

Abstract
Hepatic ischemia/reperfusion injury is initiated by the activation of Kupffer cells and their subsequent release of proinflammatory mediators, including tumor necrosis factor-α (TNFα). These mediators stimulate a cascade of events including up-regulation of CXC chemokines and vascular endothelial adhesion molecules, leading to hepatic neutrophil recruitment and tissue injury. Interleukin-13 (IL-13) is a cytokine that has been shown to suppress macrophage production of proinflammatory mediators. The objective of the current study was to determine whether IL-13 could regulate the liver inflammatory injury induced by ischemia and reperfusion. C57BL/6 mice underwent 90 minutes of partial hepatic ischemia followed by reperfusion with or without intravenous administration of recombinant murine IL-13. Hepatic ischemia/reperfusion increased expression of TNFα and macrophage inflammatory protein-2 (MIP-2), leading to hepatic neutrophil recruitment, hepatocellular injury, and liver edema. Administration of IL-13 reduced the production of TNFα and MIP-2 mRNA and protein. IL-13 suppressed liver neutrophil recruitment by up to 72% and hepatocellular injury and liver edema were each reduced by >60%. Administration of IL-13 had no effect on liver NFκB activation, but greatly increased the activation of STAT6. The data suggest that the hepatoprotective effects of IL-13 may be a result of STAT6 activation.
Liver injury caused by hepatic ischemia and reperfusion is a complication of hepatic resectional surgery, liver transplantation, and hemorrhagic shock with fluid resuscitation. 1-4 Experimental models of hepatic ischemia/reperfusion (I/R) injury have defined two distinct phases during the development of organ injury. During the initial phase, Kupffer cells are activated and release reactive oxygen species and proinflammatory cytokines, including tumor necrosis factor-α (TNFα). 5-7 The later phase of injury is dependent upon neutrophils. Enhanced hepatic production of TNFα plays a critical role in the initiation of a mediator cascade responsible for hepatic recruitment of neutrophils and subsequent liver injury. TNFα up-regulates intercellular adhesion molecule-1 (ICAM-1) expression on the hepatic vascular endothelium. 8,9 In addition, TNFα induces the hepatic expression of CXC chemokines, including macrophage inflammatory protein-2 (MIP-2) and epithelial neutrophil-activating protein-78 (ENA-78). 10,11 The combined effects of ICAM-1 and CXC chemokines result in adhesion and transmigration of neutrophils from the intravascular space to liver parenchyma. Neutrophil accumulation in hepatic sinusoids obstructs blood flow and contributes to hepatic hypoperfusion. 12 The ensuing liver injury is caused by neutrophil-derived oxidants and proteases, which directly damage endothelial cells and hepatocytes. 13
Interleukin-13 (IL-13) was originally identified as a product of activated Th2 cells which suppressed in vitro monocyte production of proinflammatory cytokines, including TNFα, IL-1, IL-6, IL-8, and MIP-1α. 14-17 In vivo, IL-13 prevents lipopolysaccharide-induced lethality and IgG immune complex-induced lung injury. 18-20 The mechanism(s) of the anti-inflammatory effects of IL-13 have been linked with inhibition of the transcription factor nuclear factor κB (NFκB). 21,22 However, in vitro, IL-13 potently activates the transcription factor, signal transducer and activator of transcription 6 (STAT6). 23,24 In the current studies, we evaluated the effects of IL-13 on the activation state of NFκB and STAT6 in liver during I/R injury and assessed whether these effects of IL-13 precluded development of liver inflammatory injury induced by hepatic ischemia and reperfusion.
Materials and Methods
Hepatic Ischemia/Reperfusion Injury Model
Male C57BL/6 mice (Charles River Laboratories, Wilmington, MA) weighing 22–28 g were used in all experiments. Partial hepatic ischemia was induced as described previously. 10 Briefly, mice were anesthetized with sodium pentobarbital (60 mg/kg i.p.). Mice received either sterile saline or recombinant murine IL-13 (1 μg; R&D Systems Inc., Minneapolis, MN) via the lateral tail vein before the induction of ischemia. A midline laparotomy was performed and an atraumatic clip was used to interrupt blood supply to the cephalad lobes of the liver. After 90 minutes of partial hepatic ischemia, mice again received either sterile saline or IL-13 (1 μg) via the lateral tail vein, and the clip was removed, initiating hepatic reperfusion. Sham control mice underwent the same protocol, but without vascular occlusion. Mice were sacrificed after 4 hours of reperfusion, and liver tissues and blood samples were taken for analysis. This project was approved by the University of Louisville Animal Care and Use Committee and was in compliance with National Institutes of Health guidelines.
Electrophoretic Mobility Shift Assay
Nuclear extracts of liver tissue were prepared by the method of Deryckere and Gannon 25 and analyzed by electrophoretic mobility shift assay (EMSA). Briefly, double-stranded NFκB consensus oligonucleotide (Promega, Madison, WI) or STAT6 consensus oligonucleotide (Santa Cruz Biotechnology, Santa Cruz, CA) were end-labeled with γ[ 32 P] ATP (3,000 Ci/mmol at 10 mCi/ml, Amersham, Arlington Heights, IL). Binding reactions containing equal amounts of nuclear protein extract (10 μg) and 35 fmol (∼50,000 cpm, Cherenkov counting) of oligonucleotide were incubated at room temperature for 30 minutes. Reaction volumes were held constant at 15 μl. For competition and supershift assays of STAT6 binding, unlabeled NFκB or STAT6 oligonucleotide (50-molar excess) or antibodies to STAT4 or STAT6 (Santa Cruz Biotechnology) were added to the reaction mixtures. Binding reaction products were separated in a 4% polyacrylamide gel and analyzed by autoradiography.
Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
Total RNA from liver tissue was extracted using RNeasy Mini Kit (Qiagen Inc., Valencia, CA). RNA (1 μg) was reverse transcribed to cDNA using random hexamers. cDNA products were coamplified by PCR (30 cycles of 95°C for 60 seconds, 59°C for 90 seconds, and 72°C for 10 seconds). Primers for TNFα (446-bp product), MIP-2 (205-bp product), and β-actin (245-bp product) have been described elsewhere. 26 PCR products were electrophoresed in a 3.5% agarose gel, stained with ethidium bromide, and photographed.
Liver Neutrophil Accumulation
Liver myeloperoxidase (MPO) content was determined by methods described elsewhere. 10 Briefly, liver tissue (50 mg) was homogenized, sonicated, and centrifuged for 20 minutes at 10,000 × g. Supernatants were reacted with 3,3′, 3,5′-tetramethylbenzidine (Sigma Chemical Co., St. Louis, MO) and read at 655 nm.
Liver neutrophil morphometric analysis was performed on frozen liver sections fixed in 0.5% glutaraldehyde and stained with hematoxylin and eosin. The number of neutrophils were counted in 5 separate high-power fields for each liver section. The results are expressed as neutrophils/high-power field.
Liver Edema
The extent of liver edema was measured by tissue wet-to-dry weight ratios. After dissection, liver samples were weighed and placed in a drying oven at 55°C until a constant weight was obtained. In this determination, liver edema is represented by an increase in the wet-to-dry weight ratios.
Blood Analyses
Blood was obtained by cardiac puncture at the time of sacrifice. Serum was analyzed for TNFα and MIP-2 by enzyme-linked immunosorbent assays (ELISA) according to manufacturer’s instructions (BioSource International, Camarillo, CA). Serum was also analyzed for alanine aminotransferase (ALT) as an indicator of hepatocellular injury. Measurements of serum ALT were made using a diagnostic kit from Sigma Chemical Co.
Statistical Analyses
All data are expressed as mean ± SE. Data were analyzed with a one-way analysis of variance with subsequent Student-Newman-Keuls test. Differences were considered significant when P < 0.05. To calculate percentage change, negative control values were subtracted from positive control and treatment group values.
Results
Effects of IL-13 on the Activation of NFκB and STAT6 in Liver
Because IL-13 is known to suppress activation of NFκB, 21 and because IL-13-induced activation of STAT6 is associated with anti-inflammatory effects of this cytokine, 24 we sought to determine whether IL-13 altered activation of NFκB and/or STAT6 in the liver during I/R injury. Nuclear extracts from liver tissue obtained from sham controls and livers undergoing ischemia and 1 or 4 hours of reperfusion were analyzed by EMSA. NFκB activation was greatly increased in liver after hepatic ischemia and 1 or 4 hours of reperfusion, compared to that in liver from sham controls (Figure 1A) ▶ . The administration of IL-13 had no effect on NFκB activation at either time point. In contrast, hepatic ischemia and reperfusion did not result in detectable increases in STAT6 activation (Figure 1A) ▶ . However, intravenous treatment with IL-13 resulted in a large increase in activation of STAT6 after 1 hour of reperfusion. IL-13-induced STAT6 activation was also present after 4 hours of reperfusion.
Figure 1.
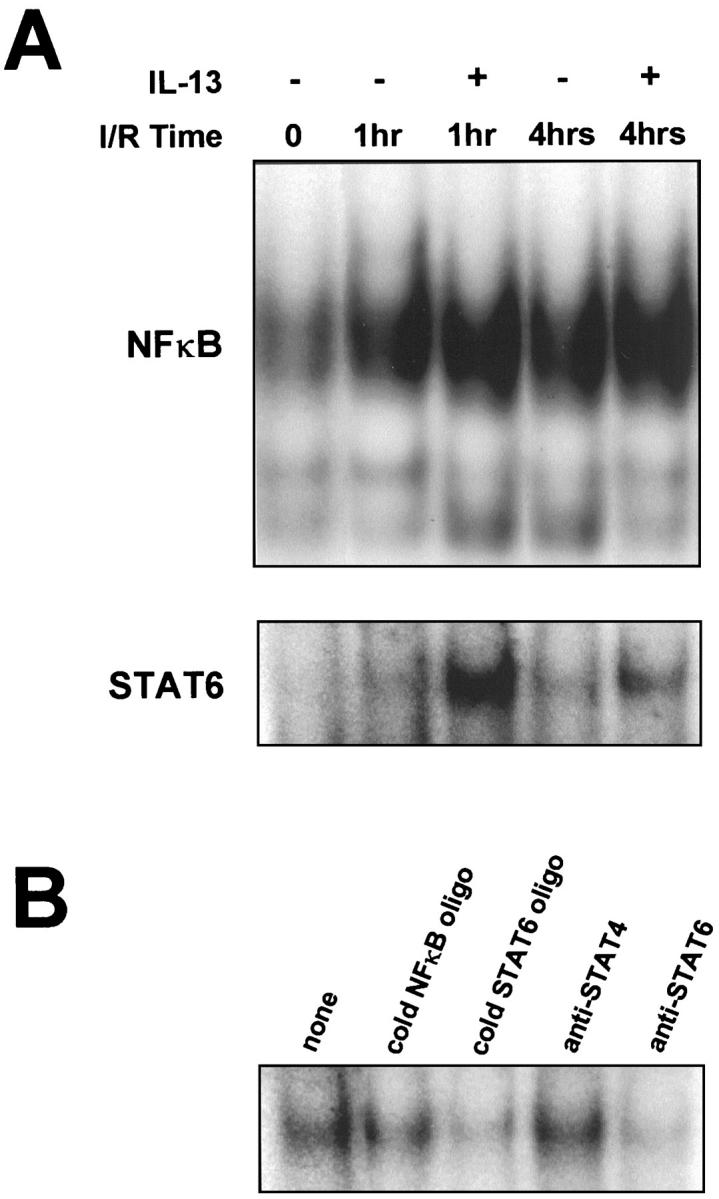
Effects of IL-13 on activation of NFκB and STAT6 in liver during hepatic ischemia/reperfusion (I/R) injury. A: EMSA analysis of NFκB and STAT6 in whole liver nuclear extracts from sham controls (I/R time: 0), ischemia and 1 hour reperfusion, and ischemia and 4 hours reperfusion in the presence or absence of IL-13. B: Competition and supershift assays were performed to verify the specificity of the STAT6 consensus oligonucleotide probe. Whole liver extracts obtained from mice treated with IL-13 and undergoing ischemia and 1 hour of reperfusion were incubated with 32P-labeled STAT6 oligonucleotide in the absence (none) or presence of unlabeled (cold) NFκB oligonucleotide, unlabeled (cold) STAT6 oligonucleotide, or antibodies to STAT4 or STAT6.
The specificity of the STAT6 consensus oligonucleotide probe was confirmed by experiments using nuclear extracts from liver undergoing ischemia and 1 hour of reperfusion (Figure 1B) ▶ . In DNA binding reactions, liver nuclear extracts that were incubated with only 32P-labeled STAT6 consensus oligonucleotide probe showed typical binding to the labeled oligonucleotide (Figure 1B) ▶ . Competition with 50-fold molar excess of unlabeled NFκB oligonucleotide had no effect on DNA binding to the STAT6 oligonucleotide. Competition with excess unlabeled STAT6 oligonucleotide completely abolished DNA binding to the labeled STAT6 oligonucleotide. Addition of polyclonal rabbit anti-STAT6, but not anti-STAT4, resulted in almost complete disappearance of the STAT6 band. These data demonstrate that IL-13 administration causes hepatic activation of STAT6.
Effects of IL-13 on Production of TNFα and MIP-2
To further investigate the effects of IL-13, including activation of STAT6, on the development of liver injury induced by I/R, we assessed whether IL-13 administration altered the generation of TNFα and MIP-2 mRNA in liver tissue. Liver RNA extracts were analyzed by RT-PCR. Hepatic ischemia and 4 hours of reperfusion resulted in large increases in the amount of TNFα (Figure 2A) ▶ and MIP-2 mRNA (Figure 2B) ▶ . Administration of IL-13 greatly reduced the generation of TNFα and MIP-2 mRNA in liver tissue. Results shown are representative of three independent experiments.
Figure 2.

Effects of IL-13 on mRNA expression for TNFα (A) and MIP-2 (B) as well as serum levels of TNFα (C) and MIP-2 (D) protein. Liver RNA extracts and serum samples were obtained from sham control mice and mice undergoing ischemia and 4 hours of reperfusion treated with saline (I/R) or IL-13 (I/R + IL-13). Liver RNA extracts were assessed by RT-PCR (A and B). Results are representative of three independent experiments. Serum samples were analyzed by ELISA (C and D). Values represent mean ± SE with n = 10 per group.
In this model, serum levels of TNFα and MIP-2 have been shown to be closely associated with the hepatic production of these mediators. 6,7,10 ELISA analysis of serum demonstrated that TNFα was significantly increased after ischemia and 4 hours of reperfusion (Figure 2C) ▶ . Administration of IL-13 reduced the amount of TNFα in serum by 75% (P < 0.001). Serum levels of MIP-2 were also increased after hepatic ischemia and reperfusion (Figure 2D) ▶ . In the presence of IL-13, serum levels of MIP-2 were decreased by 45% (P = 0.011).
Effects of IL-13 on Hepatic Neutrophil Accumulation and Inflammatory Injury
Because TNFα and MIP-2 have been shown to be critical mediators for the hepatic recruitment of neutrophils, we assessed whether the suppressive effects of IL-13 on TNFα and MIP-2 production were associated with reduced liver accumulation of neutrophils. Hepatic neutrophil accumulation was measured by liver MPO content. After hepatic ischemia and 4 hours of reperfusion there was a significant increase in liver MPO content compared to the sham controls (Figure 3A) ▶ . In the presence of IL-13, liver MPO content was reduced by 72% (P = 0.019). Similarly, hepatic I/R caused a significant increase in the number of neutrophils in liver sections assessed by morphometric analysis (Figure 3B) ▶ . The administration of IL-13 resulted in a 62% decrease in the number of neutrophils observed in liver sections (P = 0.003).
Figure 3.

Effects of IL-13 on liver neutrophil accumulation (A and B), hepatocellular injury (C), and liver edema (D) in sham control mice and mice undergoing ischemia and 4 hours of reperfusion treated with saline (I/R) or IL-13 (I/R + IL-13). Liver neutrophil accumulation was determined by measuring liver myeloperoxidase (MPO) content (A), as well as by liver tissue neutrophil morphometrics (PMN/high power field; B). Hepatocellular injury was assessed by determining serum levels of alanine aminotransferase (ALT). Liver edema was quantitated by liver wet-to-dry weight ratios. Values represent mean ± SE with n = 10 per group.
To investigate the effects of IL-13 on liver injury induced by hepatic ischemia/reperfusion, we assessed hepatocellular injury and liver edema in the presence or absence of IL-13. Hepatocellular injury was quantified by measuring serum levels of ALT and liver edema was determined by measuring liver wet-to-dry weight ratios. After hepatic ischemia and 4 hours of reperfusion there were significant increases in hepatocellular injury compared to the sham controls (Figure 3C) ▶ . In the presence of IL-13, serum ALT levels were decreased by 60% (P = 0.002). Significant liver edema was induced by hepatic ischemia and reperfusion (Figure 3D) ▶ . Treatment with IL-13 reduced liver edema by 66% (P = 0.004).
Discussion
The pathogenesis of hepatic I/R injury begins with activation of Kupffer cells during ischemia and results in the subsequent release of proinflammatory cytokines. 5-7 These proinflammatory cytokines, including TNFα, then cause the up-regulation of CXC chemokines and adhesion molecules, leading to hepatic recruitment of neutrophils and ensuing inflammatory tissue injury. 7,9,11 The administration of IL-13 reduced I/R-induced production of TNFα and MIP-2. Reduction of these mediators resulted in reduced accumulation of neutrophils in the liver and limited the development of liver injury. To determine the mechanism(s) of the protective effects of IL-13, we assessed the effects of IL-13 on the activation of NFκB and STAT6 in the liver.
Although a distinct role for STAT6 in the inflammatory response has not been defined, it has recently been shown that STAT6 activation by IL-13 enhances the production of IL-1 receptor antagonist production in hepatocytes. 27 Furthermore, the anti-inflammatory activity of IL-13 is compromised in macrophages from STAT6-deficient mice. 24 In the current studies, hepatic ischemia and up to 4 hours of reperfusion did not result in detectable activation of STAT6 in liver. Similarly, others have reported that in a murine model of partial hepatectomy STAT6 is not activated in liver during or after liver resection. 28 However, we show that administration of IL-13 caused substantial activation of STAT6 within 1 hour after hepatic reperfusion. This activation was associated with greatly reduced production of TNFα and MIP-2, suppressed neutrophil accumulation, and decreased liver injury. Although our data provide evidence suggesting an anti-inflammatory role for STAT6, little is known about the manner in which this transcription factor may exert these effects. Activation of STAT6 has been reported to suppress TNFα-induced adhesion molecule expression by antagonizing the DNA-binding of NFκB. 29 More recently, it has been shown that STAT6 regulates the expression of FcɛRI in mast cells 30 as well as an inhibitor of cyclin-dependent kinase, p27Kip1. 31 Thus, there is evidence that STAT6 may function as a negative regulator of transcription. Furthermore, a recent report demonstrated that STAT6 activation was required for IL-4-induced suppression of TNFα production in macrophages. 32 The precise mechanism by which STAT6 may suppress the production of proinflammatory mediators, such as TNFα and MIP-2, is currently unknown.
We have previously shown that IL-13 suppresses NFκB activation by preventing the degradation of the NFκB-inhibiting protein IκBα. 21 However, it was recently demonstrated that liver activation of NFκB during hepatic ischemia/reperfusion occurs without degradation of IκBα. 33 Furthermore, those studies showed that tyrosine phosphorylation of IκBα occurred, a mechanism of NFκB activation that does not require IκBα degradation. 34 Our current studies suggest that the protective effects of IL-13 in liver injury induced by I/R are unrelated to its effects on NFκB. The administration of IL-13 greatly suppressed proinflammatory cytokine and chemokine production, liver neutrophil recruitment and development of liver injury induced by ischemia and reperfusion. These data suggest that IL-13 may represent a potential therapeutic strategy for the treatment of inflammatory liver disease. The protective effects of IL-13 were preceded by activation of STAT6 in liver, suggesting that STAT6 may operate as a negative regulator of proinflammatory mediator production. Combined with the recent findings of others, 29-32 these studies provide evidence for an antiinflammatory role for STAT6 in vivo. Further studies of the function of STAT6 should provide a better understanding of the role of this transcription factor in the regulation of acute inflammatory responses.
Footnotes
Address reprint requests to Alex B. Lentsch, Ph.D., Department of Surgery, University of Louisville School of Medicine, 529 South Jackson Street, J.G. Brown Cancer Center, Room 426, Louisville, Kentucky 40202. E-mail: alentsch@louisville.edu.
Supported by The Jewish Hospital Foundation.
References
- 1.Huguet C, Gavelli A, Bona S: Hepatic resection with ischemia of the liver exceeding one hour. J Am Coll Surg 1994, 178:454-458 [PubMed] [Google Scholar]
- 2.Liu DL, Jeppsson B, Hakansson CH, Odselius R: Multiple-system organ damage resulting from prolonged hepatic inflow interruption. Arch Surg 1996, 131:442-447 [DOI] [PubMed] [Google Scholar]
- 3.Lemasters JJ, Thurman RG: Reperfusion injury after liver preservation for transplantation. Annu Rev Pharmacol Toxicol 1997, 37:327-338 [DOI] [PubMed] [Google Scholar]
- 4.Vedder NB, Fouty BW, Winn RK, Harlan JM, Rice CL: Role of neutrophils in generalized reperfusion injury associated with resuscitation from shock. Surgery 1989, 106:509-516 [PubMed] [Google Scholar]
- 5.Jaeschke H, Farhood A: Neutrophil and Kupffer cell-induced oxidant stress and ischemia-reperfusion injury in rat liver. Am J Physiol 1991, 260:G355-G362 [DOI] [PubMed] [Google Scholar]
- 6.Wanner GA, Ertel W, Muller P, Hofer Y, Leiderer R, Menger MD, Messmer K: Liver ischemia and reperfusion induces a systemic inflammatory response through Kupffer cell activation. Shock 1996, 5:34-40 [DOI] [PubMed] [Google Scholar]
- 7.Colletti LM, Remick DG, Burtch GD, Kunkel SL, Strieter RM, Campbell DA, Jr: Role of tumor necrosis factor-α in the pathophysiologic alternations after hepatic ischemia/reperfusion injury in the rat. J Clin Invest 1990, 85:1936-1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Farhood A, McGuire GM, Manning AM, Miyasaka M, Smith CW, Jaeschke H: Intercellular adhesion molecule 1 (ICAM-1) expression and its role in neutrophil-induced ischemia-reperfusion injury in rat liver. J Leukoc Biol 1995, 57:368-374 [PubMed] [Google Scholar]
- 9.Colletti LM, Cortis A, Lukacs N, Kunkel SL, Green M, Strieter RM: Tumor necrosis factor up-regulates intercellular adhesion molecule 1, which is important in the neutrophil-dependent lung and liver injury associated with hepatic ischemia and reperfusion in the rat. Shock 1998, 10:182-191 [DOI] [PubMed] [Google Scholar]
- 10.Lentsch AB, Yoshidome H, Cheadle WG, Miller FN, Edwards MJ: Chemokine involvement in hepatic ischemia/reperfusion injury in mice: roles for macrophage inflammatory protein-2 and KC. Hepatology 1998, 27:1172-1177 [DOI] [PubMed] [Google Scholar]
- 11.Colletti LM, Kunkel SL, Walz A, Burdick MD, Kunkel RG, Wilke CA, Strieter RM: Chemokine expression during hepatic ischemia/reperfusion-induced lung injury in the rat: the role of epithelial neutrophil activating protein. J Clin Invest 1995, 95:134-141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vollmar B, Richter S, Menger MD: Leukocyte stasis in hepatic sinusoids. Am J Physiol 1996, 270:G798-G803 [DOI] [PubMed] [Google Scholar]
- 13.Jaeschke H, Smith CW: Mechanisms of neutrophil-induced parenchymal cell injury. J Leukoc Biol 1997, 61:647-653 [DOI] [PubMed] [Google Scholar]
- 14.Minty A, Chalon P, Derocq JM, Dumont X, Guillemot JC, Kaghad M, Labit C, Leplatosis P, Liauzun P, Miloux B, Minty C, Casellas P, Loison G, Lupker J, Shire D, Ferrara P, Caput D: Interleukin-13 is a new human lymphokine regulating inflammatory and immune responses. Nature 1993, 362:248-250 [DOI] [PubMed] [Google Scholar]
- 15.de Waal Malefyt R, Figdor CG, Huijbens R, Mohan-Peterson S, Bennett B, Culpepper J, Dang W, Zurawski G, de Vries JE: Effects of IL-13 on phenotype, cytokine production, and cytotoxic function of human monocytes: comparison with IL-4 and modulation by IFN-γ or IL-10. J Immunol 1993, 151:6370-6381 [PubMed] [Google Scholar]
- 16.Deleuran B, Iversen L, Deleuran M, Yssel H, Kragballe K, Stengaard-Pedersen K, Thestrup-Pedersen K: Interleukin 13 suppresses cytokine production and stimulates the production of 15-HETE in PBMC: a comparison between IL-4 and IL-13. Cytokine 1995, 7:319-324 [DOI] [PubMed] [Google Scholar]
- 17.Berkman N, John M, Roesems G, Jose P, Barnes PJ, Chung KF: Interleukin 13 inhibits macrophage inflammatory protein-1α production from human alveolar macrophages and monocytes. Am J Respir Cell Mol Biol 1996, 15:382-389 [DOI] [PubMed] [Google Scholar]
- 18.Muchamuel T, Menon S, Pisacane P, Howard MC, Cockayne DA: IL-13 protects mice from lipopolysaccharide-induced lethal endotoxemia: correlation with down-modulation of TNF-α, IFN-γ, and IL-12 production. J Immunol 1997, 158:2898-2903 [PubMed] [Google Scholar]
- 19.Doyle AG, Herbein G, Montaner LJ, Minty A, Caput D, Ferrara P, Gordon S: Interleukin-13 alters the activation state of murine macrophages in vitro: comparison with interleukin-4 and interferon-γ. Eur J Immunol 1994, 24:1441-1445 [DOI] [PubMed] [Google Scholar]
- 20.Mulligan MS, Warner RL, Foreback JL, Shanley TP, Ward PA: Protective effects of IL-4, IL-10, IL-12, and IL-13 in IgG immune complex-induced lung injury. J Immunol 1997, 159:3483-3489 [PubMed] [Google Scholar]
- 21.Lentsch AB, Shanley TP, Sarma V, Ward PA: In vivo suppression of NF-κB and preservation of IκBα by interleukin-10 and interleukin-13. J Clin Invest 1997, 100:2443-2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manna SK, Aggarwal BB: IL-13 suppresses TNF-induced activation of nuclear factor-κB, activation protein-1, and apoptosis. J Immunol 1998, 161:2863-2872 [PubMed] [Google Scholar]
- 23.Izuhara K, Heike T, Otsuka T, Yamaoka K, Mayumi M, Imamura T, Niho Y, Harada N: Signal transduction pathway of interleukin-4 and interleukin-13 in human B cells derived from X-linked severe combined immunodeficiency patients. J Biol Chem 1996, 271:619-622 [DOI] [PubMed] [Google Scholar]
- 24.Takeda K, Kamanaka M, Tanaka T, Kishimoto T, Akira S: Impaired IL-13-mediated functions of macrophages in STAT6-deficient mice. J Immunol 1996, 157:3220-3222 [PubMed] [Google Scholar]
- 25.Deryckere F, Gannon F: A one-hour minipreparation technique for extraction of DNA-binding proteins from animal tissues. Biotechniques 1994, 16:405. [PubMed] [Google Scholar]
- 26.Yoshidome H, Kato A, Edwards MJ, Lentsch AB: Interleukin-10 suppresses hepatic ischemia/reperfusion injury in mice: implications for a central role of NFκB. Hepatology 1999, 30:203-208. [DOI] [PubMed] [Google Scholar]
- 27.Gabay C, Porter B, Guenette D, Billir B, Arend WP: Interleukin-4 (IL-4) and IL-13 enhance the effect of IL-1β on production of IL-1 receptor antagonist by human primary hepatocytes and hepatoma HepG2 cells: differential effect on C-reactive protein production. Blood 1999, 93:1299-1307 [PubMed] [Google Scholar]
- 28.Heim MH, Gamboni G, Beglinger C, Gyr K: Specific activation of AP-1 but not Stat3 in regenerating liver in mice. Eur J Clin Invest 1997, 27:948-955 [DOI] [PubMed] [Google Scholar]
- 29.Bennett BL, Cruz R, Lacson RG, Manning AM: Interleukin-4 suppression of tumor necrosis factor α-stimulated E-selectin gene transcription is mediated by STAT6 antagonism of NF-κB. J Biol Chem 1997, 272:10212-10219 [DOI] [PubMed] [Google Scholar]
- 30.Ryan JJ, DeSimone S, Klisch G, Shelburne C, McReynolds LJ, Han K, Kovacs R, Mirmonsef P, Huff TF: IL-4 inhibits mouse mast cell FcɛRI expression through a STAT6-dependent mechanism. J Immunol 1998, 161:6915-6923 [PubMed] [Google Scholar]
- 31.Kaplan MH, Daniel C, Schindler U, Grusby MJ: Stat proteins control lymphocyte proliferation by regulating p27Kip1 expression. Mol Cell Biol 1998, 18:1996-2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levings MK, Schrader JW: IL-4 inhibits the production of TNF-α and IL-12 by STAT6-dependent and -independent mechanisms. J Immunol 1999, 162:5224-5229 [PubMed] [Google Scholar]
- 33.Zwacka RM, Zhang Y, Zhou W, Halldorson J, Engelhardt JF: Ischemia/reperfusion injury in the liver of BALB/c mice activates AP-1 and nuclear factor κB independently of IκB degradation. Hepatology 1998, 28:1022-1030 [DOI] [PubMed] [Google Scholar]
- 34.Imbert V, Rupec RA, Livolsi A, Pahl HL, Traenckner EB, Mueller-Dieckmann C, Farahifar D, Rossi B, Auberger P, Baeuerle PA, Peyron JF: Tyrosine phosphorylation of IκB-α activates NF-κB without proteolytic degradation of IκB-α. Cell 1996, 86:787-798 [DOI] [PubMed] [Google Scholar]