

Abstract
Most colorectal cancers have loss of function mutations in the adenomatosis polyposis coli (APC) tumor suppressor gene. This leads to accumulation of β-catenin, which together with the DNA binding protein TCF-4 functions as a transcriptional activator. Recently defined target genes are c-myc and cyclin D1, linking the APC gene defect to the capacity for autonomous proliferation of colon tumors. Here we report the identification of the matrix metalloproteinase MMP-7 as another target gene of β-catenin/TCF-4. MMP-7 is overexpressed in 80% of human colorectal cancers and known to be an important factor for early tumor growth, with a potential function also for later progression steps, like invasion and metastasis. Our results explain the high percentage of MMP-7 overexpression in colon tumors. Moreover they indicate that defects in the APC tumor suppressor gene may also have an influence on later steps of colon tumor progression.
Loss of function mutations in the adenomatosis polyposis coli (APC) tumor suppressor gene are an early event in colorectal carcinogenesis and found in up to 80% of human colon tumors. 1 Normal APC protein promotes degradation of β-catenin. As an effector of the (wingless) WNT-signaling pathway, β-catenin is able to bind to the T cell factor) (TCF) family of transcription factors and subsequently activate target genes. 2 Accordingly, in colorectal cancer cells with mutated APC gene β-catenin accumulates in the nuclear TCF bound fraction leading to a constitutive target gene activation. 3,4 Recently defined genes activated by β-catenin/TCF are c-myc 5 and cyclin D1. 6 Both proteins have an essential function in cellular proliferation and are known to be overexpressed in colorectal cancers. 7,8 These findings directly link mutations in the APC gene or other effectors of the APC controlled WNT-pathway to the autonomous growth of colorectal tumors.
Autonomous growth is found in both benign colorectal adenomas and malignant colorectal carcinomas. The essential step in the progression from adenomas to carcinomas is invasive growth, whereby basal membranes and extracellular matrix are degraded. One class of proteins participating in this process are matrix matalloproteinases (MMPs). However, recent evidence suggests that MMPs play a more complex role in tumor progression. They are necessary to create a microenvironment supporting the initiation and maintenance of growth of primary tumor and metastasis. 9 Their mechanisms are not yet fully understood; however, they could also participate in processes like tumor angiogenesis. In contrast to other MMPs such as gelatinase A (MMP-2), stromelysin 1 (MMP-3), and stromelysin 3 (MMP-11), which are produced by stromal cells, MMP-7 (also named matrilysin) is expressed in the tumor cells of colorectal carcinomas and other epithelial tumors. 10 Overexpression of MMP-7 has been demonstrated for about 80% of colorectal carcinomas. 10,11 MMP-7 was shown to be important for the growth of colon adenoma 12 and the tumorigenicity of colon cancer cells. 13 Additionally it was shown that MMP-7 plays an important role for the invasive and metastatic potential of cancer cells. 14,15
In APC gene mutated colorectal cancers overexpression of β-catenin is often found predominantly at the invasion front. 16 This led us to the hypothesis that β-catenin/TCF are directly involved in invasion processes by activating invasion associated genes. A GenBank search for the TCF consensus binding sequence (5′-A/T A/T CAAAG-3′) in the promoters of such genes identified several MMPs. Here we demonstrate that the promoter of the human MMP-7 gene contains two TCF binding sites and is activated by β-catenin/TCF. Moreover, MMP-7 expression is correlated with β-catenin expression in hereditary and sporadic human colon carcinomas. We conclude that overexpression of the tumor progression factor MMP-7 in up to 80% of colorectal carcinomas can be caused by mutations in the APC gene.
Materials and Methods
Electromobility Shift Assays (EMSAs)
The following double stranded oligonucleotides were used as probes or for competition: TCF(1.): 5′-ACATACTTTCAAAGTTCTGTA; TCF(1.m): 5′-ACATACTGCCAAAGTTCTGTA; TCF(2.): 5′-AAAAATCCTTTGAAAGACA-AA; TCF(2.m): 5′-AAAAATCCTTTGGCAGACAAA; TBE1: 5′-CGCACCTTTGATTTCTGCACCTTTGATTTCT. Probes were end-labeled to 3 × 10 8 dpm/μg 0.5 ng probe was incubated with 0.5 μg of bacterially expressed GST-TCF-4 (codons 265–496) as described. 5 For specific competition 500× amounts (250 ng) of unlabeled oligonucleotides were used.
Cells and Cell Culture
HeLa and HT29 cells were obtained from ATCC. All cell lines were cultivated under standard conditions in DMEM + 10% fetal bovine serum.
DNA Clones
phMMP-7prLuc was constructed by cloning the XhoI/HinDIII fragment (encoding nucleotides −933 to +35 of the human MMP-7 promoter) of the plasmid p-933HPCAT (gift from L. Matrisian, Vanderbilt University, Nashville, TN) into the XhoI/HinDIII opened vector pGL3-Basic (Promega, Mannheim, Germany). For mutation of phMMP-7prLuc (wild type; WT) with the QuickChange mutagenesis kit (Stratagene, Heidelberg, Germany) we used the following primers: pTCF(1.m): 5′-GTTAATGAAAAATAACACATACTGCCAAAGTTCTGTAGACTC; pTCF(2.m): 5′-GACAGAAAAAAAAATCCTTTGGCAGACAAATACATTGTG.To construct the four times tandem repeat clones p4xTCF(1.)-Luc, p4xTCF(1.m)-Luc, p4xTCF(2.)-Luc and p4xTCF(2.m)-Luc, we polymerized the corresponding double-stranded oligonucleotides used for EMSAs and cloned them into the HinDIII cut and blunted vector ptkLuc (gift from E. Serfling, Department of Pathology, Wuerzburg, Germany), containing a thymidine kinase minimal promoter. Following plasmids were gifts from other researchers: pGST-TCF4(DBD), pcDNA/myc-hTCF4 and pcDNA/myc-DN-TCF4 from K. Kinzler and B. Vogelstein (Johns Hopkins Oncology Center, Baltimore, MD), pcDNAhβ-catenin was a gift from H. C. Clevers (University Hospital, Utrecht, The Netherlands), pSV40Ets1 from J. Ghysdael (Institute Curie, Orsay, France), pMSc-fos from M. Schuermann (Marburg, Germany) and pRSVc-jun from P. Angel (Karlsruhe, Germany).
Transfections and Reporter Assays
HeLa and HT29 cells were grown in 60-mm tissue culture dishes to a confluency of 50% and transfected with 2 μg of reporter construct, 0.5 μg of pCMVβGal-control, and 3 μg of expression vectors or empty vector pCNA3 as indicated, using Lipofectamine Plus (Life Technologies, Karlsruhe, Germany). Cells were harvested after 40 hours, luciferase assays were carried out and normalized for transfection efficiency through β-galactosidase activity using Dual Light Kit (Perkin Elmer, Weiterstadt, Germany). Assays were performed in triplicate.
Immunohistochemistry
Formalin-fixed, paraffin-embedded tissue blocks of colorectal cancers with variable expression of β-catenin were used. Three tumors were from genetically defined familial adenomatosis polyposis (FAP) patients and two from patients with hereditary non-polyposis colorectal cancers (HNPCC). APC gene status of the HNPCC cases was considered as normal, based on loss of heterozygosity analyses (marker D5S107). However, exact sequencing data of the APC gene were not available for these cases. The other cases were sporadic carcinomas. 3-μm serial sections were deparaffinized, rehydrated, and incubated with the different mouse monoclonal antibodies for 2 hours. Antigen retrieval was used for α-MMP-7, mib-1 (microwave treatment), and CEA (protease XIV). Dilutions were 1:300 for α-MMP-7 (Chemicon, Hofheim, Germany), 1:200 for α-β-catenin (Transduction, Lexington, KY), 1:30 for α-Mib-1 and α-CEA (Dako, Hamburg, Germany). Biotinylated rabbit α-mouse Ig antiserum diluted 1:50 was used as the secondary antibody. After washing, slides were incubated for 30 minutes at room temperature with streptavidin-coupled alkaline phosphatase (Dako, Hamburg, Germany) and developed for 12 minutes at 37°C using Fast Red (Sigma, Deisenhofen, Germany) as substrate. After rinsing in water, sections were counterstained with hemalum (Merck, Darmstadt, Germany), dehydrated, and coverslipped.
Results
TCF4 Binds to Two Elements in the Human MMP-7 Promoter
By searching the GenBank, we identified potential TCF binding sites in the promoters of several human invasion associated genes including MMP-1, MMP-3, and MMP-7. We focused on MMP-7 because of its overexpression in most colorectal cancers. Three transforming growth factor (TGF)-β inhibitory elements (TIEs), two binding sites for ets transcription factors (PEA3), and one AP-1 binding site were already described in the human MMP-7 promoter. 17 We identified two potential TCF binding elements: TCF(1.) from −109 to −103 and TCF(2.) from −194 to −188, which matched the consensus sequence 5′-A/T A/T CAAAG-3′ (Figure 1a) ▶ .
Figure 1.
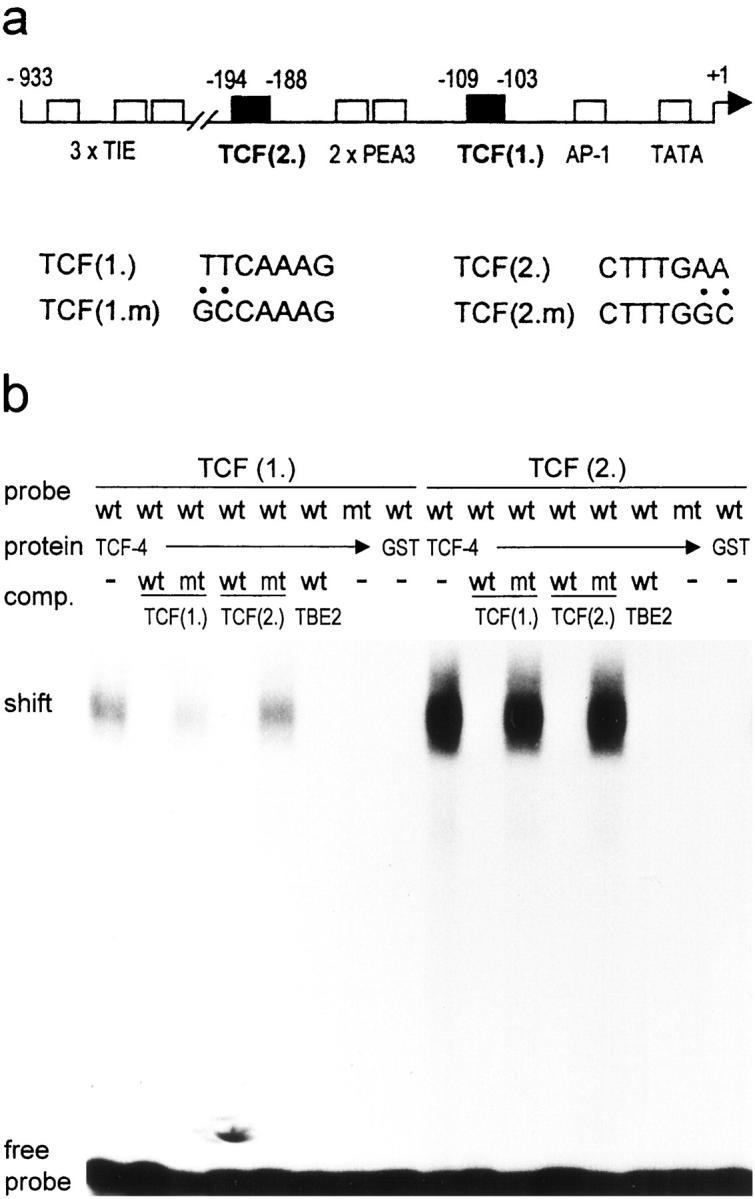
Identification of two TCF binding sites in the human MMP-7 promoter. a: Localization of the two identified TCF binding sites in the human MMP-7 promoter, TCF(1.) and TCF(2.) (▪). Also indicated are the previously defined TGF-β inhibitory element-like sequences (TIE), the ets (PEA3) and the AP-1 binding sites (□). Numbers indicate nucleotide positions relative to the transcription initiation site (+1). The diagram is not to scale. Also shown are the nucleotide sequences of both sites and the mutated sequences used in the following experiments (TCF(1.m) and TCF(2.m)). b: TCF-4 binds specifically to both TCF sites. EMSA with TCF(1.) and TCF(2.) sequences (wt) or their mutants (mt) as probes, incubated with GST-TCF-4 (TCF-4) or GST alone. For specific competition the unlabeled oligonucleotides used as probes or TBE2 (TCF binding site of the c-myc promoter) were used.
To verify TCF-4 binding we used both potential TCF elements as probes in an electromobility shift assay and incubated with recombinant GST-TCF-4. TCF-4 bound to both probes, but complex formation was much stronger with TCF(2.) (Figure 1b) ▶ . GST protein alone did not form a complex. Specificity was confirmed by using the two mutated TCF sites as probes, which then were not bound by TCF-4. Moreover, we used wild-type or mutated TCF(1.), TCF(2.) and TBE1, a TCF site of the c-myc promoter, 5 as specific competitors.
β-Catenin and TCF4 Activate the Human MMP-7 Promoter
To determine whether TCF-4 binding to the defined elements has a functional consequence, we transiently transfected luciferase reporter constructs controlled by the human MMP-7 promoter together with different expression constructs into HeLa cells (Figure 2b) ▶ . Cotransfections of TCF-4 or β-catenin expression clones led to a 3.2-fold or 1.88-fold activation, respectively, compared to the empty vector pcDNA3. This was similar to the known stimulatory effect of TPA on the MMP-7 promoter (2.36- fold). 17 A synergistic activation (9.4-fold) was found after cotransfection of TCF-4 and β-catenin expression constructs. We further tested the effect of overexpressed Ets1 and c-Jun/c-Fos, binding to the PEA3 and the AP-1 site in the MMP-7 promoter. Isolated stimulation of the promoter through only one of these elements had only a weak effect in HeLa cells. However combined expression of both TCF4/β-catenin and Ets1, c-Jun/c-Fos led to a maximum activation of the MMP-7 promoter. This was similar to a combined activation by TCF4/β-catenin and TPA, which mimics AP-1 activation. The empty luciferase vector did not respond to either activator (not shown). Thus TCF/β-catenin contributed to a high extent (∼60%) to the maximum activation of the human MMP-7 promoter achieved through all defined elements.
Figure 2.

β-catenin/TCF activate the human MMP-7 promoter. a: Reporter constructs used in transient transfection assays. The phMMP-7prLuc (WT) was mutated (crossed boxes) at only one of the two TCF binding sites (TCF(1.m), and TCF(2.m)), leaving the other intact, or at both sites (TCF1.m/2.m). For both TCF sites four copies of the wild-type (4xTCF) or mutated (4xTCFm) sequences were cloned upstream of a thymidine kinase minimal promoter. All constructs contain luciferase (Luc) cDNA as reporter. b: TCF4 and β-catenin activate the human MMP-7 promoter. Hela cells were cotransfected with WT reporter construct and one μg of the indicated expression vectors (act.) or stimulated with TPA (100 ng/ml). c: Mutations in the TCF binding sites reduce activation of the MMP-7 promoter. HeLa cells were cotransfected with the indicated reporter constructs (rep.) and expression constructs for TCF4 and β-catenin. d: β-catenin/TCF4 activate via isolated TCF sites. HeLa cells were cotransfected with the indicated reporter constructs (rep.) and expression constructs for TCF4 and β-catenin. Mutations in the TCF elements abolished stimulation. In all diagrams columns show activation values compared to cotransfection with the empty expression vector. Data are presented as the mean with standard deviation from three separate experiments. e: Dominant negative TCF-4 (DNTCF4) reduces activity of the human MMP-7 promoter in colon carcinoma cells. HT29 colon carcinoma cells were cotransfected with the indicated reporters (phMMP-7prLuc (WT) or TCF(1.m/2.m)) and expression constructs (vector control, β-catenin +TCF, dNTCF). Note that DNTCF4 leads to a significant reduction only of the WT MMP-7 promoter activity. Shown is the percentage related to the basal activity (100%) of the reporter constructs in HT29 cells.
To test the functional significance of each individual TCF site we created reporter constructs in which one or both elements were mutated, such that TCF-4 could no longer bind (same mutations as used in EMSAs) (Figure 2a) ▶ . Mutation in TCF(1.) reduced the stimulatory effect of TCF-4 plus β-catenin to about 30% compared to the wild-type promoter (Figure 2c) ▶ . The promoter mutated only in TCF(2.) could be activated by up to 44% compared to the wild-type promoter by both activators. Interestingly, mutations in both sites could not fully suppress stimulation by β-catenin/TCF-4 (1.81-fold). Recent data demonstrating that β-catenin/TCF also activates c-jun gene transcription 18 indicate that this residual stimulation could be due to indirect stimulation through the AP-1 site (−67 to −61) of the human MMP-7 promoter. 17
We next tested isolated TCF(1.) and TCF(2.) by cloning four copies of each wild-type and mutated site upstream of a thymidine kinase-minimal promoter luciferase construct (Figure 2d) ▶ . Both polymerized TCF sites were responsive to β-catenin/TCF-4. TCF(2.) was activated stronger, which is consistent with our observation of a higher binding affinity of TCF-4 to this site (Figure 1b) ▶ . Nucleotide substitutions, which abolished TCF-4 binding, prevented activation.
Dominant Negative TCF Suppresses the Activity of the MMP-7 Promoter in Colon Cancer Cells
The HT29 colon cancer cell line has no functional APC protein due to mutations on both alleles. Thus the MMP-7 promoter should be constitutively activated by endogenous β-catenin/TCF-4. Accordingly, cotransfection of expression vectors for both factors had only a weak additional stimulatory effect on the wild-type promoter (Figure 2e) ▶ . Similar results were obtained with the APC gene deficient colon cancer cell lines SW480 and SW620, which showed an even weaker MMP-7 promoter activation by a combination of TCF4 and β-catenin expression vectors (data not shown). To further confirm the importance of β-catenin/TCF-4 for the activation of the human MMP-7 promoter, we tried to inhibit its endogenous activity in HT29 by a dominant-negative TCF-4 mutant (DN-TCF-4). 4 DN-TCF4 lacks the β-catenin binding domain, thereby binding to the TCF elements without activating transcription. Transfection of the DN-TCF-4 construct suppressed the promoter activity to 18%. In contrast DN-TCF-4 had only a weak inhibitory effect on the promoter construct with mutations in both TCF sites (endogenous activity of this construct was 40% compared to WT promoter).
Correlation of Nuclear β-Catenin and MMP-7 Expression in Human Colorectal Carcinomas
β-Catenin is overexpressed in a high percentage of sporadic colorectal carcinomas. If MMP-7 activation is highly dependent on β-catenin, one would expect a correlated expression of both proteins. Serial sections of 16 representative colorectal cancers with strong, weak, or no immunohistochemically detectable β-catenin were stained with α-MMP-7 antibodies. Included were both sporadic and hereditary forms of colon carcinomas. All cases showed a correlation in the expression pattern of both proteins. Especially, all three included tumors from FAP patients with defects in the APC gene expressed both β-catenin and MMP-7. Two investigated tumors from HNPCC patients with high grade microsatellite instability did not express nuclear β-catenin and MMP-7. However, we had not included HNPCC tumors with mutations in the APC or β-catenin gene, which occur in >50% of these tumors. Representative results for a β-catenin high and low expressing tumor are shown (Figure 3) ▶ . Immunohistochemistry for the colon tumor marker CEA and the proliferation marker mib-1 were used to control the staining efficiency of the different tumors. Interestingly, in all investigated cases we found no correlation of β-catenin expression and the proliferation activity, measured by the percentage of mib-1 stained cells (compare Figure 3, d and h ▶ ).
Figure 3.
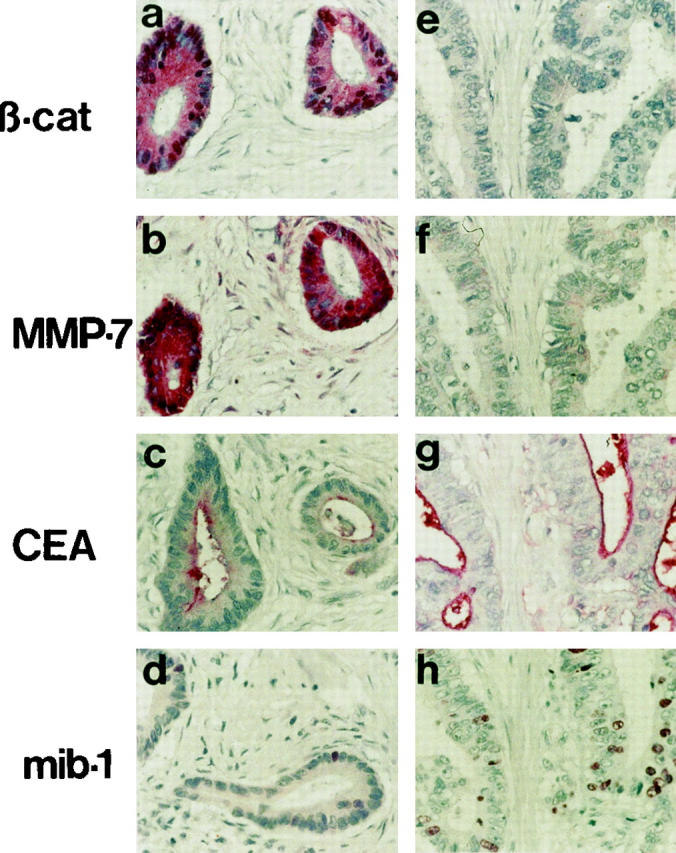
Correlation of β-catenin and MMP-7 expression in sporadic colorectal carcinomas (red staining). Immunohistochemistry of two representative colorectal carcinomas with strong (a–d) or very weak (e–h) expression of β-catenin. Serial sections were stained with the indicated antibodies. Note the correlation of strong (a, b) and weak (e, f) expression of β-catenin and MMP-7. The staining efficiencies of the tumor marker CEA and the proliferation marker mib-1 are comparable in both tumors (only few cells are mib-1-positive in the β-catenin strong expressing tumor) (d). Magnification, ×400.
Discussion
We have shown that β-catenin activates the human MMP-7 promoter via TCF4 binding to two consensus elements. Promoter activation in HT-29 colon cancer cells could be suppressed by transfection of a dominant negative TCF-4 mutant. Moreover, the expression of β-catenin and MMP-7 is correlated in human colorectal carcinomas. These results indicate that MMP-7 expression is regulated by β-catenin/TCF. We suggest that APC gene mutations in colorectal cancers, correlated with a constitutive activation of this transcriptional activator complex, lead to an overexpression of MMP-7. The same regulatory mechanism was shown to be responsible for the activation of c-myc 5 and cyclin D1, 6, enabling autonomous growth of colorectal tumor cells. During the preparation of this manuscript, similar results were published for the mouse MMP-7 promoter, which confirm our results. 19 A conserved regulation of both the human and the mouse MMP-7 gene by β-catenin/TCF underlines the importance of this regulatory mechanism. However, there are some striking differences in the promoters’ structures: we identified two TCF sites upstream of the transcription start site, surrounding two Ets sites in the human promoter. The mouse promoter contains only one TCF site, which lies downstream of the TATA box. This could be of functional relevance, for instance for the interaction of the TCFs with other transcription factors.
Our data can explain important previous observations in intestinal tumors of mice heterozygous for the ApcMin allele (Min/+), the animal model mimicking the human FAP syndrome. 12 There, Wilson et al showed that the majority of these adenomas (88%) overexpressed MMP-7 mRNA. Furthermore they demonstrated that intestinal tumorigenesis in these mice was suppressed by an additional targeted disruption of the MMP-7 gene. The authors suggested a cooperative mechanism of the APC gene defects and MMP-7 expression for tumorigenesis. Our data, defining MMP-7 as a target gene of β-catenin/TCF, support these findings and can also explain the high frequency of MMP-7 overexpression (up to 80%) in human colorectal cancers, 10,20 which is similar to the frequency of APC gene mutations. 1
In contrast to other MMPs, MMP-7 was shown to be an important factor already for the growth of early colon adenomas. 12 Accordingly, low amounts of MMP-7 are also found in earlier adenomas. 21 However there is an increase in MMP-7 expression, correlated with tumor progression. Late, highly dysplastic colon adenomas, the first progression stage with invasive potential, show a high MMP-7 expression level. 20-22 This is consistent with a postulated role of MMP-7 also for later tumor progression steps. 14,15 We also detected potential TCF binding sites in the promoters of other MMPs (data not shown), one of which (MMP-1) is also overexpressed in human colorectal cancer and associated with poor prognosis. 23 Recent work also demonstrated an indirect effect of β-catenin on the expression of uPAR, 18 another molecule involved in invasion. These data extend the effect of APC gene mutations from autonomous proliferation, which is already a characteristic of benign colon adenomas, to later steps in colon cancer development.
An important question to be discussed is why high MMP-7 expression is restricted to later progression steps, whereas APC gene mutations are probably the earliest genetic defect in colorectal carcinogenesis. Previous analysis of the MMP-7 promoter also defined binding sites for the transcription factors AP-1 and PEA3. 17 Both factors are downstream effectors of the ras signaling pathway 24-26 and activated K-ras was shown to enhance MMP-7 expression in colon cancer cell lines. 27 Activating mutations in the K-ras oncogene are accumulated in a high percentage of late, highly dysplastic colon adenomas and colorectal carcinomas. 1,28 Thus, APC and K-ras mutations could have an additive effect in activating MMP-7 gene transcription in colorectal tumors. This is supported by our results that maximum activation of the MMP-7 promoter is achieved by a combination of TCF4, β-catenin, ets-1, c-jun, and c-fos expression vectors (Figure 2b) ▶ . Further work will investigate the relative significance of the different transcriptional activators. In common with other MMP genes, the regulatory elements of MMP-7 possess TGF-β inhibitory elements, negatively regulating its transcription. 17 It is now clear that inactivating mutations in molecules of the TGF-β signaling pathway are additional important genetic alterations during colon tumor progression, and also often occur during the step from benign to malignant growth. 29 Moreover, mice with a combined defect in the APC gene and the TGF-β pathway develop more aggressive malignant tumors compared to mice with an isolated APC gene defect. 30 This indicates functional cooperation of both pathways. One could speculate that both constitutive activation of β-catenin/TCF and inactivating mutations in the TGF-β pathway are necessary for efficient activation of MMP-7 transcription. Thus MMP-7 could be a model for an effector gene simultaneously affected by mutations in the two main oncogenic pathways in colorectal carcinogenesis, the WNT and the TGF-β pathway. Further work will address this point.
Our results, defining MMP-7 as a target gene of β-catenin, explain the high percentage of MMP-7 overexpressing colorectal cancers. The possible functions of MMP-7, not only in early but also in late tumorigenesis, 14,15 extend the effects of APC gene defects in colon tumors.
Acknowledgments
We thank K. W. Kinzler, B. Vogelstein, H. Clevers, L. M. Matrisian, J. Ghysdael, M. Schuermann, P. Angel, and E. Serfling for providing DNA clones and L. McKenna for critical reading of the manuscript. For expert technical assistance we thank U. Suchy and C. Knoll.
Footnotes
Address reprint requests to Thomas Brabletz, Department of Pathology, University of Erlangen-Nürnberg, Krankenhausstrasse 8–10, D-91054 Erlangen, Germany. E-mail: thomas.brabletz@patho.med.uni-erlangen.de.
References
- 1.Kinzler KW, Vogelstein B: Lessons from hereditary colorectal cancer. Cell 1996, 87:159-170 [DOI] [PubMed] [Google Scholar]
- 2.Molenaar M, van de Wetering M, Oosterwegel M, Peterson-Maduro J, Godsave S, Korinek V, Roose J, Destree O, Clevers H: XTcf-3 transcription factor mediates β-catenin-induced axis formation in Xenopus embryos. Cell 1996, 86:391-399 [DOI] [PubMed] [Google Scholar]
- 3.Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW: Activation of β-catenin-Tcf signaling in colon cancer by mutations in β-catenin or APC. Science 1997, 275:1787-1790 [DOI] [PubMed] [Google Scholar]
- 4.Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H: Constitutive transcriptional activation by a β-catenin-Tcf complex in APC−/− colon carcinoma. Science 1997, 275:1784-1787 [DOI] [PubMed] [Google Scholar]
- 5.He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW: Identification of c-myc as a target of the APC pathway. Science 1998, 281:1509-1512 [DOI] [PubMed] [Google Scholar]
- 6.Tetsu O, McCormick F: Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells (in process citation). Nature 1999, 398:422-426 [DOI] [PubMed] [Google Scholar]
- 7.Sikora K, Chan S, Evan G, Gabra H, Markham N, Stewart J, Watson J: c-myc oncogene expression in colorectal cancer. Cancer 1987, 59:1289-1295 [DOI] [PubMed] [Google Scholar]
- 8.Arber N, Doki Y, Han EK, Sgambato A, Zhou P, Kim NH, Delohery T, Klein MG, Holt PR, Weinstein IB: Antisense to cyclin D1 inhibits the growth and tumorigenicity of human colon cancer cells. Cancer Res 1997, 57:1569-1574 [PubMed] [Google Scholar]
- 9.Chambers AF, Matrisian LM: Changing views of the role of matrix metalloproteinases in metastasis. J Natl Cancer Inst 1997, 89:1260-1270 [DOI] [PubMed] [Google Scholar]
- 10.Newell KJ, Witty JP, Rodgers WH, Matrisian LM: Expression and localization of matrix-degrading metalloproteinases during colorectal tumorigenesis. Mol Carcinog 1994, 10:199-206 [DOI] [PubMed] [Google Scholar]
- 11.Yoshimoto M, Itoh F, Yamamoto H, Hinoda Y, Imai K, Yachi A: Expression of MMP-7(PUMP-1) mRNA in human colorectal cancers. Int J Cancer 1993, 54:614-618 [DOI] [PubMed] [Google Scholar]
- 12.Wilson CL, Heppner KJ, Labosky PA, Hogan BL, Matrisian LM: Intestinal tumorigenesis is suppressed in mice lacking the metalloproteinase matrilysin. Proc Natl Acad Sci USA 1997, 94:1402-1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Witty JP, McDonnell S, Newell KJ, Cannon P, Navre M, Tressler RJ, Matrisian LM: Modulation of matrilysin levels in colon carcinoma cell lines affects tumorigenicity in vivo. Cancer Res 1994, 54:4805-4812 [PubMed] [Google Scholar]
- 14.Powell WC, Knox JD, Navre M, Grogan TM, Kittelson J, Nagle RB, Bowden GT: Expression of the metalloproteinase matrilysin in DU-145 cells increases their invasive potential in severe combined immunodeficient mice. Cancer Res 1993, 53:417-422 [PubMed] [Google Scholar]
- 15.Yamamoto H, Itoh F, Hinoda Y, Imai K: Suppression of matrilysin inhibits colon cancer cell invasion in vitro. Int J Cancer 1995, 61:218-222 [DOI] [PubMed] [Google Scholar]
- 16.Brabletz T, Jung A, Hermann K, Gunther K, Hohenberger W, Kirchner T: Nuclear overexpression of the oncoprotein β-catenin in colorectal cancer is localized predominantly at the invasion front. Pathol Res Pract 1998, 194:701-704 [DOI] [PubMed] [Google Scholar]
- 17.Gaire M, Magbanua Z, McDonnell S, McNeil L, Lovett DH, Matrisian LM: Structure and expression of the human gene for the matrix metalloproteinase matrilysin. J Biol Chem 1994, 269:2032-2040 [PubMed] [Google Scholar]
- 18.Mann B, Gelos M, Siedow A, Hanski ML, Gratchev A, Ilyas M, Bodmer WF, Moyer MP, Riecken EO, Buhr HJ, Hanski C: Target genes of β-catenin-T cell-factor/lymphoid-enhancer-factor signaling in human colorectal carcinomas. Proc Natl Acad Sci USA 1999, 96:1603-1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crawford HC, Fingleton BM, Rudolph-Owen LA, Heppner Goss KJ, Rubinfeld B, Polakis P, Matrisian LM: The metalloproteinase matrilysin is a target of β-catenin transactivation in intestinal tumors. Oncogene 1999, 18:2883-2891 [DOI] [PubMed] [Google Scholar]
- 20.Mori M, Barnard GF, Mimori K, Ueo H, Akiyoshi T, Sugimachi K: Overexpression of matrix metalloproteinase-7 mRNA in human colon carcinomas. Cancer 1995, 75:1516-1519 [DOI] [PubMed] [Google Scholar]
- 21.Takeuchi N, Ichikawa Y, Ishikawa T, Momiyama N, Hasegawa S, Nagashima Y, Miyazaki K, Koshikawa N, Mitsuhashi M, Shimada H: Matrilysin gene expression in sporadic and familial colorectal adenomas. Mol Carcinog 1997, 19:225-229 [PubMed] [Google Scholar]
- 22.Ishikawa T, Ichikawa Y, Mitsuhashi M, Momiyama N, Chishima T, Tanaka K, Yamaoka H, Miyazakic K, Nagashima Y, Akitaya T, Shimada H: Matrilysin is associated with progression of colorectal tumor. Cancer Lett 1996, 107:5-10 [DOI] [PubMed] [Google Scholar]
- 23.Murray GI, Duncan ME, O’Neil P, Melvin WT, Fothergill JE: Matrix metalloproteinase-1 is associated with poor prognosis in colorectal cancer. Nat Med 1996, 2:461-462 [DOI] [PubMed] [Google Scholar]
- 24.O’Hagan RC, Tozer RG, Symons M, McCormick F, Hassell JA: The activity of the Ets transcription factor PEA3 is regulated by two distinct MAPK cascades. Oncogene 1996, 13:1323-1333 [PubMed] [Google Scholar]
- 25.Johnson R, Spiegelman B, Hanahan D, Wisdom R: Cellular transformation and malignancy induced by ras require c-jun. Mol Cell Biol 1996, 16:4504-4511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Silberman S, Janulis M, Schultz RM: Characterization of downstream Ras signals that induce alternative protease-dependent invasive phenotypes [published erratum appears in J Biol Chem 1997, 272: 11670]. J Biol Chem 1997, 272:5927-5935 [DOI] [PubMed] [Google Scholar]
- 27.Yamamoto H, Itoh F, Senota A, Adachi Y, Yoshimoto M, Endoh T, Hinoda Y, Yachi A, Imai K: Expression of matrix metalloproteinase matrilysin (MMP-7) was induced by activated Ki-ras via AP-1 activation in SW1417 colon cancer cells. J Clin Lab Anal 1995, 9:297-301 [DOI] [PubMed] [Google Scholar]
- 28.Forrester K, Almoguera C, Han K, Grizzle WE, Perucho M: Detection of high incidence of K-ras oncogenes during human colon tumorigenesis. Nature 1987, 327:298-303 [DOI] [PubMed] [Google Scholar]
- 29.Grady WM, Rajput A, Myeroff L, Liu DF, Kwon K, Willis J, Markowitz S: Mutation of the type II transforming growth factor-β receptor is coincident with the transformation of human colon adenomas to malignant carcinomas. Cancer Res 1998, 58:3101-3104 [PubMed] [Google Scholar]
- 30.Takaku K, Oshima M, Miyoshi H, Matsui M, Seldin MF, Taketo MM: Intestinal tumorigenesis in compound mutant mice of both Dpc4 (Smad4) and Apc genes. Cell 1998, 92:645-656 [DOI] [PubMed] [Google Scholar]