = 15 animals/group). Irrespective of staining protocol or method of quantification used, the neovascular response was reduced in L-NAME-treated mice relative to those treated with D-NAME as indicated by the P value in each panel: A, P < 0.003; B, P < 0.001; C, P < 0.0099; D, P < 0.0009. BVC, blood vessel count.
Quantification of Histologically Distinct Areas within Implants
Quantification of the various tissue compartments contained within tumor cell-inclusive implants indicated that the quantity of peripherally located stromal tissue was reduced in L-NAME relative to D-NAME-treated animals (L-NAME, 4998.5 ± 1055.2 pixels; D-NAME, 9758.3 ± 1515.4 pixels, P < 0.02), and the quantity of necrotic tissue was higher in L-NAME relative to D-NAME-treated animals (L-NAME, 73709.5 ± 8638.0 pixels; D-NAME, 38434.5 ± 7918.3 pixels, P < 0.007). In addition, the mass of viable tissue (ie, stroma and tumor cells), was lower in L-NAME relative to D-NAME-treated animals (L-NAME, 17.1 ± 3.5%; D-NAME, 28.8 ± 4.0%, P < 0.04). Values for the mass of viable tissue were calculated using the following formula: weight of implant (mg) × (1 − necrotic fraction of the implant).
Discussion
Angiogenesis, the development of new blood vessels from the pre-existing vascular bed, is an essential feature of many physiological conditions including wound healing, embryonic development, and endometrial proliferation. Numerous pathological conditions, such as diabetic retinopathy, rheumatoid arthritis, and tumor growth are also characterized by abnormal neovascularization. Growth of solid tumors cannot proceed beyond a microscopic size without the development of an extensive vascular system. 27 Furthermore, because the degree of vascularization often correlates with poor clinical prognosis and increased likelihood of metastasis of a number of human tumors, 28,29 targeting angiogenesis in the therapeutic intervention of cancer has received substantial attention. Although a number of compounds characterized as inhibitors of angiogenesis have entered clinical trials, intense efforts to identify potent angiogenesis inhibitors with improved selectivity continue. However, a consistent limitation of these investigations has been the availability of simple, reproducible, reliable, and easily quantifiable assays that reflect in vivo systems for tumor-induced angiogenesis. Overly simplistic cellular in vitro systems and technical difficulties of currently used in vivo angiogenesis assays are important limiting factors.
In vitro models of angiogenesis 22,30 may not be ideal for measuring tumor-induced angiogenesis because of the inability in providing all of the necessary cells and/or factors that may interact in the in vivo tumor environment. The most widely used in vivo systems are the rabbit cornea and the chick chorioallantoic membrane (CAM) assays, in which variability of results and subjectivity in quantification remain important limitations. 31 A major concern of the rabbit cornea assay is the potential development of xenograft reactions after tumor implantation. Although the cornea is an immunoprivileged site, as it gradually becomes vascularized, the possible contribution of xenograft reactions to the angiogenic response cannot be disregarded. The CAM assay does not present this problem, because the host is naturally immunodeficient. However, major disadvantages of this assay are the time limit of 7 to 10 days imposed by embryo growth and acquisition of immunocompetence 32 and difficulties in objectively quantifying the neovascular response. 31,33 Another potential concern is inherent with properties of the chick chorioallantoic membrane, which is a growing and developing embryonic structure; the relative contribution of vasculogenesis and/or angiogenesis, two discrete processes which are differentially regulated, to the development of new blood vessels may not be clear.
The present in vivo model of tumor-induced angiogenesis is devoid of these limitations. The assay is simple, reproducible, and objectively quantifiable. Suspension of tumor cells within the Matrigel matrix serves to effectively immobilize tumor cells; the ensuing angiogenic response is organized, and all stages of the neovascular response can be quantified. Therefore, the kinetics of tumor development and neovascularization may be followed for a considerable length of time (eg, 2 weeks in the present experiment); in separate experiments, we have established that the experimental procedure can be extended (eg, up to 6 weeks). Furthermore, because the developing blood vessels converge within a discrete area, retrieval and quantification of developing blood vessels are simple and complete. Use of a specific inoculation site in the murine model minimizes variation of the angiogenic response. Although we used an inbred mouse strain and its syngeneic tumor, nude mice could appropriately serve as the host for xenografts in other applications of this assay, and in particular, for human tumors.
We used the present assay to evaluate the effects of NO on the angiogenic response by administering L-NAME or D-NAME to mice using osmotic minipumps in two separate experiments. Results from both clearly showed that, irrespective of method of quantification, NOS inhibition dramatically reduced the neovascular response. The growth patterns of histologically distinct areas present within the implants were differentially affected by NOS inhibition. The stromal component of the implants, which supports the vascular supply, was reduced in L-NAME- relative to D-NAME-treated animals. In addition, there was increased necrosis and reduced viable tissue mass within implants obtained from L-NAME relative to D-NAME-treated animals, supporting prior observations of antitumor effects of NOS inhibition in mice transplanted with C3L5 mammary adenocarcinomas. 14,15 Therefore, the antitumor and antimetastatic effects of NOS inhibition, previously attributed in part to reduced tumor cell invasiveness, 16 may also be explained by reduced neovascularization.
Inherent NOS activity of the eNOS-expressing mammary adenocarcinoma cells used in the present research is likely the major source of NO contributing to the NO-mediated induction of angiogenesis. eNOS expression by endothelial cells, as well as iNOS expression by some macrophages and endothelial cells, may serve as additional minor sources of NO in these tumor implants.
The precise molecular mechanisms responsible for reduced angiogenesis with NOS inhibition in our model remain to be determined. NO is required for endothelial cell proliferation, migration, and organization, key components of the angiogenic cascade. 22,34,35 Using a rabbit cornea assay, it has been shown that NO is a downstream mediator of vascular endothelial growth factor (VEGF)-induced angiogenesis, since it could be blocked by administering L-NAME. 35 Further evidence supports this notion; angiogenesis in response to tissue ischemia (an inducer of VEGF) was reduced in eNOS −/− mice. 36 VEGF-stimulated proliferation of endothelial cells, triggered by NO, was shown to require intracellular signaling via cGMP-dependent protein kinase, 37 Raf-1 kinase, 37 and mitogen- activated protein kinase. 35 Our preliminary results (data not shown) indicate that the C3L5 mammary adenocarcinoma cells used in the present research express VEGF protein in vitro. Whether VEGF expression in these cells is induced by endogenous NO remains to be determined; an up-regulation of VEGF mRNA by NO was reported for rat mesangial cells. 38 Co-expression of eNOS and VEGF in C3L5 cells may equip them with a dual advantage in inducing NO-mediated angiogenesis from the host vasculature, VEGF-mediated stimulation of NO in the vascular endothelium and NO produced by tumor cells by activation of eNOS.
Footnotes
Address reprint requests to Peeyush K. Lala, Department of Anatomy and Cell Biology, Medical Science Building, The University of Western Ontario, London, Ontario, Canada N6A 5C1. E-mail: pklala@julian.uwo.ca.
Supported by U.S. Army Grant DMAD-17-96-6096.
References
- 1.Moncada S, Higgs A: The L-arginine-nitric oxide pathway. N Engl J Med 1993, 329:2002-2012 [DOI] [PubMed] [Google Scholar]
- 2.Knowles RG, Moncada S: Nitric oxide synthases in mammals. Biochem J 1994, 298:249-258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thomsen LL, Lawton FG, Knowles RG, Beesley JE, Riveros-Moreno V, Moncada S: Nitric oxide synthase activity in human gynecological cancer. Cancer Res 1994, 54:1352-1354 [PubMed] [Google Scholar]
- 4.Thomsen LL, Miles DW, Happerfield L, Bobrow LG, Knowles RG, Moncada S: Nitric oxide synthase activity in human breast cancer. Br J Cancer 1995, 72:41-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dueñas-Gonzalez A, Isales CM, del Mar Abad-Hernandez M, Gonzalez-Sarmiento R, Sangueza O, Rodriguez-Commes J: Expression of inducible nitric oxide synthase in breast cancer correlates with metastatic disease. Mod Pathol 1997, 10:645-649 [PubMed] [Google Scholar]
- 6.Cobbs CS, Brenman JE, Aldape KD, Bredt DS, Israel MA: Expression of nitric oxide synthase in human central nervous system tumors. Cancer Res 1995, 55:727-730 [PubMed] [Google Scholar]
- 7.Thomsen LL, Miles DW: Role of nitric oxide in tumor progression: lessons from human tumors. Cancer Metastasis Rev 1998, 17:107-118 [DOI] [PubMed] [Google Scholar]
- 8.Klotz T, Bloch W, Volberg C, Engelmann U, Addicks K: Selective expression of inducible nitric oxide synthase in human prostate carcinoma. Cancer 1998, 82:1897-1903 [PubMed] [Google Scholar]
- 9.Gallo O, Masini E, Morbidelli L, Franchi A, Fini-Storchi I, Vergari WA, Ziche M: Role of nitric oxide in angiogenesis and tumor progression in head and neck cancer. J Natl Cancer Inst 1998, 90:587-596 [DOI] [PubMed] [Google Scholar]
- 10.Fujimoto H, Ando Y, Yamashita T, Terazaki H, Tanaka Y, Sasaki J, Matsumoto M, Suga M, Ando M: Nitric oxide synthase activity in human lung cancer. Jpn J Cancer Res 1997, 88:1190-1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kennovin GD, Hirst DG, Stratford MRL, Flitney FW: Inducible nitric oxide synthase is expressed in tumor-associated vasculature: inhibition retards tumor in vivo. Moncada S Feelisch M Busse R Higgs EA eds. Biology of Nitric Oxide, Part 4: Enzymology, Biochemistry, and Immunology. 1994, :pp 473-479 Portlant Press, London [Google Scholar]
- 12.Edwards P, Cendan JC, Topping DB, Moldawer LL, MacKay S, Copeland EM, Lind DS: Tumor cell nitric oxide inhibits cell growth in vitro, but stimulates tumorigenesis and experimental lung metastasis in vivo. J Surg Res 1996, 63:49-52 [DOI] [PubMed] [Google Scholar]
- 13.Lala PK, Orucevic A: Role of nitric oxide in tumor progression: lessons from experimental tumors. Cancer Metastasis Rev 1998, 17:91-106 [DOI] [PubMed] [Google Scholar]
- 14.Orucevic A, Lala PK: NG-nitro-L-arginine methyl ester, an inhibitor of nitric oxide synthesis, ameliorates interleukin-2-induced capillary leakage, and reduces tumor growth in adenocarcinoma-bearing mice. Br J Cancer 1996, 73:189-196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Orucevic A, Lala PK: Effects of NG-methyl-L-arginine, an inhibitor of nitric oxide synthesis, on IL-2-induced capillary leakage and antitumor responses in healthy and tumor-bearing mice. Cancer Immunol Immunother 1996, 42:38-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Orucevic A, Bechberger J, Green AM, Shapiro RA, Billiar TR, Lala PK: Nitric oxide production by murine mammary adenocarcinoma cells promotes tumor cell invasiveness. Int J Cancer 1999, 81:889-896 [DOI] [PubMed] [Google Scholar]
- 17.Chhatwal VJ, Ngoi SS, Chan ST, Chia YW, Moochhala SM: Aberrant expression of nitric oxide synthase in human polyps, neoplastic colonic mucosa and surrounding peritumoral normal mucosa. Carcinogenesis 1994, 15:2081-2085 [DOI] [PubMed] [Google Scholar]
- 18.Ambs S, Merriam WG, Bennett WP, Felley-Bosco E, Ogunfusika MO, Oser SM, Klein S, Shields PG, Billiar TR, Harris CC: Frequent nitric oxide synthase-2 expression in human colon adenomas: implication for tumor angiogenesis and colon cancer progression. Cancer Res 1998, 58:334-341 [PubMed] [Google Scholar]
- 19.Dong Z, Staroselsky AH, Qi X, Xie K, Fidler IJ: Inverse correlation between expression of inducible nitric oxide synthase activity and production of metastasis in K-1735 murine melanoma cells. Cancer Res 1994, 54:789-793 [PubMed] [Google Scholar]
- 20.Juang SH, Xie K, Xu L, Shi Q, Wang Y, Yoneda J, Fidler IJ: Suppression of tumorigenicity and metastasis of human renal carcinoma cells by infection with retroviral vectors harboring the murine inducible nitric oxide synthase gene. Hum Gene Ther 1998, 9:845-854 [DOI] [PubMed] [Google Scholar]
- 21.Ziche M, Morbidelli L, Masini E, Granger HJ, Geppetti G, Ledda F: Nitric oxide promotes DNA synthesis and cyclic GMP formation in endothelial cells from postcapillary venules. Biochem Biophys Res Commun 1993, 192:1198-1203 [DOI] [PubMed] [Google Scholar]
- 22.Ziche M, Morbidelli L, Masini E, Amerini S, Granger HJ, Maggi CA, Geppetti P, Ledda F: Nitric oxide mediates angiogenesis in vivo and endothelial cell growth and migration in vitro promoted by substance P. J Clin Invest 1994, 94:2036-2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Konturek SJ, Brzozowski T, Majka J, Pytko-Polonczyk J, Stachura J: Inhibition of nitric oxide synthase delays healing of chronic gastric ulcers. Eur J Pharmacol 1993, 239:215-217 [DOI] [PubMed] [Google Scholar]
- 24.Jenkins DC, Charles IG, Thomsen LL, Moss DW, Holmes LS, Baylis SA, Rhodes P, Westmore K, Emson PC, Moncada S: Roles of nitric oxide in tumor growth. Proc Natl Acad Sci USA 1995, 92:4392-4396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lala PK, Parhar RS: Eradication of spontaneous and experimental adenocarcinoma metastases with chronic indomethacin and intermittent IL-2 therapy. Int J Cancer 1993, 54:677-684 [DOI] [PubMed] [Google Scholar]
- 26.Kibbey MC, Grant DS, Kleinman HK: Role of the SIKVAV site of laminin in promotion of angiogenesis and tumor growth: an in vivo Matrigel model. J Nat Cancer Inst 1992, 84:1633-1637 [DOI] [PubMed] [Google Scholar]
- 27.Folkman J: Tumor angiogenesis: therapeutic applications. N Engl J Med 1971, 285:82-86 [DOI] [PubMed] [Google Scholar]
- 28.Weidner N, Semple JP, Welch WR, Folkman J: Tumor angiogenesis and metastasis-correlation in invasive breast carcinoma. N Engl J Med 1991, 324:1-8 [DOI] [PubMed] [Google Scholar]
- 29.Wiggins DL, Granai CO, Steinhoff MM, Calabresi P: Tumor angiogenesis as a prognostic factor in cervical carcinoma. Gynecol Oncol 1995, 56:353-356 [DOI] [PubMed] [Google Scholar]
- 30.Brown KJ, Maynes SF, Bezos A, Maguire DJ, Ford MD, Parish CR: A novel in vitro assay for human angiogenesis. Lab Invest 1996, 75:539-555 [PubMed] [Google Scholar]
- 31.Auerbach R, Auerbach W, Polakowski I: Assays for angiogenesis: a review. Pharmacol Ther 1991, 51:1-11 [DOI] [PubMed] [Google Scholar]
- 32.Leighton J: Invasion and metastasis of heterologous tumors in the chick embryo. Prog Exp Tumor Res 1964, 4:98-125 [DOI] [PubMed] [Google Scholar]
- 33.Vu MT, Smith CF, Burger PC, Klintworth GK: An evaluation of methods to quantitate the chick chorioallantoic membrane assay in angiogenesis. Lab Invest 1985, 53:499-508 [PubMed] [Google Scholar]
- 34.Papapetropoulos A, Desai KM, Rudic RD, Mayer B, Zhang R, Ruiz-Torres MP, Garcia-Cardeña G, Madri JA, Sessa WC: Nitric oxide synthase inhibitors attenuate transforming-growth-factor-β1-stimulated capillary organization in vitro. Am J Pathol 1997, 150:1835-1844 [PMC free article] [PubMed] [Google Scholar]
- 35.Ziche M, Morbidelli L, Choudhuri R, Zhang HT, Donnini S, Granger HJ, Bicknell R: Nitric oxide synthase lies downstream from vascular endothelial growth factor-induced but not fibroblast growth factor-induced angiogenesis. J Clin Invest 1997, 99:2625-2634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murohara T, Asahara T, Silver M, Bauters C, Masuda H, Kalka C, Kearney M, Chen D, Symes JF, Fishman MC, Huang PL, Isner JM: Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J Clin Invest 1998, 101:2567-2578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hood J, Granger HJ: Protein kinase G mediates vascular endothelial growth factor-induced Raf-1 activation and proliferation in human endothelial cells. J Biol Chem 1998, 273:23504-23508 [DOI] [PubMed] [Google Scholar]
- 38.Frank S, Stallmeyer B, Kampfer H, Schaffner C, Pfeilschifter J: Differential regulation of vascular endothelial growth factor and its receptor fms-like tyrosine kinase is mediated by nitric oxide in rat renal mesangial cells. Biochem J 1999, 338:367-374 [PMC free article] [PubMed] [Google Scholar]