

Abstract
A prominent feature of the clinical spectrum of multiple sclerosis (MS) is its high incidence of onset in the third decade of life and the relative rarity of clinical manifestations during childhood and adolescence, features suggestive of age-related restriction of clinical expression. Experimental allergic encephalomyelitis (EAE), a model of central nervous system (CNS) autoimmune demyelination with many similarities to MS, has a uniform rapid onset and a high incidence of clinical and pathological disease in adult (mature) animals. Like MS, EAE is most commonly seen and studied in female adults. In this study, age-related resistance to clinical EAE has been examined with the adoptive transfer model of EAE in SJL mice that received myelin basic protein-sensitized cells from animals 10 days (sucklings) to 12 weeks (young adults) of age. A variable delay before expression of clinical EAE was observed between the different age groups. The preclinical period was longest in the younger (<14 days of age) animals, and shortest in animals 6 to 8 weeks old at time of transfer. Young animals initially resistant to EAE eventually expressed well-developed clinical signs by 6 to 7 weeks of age. This was followed by a remitting, relapsing clinical course. For each age at time of sensitization, increased susceptibility of females compared to males was observed. Examination of the CNS of younger animal groups during the preclinical period showed lesions of acute EAE. Older age groups developed onset of signs coincident with acute CNS lesions. This age-related resistance to clinical EAE in developing mice is reminiscent of an age-related characteristic of MS previously difficult to study in vivo. The associated subclinical CNS pathology and age-related immune functions found in young animals may be relevant to the increasing clinical expression of MS with maturation, and may allow study of factors associated with the known occasional poor correlation of CNS inflammation and demyelination and clinical changes in this disease.
Multiple sclerosis (MS), the archetypal demyelinating disease of the human central nervous system (CNS), has a 2:1 female:male incidence, presents primarily in young adults, and exists in many forms including relapsing-remitting, chronic progressive, and relatively benign phenotypes spanning many years. 1 In general, symptoms correlate with CNS pathology. 2 It has been postulated that expression of clinical disease might occur after a latent period following a putative event (eg, viral infection, sensitization, or immune stimulation) during childhood. 3 Subsequent to such an event, epidemiological evidence had suggested a prolonged latent period before the development of clinical disease, 3-7 with a mean age of onset of 27 years. 3 The precise relationship between inflammatory and demyelinating lesion pathology and clinical symptoms in MS is not known. However, CNS studies in MS, including cerebrospinal fluid oligoclonal IgG banding and magnetic resonance imaging (MRI), have presented preclinical evidence of CNS inflammation and demyelination predating the diagnosis of clinical MS in previously unaffected twins of MS patients, 8-12 suggesting that CNS pathology may precede clinical manifestations.
Experimental allergic encephalomyelitis (EAE), an animal model with similarities to MS, exists in many forms, some of which present with clinical relapses and episodes of inflammatory demyelination. 13-20 In the present adoptive transfer model of EAE in mice, 20 young adults are commonly used and these develop a predictable clinical and pathological disease with a uniform and usually rapid onset 1 to 2 weeks after transfer of lymphoid cells sensitized to CNS antigen. Among the features of MS that have been difficult to replicate in animal models is the putative latent period of disease before clinical onset. Clinical presentation before adulthood is rare, although an early predisposition with minimal clinical expression during adolescence is postulated and at the time of initial diagnosis, pathological manifestations are usually well established. Occasionally at the time of diagnosis of MS, review of past history may show evidence of early transient clinical signs or preclinical MRI abnormalities can be found in individual cases, supportive of the possibility of a presumed early window of predisposition in childhood. 21-23 In most animal models of MS, disease induction occurs in adults and this usually leads to limited, uniform delays in onset of signs.
The present study was undertaken to determine whether certain features of MS could be documented and investigated in EAE. These include developmental, age-related resistance to clinical disease and an early preclinical predisposition for autoimmune demyelination. Our findings showed the existence of a period of time during development when animals were resistant to acute clinical manifestations yet displayed fulminant CNS pathology during the silent latent period, after which a much delayed, chronic relapsing clinical course ensued. Understanding the immunological mechanisms underlying the opening and closing of this window of early clinical resistance to EAE may provide important insights into host factors operative during the preclinical or remission phases of MS.
Materials and Methods
Animals
The study protocol was reviewed and approved by an institutional animal care and use committee and all animals were maintained in animal facilities according to the NIH Animal Research Advisory Committee guidelines in NIH Manual 3040–2. Six-week-old SJL/J mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and housed for 4 to 5 weeks before they were used as lymph node cell (LNC) donors. For each experiment, 30 mice were used as donors. Recipient SJL mice were from timed pregnancies obtained from the National Cancer Institute/Division of Cancer Treatment colony (Frederick, MD), and were maintained in the National Institute of Neurological Disorders and Stroke animal facilities. Each EAE experiment involved 25 to 45 recipient mice that were followed for up to 120 days after transfer of sensitizing cells, vide infra.
Adoptive Transfer EAE
Ten-week-old mice were anesthetized with methoxyfluorane and immunized subcutaneously at four sites with 0.025 ml of an emulsion containing 400 μg guinea pig myelin basic protein (MBP), prepared as previously described, 24 and complete Freund’s adjuvant (Difco, Detroit, MI). Ten days later, animals were killed and lymph nodes draining the immunization sites aseptically removed and pressed through a wire screen to produce a single-cell suspension. Lymph node cells (LNC) were washed 4 times in Hanks’ Balanced Salt Solution (HBSS, Biofluids, Rockville, MD) and cultured (5% CO2, 37°C, 95% humidity) for 4 days at a concentration of 4 × 10 6 cells/ml with 25 μg/ml of MBP in 24-well plates (Nunc, Roskilde, Denmark). Culture medium was RPMI-1640 (Biowhittaker, Walkersville, MD), with 10% heat-inactivated fetal bovine serum, 200 mmol/L L-glutamine, 10 mmol/L penicillin/streptomycin, 200 mmol/L nonessential amino acids, 100 mmol/L sodium pyruvate, 200 mmol/L Hepes buffer (Biowhittaker), and 50 mmol/L mercaptoethanol (Sigma, St. Louis, MO). After washing in phosphate buffered saline (PBS), 3 × 10 7 LNC in 200 μl were injected intraperitoneally (i.p.) into groups of naïve recipient mice aged 10 days to 12 weeks. For a single experiment, all age groups were injected at the same time. For consistency, the i.p. route was selected for all age groups because tail vein injection was not possible in the younger groups, whose small size (weights as low as 2 g) made venous access difficult and intravascular fluid volumes poorly tolerated.
On three occasions, instead of MBP, ovalbumin (OVA, Millipore, Freehold, NJ) was used for control purposes at the same concentrations for immunization, cell culture, and proliferation assay.
Proliferation Assay
To verify specificity to immunizing antigen, LNC were cultured at a concentration of 4 × 10 5 cells/well, plated in quadruplicate in 96-well flat-bottom plates (Nunc) with 100, 50, 25, 12.5, and 0 μg/ml MBP or 4 μg/ml Concanavalin A (Con A) (Sigma) for 4 days. 25 3H-methyl thymidine (New England Nuclear, Boston, MA) (1 μCi/well), was added at 80 hours, plates were harvested with a Tomtec cell harvester (Wallac Inc., Gaithersburg, MD) onto glass filters at 96 hours, and counts per minute (cpm) were measured with a 1205 Betaplate (Wallac, Oy, Turku, Finland). The stimulation index (S.I.) of cpm to antigen/cpm to no antigen was used to confirm specific dose-response proliferation (S.I. 30–100) to immunizing antigen and for assessment of age differences in antigen presentation.
Clinical Evaluation
Mice were observed daily for signs of EAE. 25 Neurological deficits were scored by a blinded individual as follows: 0.5 = slight tail weakness; 1.0 = weak floppy tail; 2.0 = mild hind limb weakness; 3.0 = mild hind limb and forelimb weakness; 4.0 = severe hind limb and forelimb involvement with or without paralysis; and 5.0 = moribund state or death. Because this is not a linear scale, differences between each grade were not equivalent.
Monoclonal Antibodies and Fluorescence-Activated Cell Sorter (FACS) Analysis
Spleens from animals of different ages were removed aseptically and pressed through wire screens to produce a single-cell suspension of antigen-presenting cells (APC). Spleen cells (SC) were washed in HBSS and cultured in 6-well plates (Costar, Cambridge, MA), at a concentration of 2 × 10 6 cells/ml in freshly prepared RPMI with additives, as described above. 26 SC were removed from culture after 6, 18, 24, 48, and 72 hours and prepared for FACS analysis (FACScan, Becton Dickinson, Mountain View, CA). SC were washed, incubated for 20 minutes at 18°C with 2% normal mouse serum or rat IgG in PBS before incubation with mouse or rat monoclonal antibodies (mAb, 30 minutes at 0°C). SC were then washed twice and fixed with 2% paraformaldehyde. mAb OX-6 (anti-Ias), conjugated to fluorescein isothiocyanate (FITC, Serotec, Indianapolis, IN) at 1:100 was used for the staining of MHC class II; mAb L3T4 conjugated to phycoerythrin (PE, Becton Dickinson, Mountain View, CA) at 1:100 for CD4; mAb Lyt-2 conjugated to PE (Becton Dickinson, Bedford, MA) at 1:100 for CD8; and anti-Mac-1 (Boehringer Mannheim, Indianapolis, IN), conjugated to FITC at 1:15, was used to stain macrophages. Control Ab were rat IgG-FITC at 1:200 and rat IgG-PE at 1:200 (Zymed, South San Francisco, CA), mouse IgG-PE at 1:50, and mouse IgG-FITC at 1:50 (Becton Dickinson, Bedford, MA).
Antigen Presentation Analysis
Presentation of MBP to MBP-sensitized adult LNC by splenocyte APC from 2- and 10-week-old female mice was measured by 3H-methyl thymidine incorporation. LNC were obtained from immunized animals and cultured for 4 days in 24-well plates, as described above. The medium containing MBP was then replaced with fresh medium containing no MBP and 2 × 106/ml irradiated (3500 rads) splenocytes from 12-week-old animals. Cells were cultured for 10 days in 80-cm flasks (Nunc), washed, and replated (2 × 10 5 cells/well, 25 μg/ml MBP) in quadruplicate in 96-well flat-bottom plates (Costar) with irradiated splenocytes (1 × 10 5 cells/ml) from 2- or 10-week-old animals. Each well was pulsed with 1μCi 3H-methyl thymidine and radioactivity was measured as described above.
Neuropathology
Animals from each age group were studied from 7 days post-transfer (dpt), the usual time of clinical onset postsensitization in adult animals, to 69 dpt. At time of sampling, mice were anesthetized with ether and perfused via the left cardiac ventricle with 20 to 40 ml of cold 2.5% glutaraldehyde in phosphate buffer, pH 7.4. The entire CNS was removed and thin slices taken from the cerebrum, cerebellum, medulla/pons, and cervical, thoracic, and lumbar spinal cord. In addition, optic nerves and spinal nerve roots were sampled. The tissue was postfixed in 1% osmic acid, dehydrated in ethyl alcohol, and embedded in Epon 812. Epoxy sections were cut 1 μm thick and stained with toluidine blue for light microscopy. Thin sections for electron microscopy (EM) were contrasted with lead and uranium salts, carbon-coated, and scanned in a Siemens 101. Light microscope analysis was carried out by a blinded individual. Histological scores for demyelination and inflammation were scored from 1 to 5 based on the extent of white matter involvement.
Terminal Deoxyribonucleotidyl Transferase dUTP Nick End-Labeling (TUNEL)
Terminal deoxyribonucleotidyl transferase (TdT) labeling was performed to evaluate the incidence of apoptosis. 27-29 MBP-specific LNC were transferred into groups animals (n = 5) of different ages (2, 4, 6, 10, 12 weeks). By 7 dpt, clinical signs (hind limb and tail weakness) were well developed in 10- and 12-week-old animals. At this time point, representative animals were sacrificed under methoxyfluorane and perfused with 60 ml PBS followed by Parafix (Molecular Histology, Gaithersburg, MD) via the left cardiac ventricle. The CNS was removed en bloc from 1 animal at each age point and from a normal 2-week-old and a 7-day-old animal. In addition, CNS tissue was taken 36 hours after 500 Gy total body irradiation of a normal 10-week-old animal. The other injected animals from the different age groups were followed to verify a remitting-relapsing clinical disease course. Brains and spinal cords were sectioned longitudinally as 5-μm sections, and a section adjacent to the central canal from each spinal cord was mounted on a sialinized slide. For positive controls, sections from the cervical and inguinal lymph nodes, thymus, and spleen of the 10-week-old irradiated animal were also mounted. Sections were dewaxed and hydrated in graded alcohols, washed in 2% H2O2 for 5 minutes, and digested in 20 μg/ml proteinase K. Slides were rinsed and washed in TdT buffer, 30 mmol/L Tris, pH 7.2, 140 mmol/L cacodylate, and 1 mmol/L biotinylated dUTP (Pharmacia, Piscataway, NJ) for 1 hour at 37°C. Reaction was terminated in 3× SSC (Biofluids, Rockville, MD) for 15 minutes. Sections were covered with 2% bovine serum albumin (Sigma, St. Louis, MO) for 20 minutes, rinsed in PBS, covered with 1:20 alkaline phosphatase, stained with Fast Red (Dako, Carpinteria, CA), and counterstained with hematoxylin (Sigma). Thirty light photomicrographs (each covering 0.75 mm in tissue depth) were taken of the entire length of a longitudinal section of spinal cord from each animal and the number of Fast Red-labeled cells (cells showing DNA fragmentation) per 0.75 mm recorded by a blinded individual.
Results
We present results from the clinical observations of the different age groups, followed by the pathological analysis and selected in vitro experiments conducted to correlate age-related immune mechanisms with observed key ages to explore possible mechanisms underlying the expression of clinical disease.
Clinical Course Differs between Immature and Adult Mice
Adoptive transfer of 3 × 10 7 MBP-sensitized LNC into female mice of various ages produced an age-related onset of clinical disease which was associated with the youngest recipient animals expressing well-developed clinical disease by the time of maturity or 7 to 10 weeks of life, and in animals mature at the time of transfer, by 7 to 10 dpt. Thus, younger animals had longer delays before well-developed signs were expressed as a clinical event. With increase in recipient age at time of transfer, time to development of clinical EAE decreased. The longest preclinical periods were seen in younger animals. Experiments were repeated 4 times. A representative experiment is presented in Figure 1 ▶ . All young female animal groups (1.5 to 4 weeks of age at time of transfer) had long delays before expression of well-developed or consistent disease (Figure 1, A–C) ▶ . Rarely, in about 10% of animals in the intermediate young (3- and 4-week-old) animal groups, there was the brief (1 day) appearance of very mild disease (clinical score <1) in the 3 to 5 weeks after cell transfer. However, most young animals up to 5 weeks of age at the time of transfer showed no clinical signs for many weeks (Figure 1, A–C) ▶ . The duration of the preclinical phase was greatest in the youngest groups (Figure 1, A and B) ▶ , and became shorter as the age at time of LNC transfer increased, as shown in another representative experiment in Table 1 ▶ . Ultimately, all young age groups manifested significant clinical disease (clinical scores consistently ≥2 to 5), by the time they reached adulthood.
Figure 1.

A representative experiment showing the change in time course of clinical EAE expression with increase in age of the recipient animals at time of injection of MBP-specific LNC.
Table 1.
Adoptive Transfer EAE at Different Stages of Maturation
| Age (wks) at injection | EAE incidence | Onset (dpt) | Age (wks) of onset | Days to peak score | Age (wks) at peak disease | Relapse |
|---|---|---|---|---|---|---|
| 2 | 6/7 | 34 | 7 | 64 (3) | 11 | + |
| 3 | 6/6 | 18 | 5.5 | 51 (3) | 10 | + |
| 4 | 4/5 | 19 | 7 | 32 (3) | 8.5 | + |
| 6 | 6/6 | 8 | 7 | 23 (4) | 9 | + |
| 8 | 5/5 | 8 | 9 | 17 (4) | 10 | + |
| 10 | 6/6 | 7 | 11 | 10 (4) | 11 | + |
dpt, days post-transfer.
Transfer of LNC to 1.5- to 2.5-week-old mice resulted in onset of overt clinical disease from 45 to 60 dpt, with maximum signs developing 10 to 14 days after the delayed clinical onset (Figure 1, A and B) ▶ . Initial signs began most often with a weak tail and progressed to hind limb, then forelimb involvement. With increase in age at time of cell transfer, delay in clinical onset declined such that in most mice, regardless of age at time of transfer, full expression of clinical disease occurred between 50 and 70 days of age. Despite the separation of animals by age at time of transfer, the ages of the animals at the time of peak clinical disease showed remarkable concordance, as shown in Table 1 ▶ . The severity of the clinical disease reached as high as grade 5 in some individual animals, although when compared to mean scores for adult animal groups in the same experiments, mean scores were most often 1 to 2 clinical points less than those of adult animals. Maximum clinical disease occasionally varied from experiment to experiment by about 1 clinical score point for all age groups, but within a single experiment, the relationship of delayed clinical disease in young versus adult animals remained constant.
In comparison to the youngest groups (1.5 to 2.5 weeks of age at time of sensitization), transfer of adult LNC into intermediate-aged females (5.5 to 7 weeks of age at time of sensitization), resulted in shorter or no delays in clinical disease onset and a more consistent clinical disease course (Figure 1, D and E ▶ , and Table 1 ▶ ), more closely approximating the clinical course of adult (8- to 10-week-old) animals. Although these intermediate-aged animals had onset of clinical signs at 7 to 10 days after transfer of LNC, the initial episode of clinical signs was milder than in the oldest adult age groups and was often separated in time by a return to normal baseline before onset of later episodes which were marked by a chronic accumulation of baseline neurological deficit, as seen in the oldest age groups.
Transfer of LNC into females at 8 to 12 weeks of age reproducibly resulted in an onset of clinical disease 7 to 10 dpt, as seen in Figure 1F ▶ . Initial signs were most often a weak tail at 7 dpt, followed by ascending weakness involving the hind limbs and then the forelimbs by 10 dpt. Well-developed (clinical score ≥2) disease was attained 3 to 5 days after onset. The initial episode was marked by rapid onset and rise in clinical score and more severe signs as compared with later episodes. Later episodes had a more gradual onset and scores for the episodes were often lower with less recovery for the following remissions. During the first episode, adult animals maintained a high clinical score for 5 to 7 days and then showed clinical improvement which continued for 1 to 3 weeks. Of these mice, 90% displayed residual signs during the recovery period; few recovered completely. Four to five weeks after the initial signs, adult age groups showed new clinical relapses with onset over 5 to 10 days. Early relapses were followed by a period of clinical improvement and later episodes of mild to moderate clinical worsening. Each period of remission was associated with a higher level of baseline-accumulated neurological deficit.
Gender Effects Occur across All Ages
When clinical scores of male and female animals in each age group (2 to 10 weeks at time of transfer) were compared, the clinical course had a similar remitting-relapsing pattern for both genders. In each of four experiments with both genders, within each age group, male mice commonly displayed a greater delay in clinical onset in comparison to females, though this delay varied in length from experiment to experiment. Both male and female mice had chronic relapses, but the relapses in females were more distinct and severe than those of the males, which had more gradual onset and milder clinical changes. Within mature age groups, in comparison to females, males showed resistance to clinical disease with longer delays in onset and lower clinical scores, as previously described. 30,31 The same differences were also seen in young age groups, when 3 × 10 7 LNC from adult female mice were transferred into 2- and 4-week-old male and female recipients, as shown in two (of four) experiments in Table 2 ▶ . Although males were resistant to clinical disease, once severe disease occurred, in comparison to female animals, they retained substantial clinical disease with more rapid accumulation of chronic neurological deficits in older age groups. The clinical scores in male mice, as in females, could be increased in a dose-related manner in each age group by increasing the number of transferred female LNC into naïve male mice (Figure 2A) ▶ . Interestingly, although male recipient mice were resistant to clinical EAE, male donor LNC transferred clinical EAE with a typical 7- to 8-day onset after injection into susceptible 10-week-old female recipients (Figure 2A) ▶ .
Table 2.
EAE in Male vs. Female Recipients during Development
| Sex of animals | Age (wks) at injection | EAE incidence | Onset (dpt) | Age (wks) at onset | Days to peak (score) | Age (wks)at peak disease |
|---|---|---|---|---|---|---|
| Males | 2 | 4/5 | 54.8 | 10 | 81 (1.8) | 13.5 |
| Females | 2 | 4/4 | 15 | 4 | 64 (3.25) | 11 |
| Males | 4 | 2/3 | 16 | 6 | 26 (3.0) | 7.5 |
| Females | 4 | 2/2 | 20 | 7 | 39 (3.0) | 9.5 |
| Males | 10 | 3/5 | 18 | 12.5 | 27 (1.0) | 14 |
| Females | 10 | 5/5 | 10 | 11 | 15 (3.0) | 12 |
dpt, days post-transfer.
Figure 2.

A: LNC prepared from resistant male donor animals produced clinical EAE with typical onset 7 to 8 days after injection into susceptible 10-week-old female recipient animals. 3E7 = 3 × 10. 7 B: An example of an experiment showing that LNC from both 2- and 10-week-old donor animals can transfer clinical EAE to susceptible 10-week-old recipient animals. This experiment shows an additive effect when higher numbers of cells achieved by mixing equal numbers of 2- and 10-week-old LNC are injected into 10-week-old female recipients. No inhibitory effect is produced by mixing cells from 2-week-old donors with 10-week-old donor LNC. 3E7 = 3 × 10. 7
LNC Specificity Critical for Disease at Any Age:Young LNC Capable of Transferring Typical Clinical EAE into Adult Mice
To investigate whether T cell specificity was necessary or whether merely relatively large numbers of activated T-cells could induce EAE in the youngest age groups, 3 × 10 7 adult or 6 × 10 7 OVA-sensitized LNC were transferred into recipients. OVA-sensitized LNC did not produce clinical EAE in animals of any age followed up to 100 dpt; nor did they produce CNS lesions at times typical for acute (7 dpt) or chronic (62 dpt) EAE in animals of any age.
To evaluate whether T cells from the youngest animals could participate in or prevent induction of EAE, 10-week-old females were injected with 3 × 10 7 young MBP-specific LNC produced by immunizing 9-day-old female animals, followed by culture of the LNC as for cells from adult animals. The transfer of LNC from the youngest group of mice produced a clinical disease similar to that seen after 3 × 10 7 adult MBP LNC were injected into 10-week-old females at the same time (data not shown). Mixing 10-week- and 2-week-old MBP-specific LNC 1:1 and injecting 3 × 10 7 LNC, had no effect on clinical outcome, whereas 6 × 10 7 cells of the 1:1 mixture gave higher clinical scores in 10-week-old adult female animals (Figure 2B) ▶ . No inhibition of clinical disease in adult animals was observed after injection of young LNC.
Discordant Pathological and Clinical Signs in Immature but Not Adult Mice
The CNS of animals from all groups were examined 7, 42, and 52 dpt for histopathological changes. Surprisingly, younger animal groups (2 and 3 weeks of age at transfer) showed well-developed lesions typical of acute EAE despite lack of clinical disease and delay in clinical signs (Table 3) ▶ . CNS tissue from 2- and 3-week-old mice sampled for pathology at 7 dpt displayed extensive lesion activity. This comprised large areas of white matter from all levels of the neuraxis, including cerebellum (Figure 3, A and B) ▶ , cervical spinal cord (Figure 3, C and D) ▶ , and lower lumbar cord (Figure 3, E and F) ▶ , that contained perivascular, meningeal, and parenchymal collections of small lymphocytes, large mononuclear cells, polymorphonuclear leukocytes (PMNs), macrophages containing myelin debris, and primary demyelination. In contrast to the usual picture of adoptively transferred EAE in older age groups, vide infra, 19,20 CNS lesions in 2- and 3-week-old animals sensitized for EAE were diffuse, poorly circumscribed, and not particularly limited to perivascular areas. Some of the diffuse nature of the pathology was related to changes occurring in CNS tissue that was still undergoing myelination, so that myelin sheath thickness varied from negligible to mature dimensions, giving the tissue a more edematous texture. By light microscopy, large numbers of mononuclear cells with densely staining nuclei were readily observed in the CNS of lesions from animals sensitized at 2 and 3 weeks of age and sampled 7 days later (Figure 3, C and D) ▶ . These cells with dense nuclei were suggestive of apoptosis, were more common in young sensitized animals, and were frequently seen to have been internalized by other cells, commonly astrocytes (Figure 3G) ▶ . The presence of infiltrating cells undergoing apoptosis was confirmed by EM and immunocytochemistry (vide infra).
Table 3.
Histology of Acute EAE in SJL Mice Sensitized at Different Ages
| Age (wks) at sensitization | Day of sampling | Clinical grade | Inflammation | Demye- lination |
|---|---|---|---|---|
| 2 | 7 | 0 | ++ | ++ |
| 3 | 7 | 0.5 | ++ | + |
| 4 | 7 | 0 | + | − |
| 6 | 7 | 1.0 | + | + |
| 8 | 7 | 1.0 | +/− | + |
| 10 | 7 | 2.0 | ++ | + |
++++, Total white matter involvement; +++, formation of plaques, fusion of areas of demyelination and inflammation; ++, perivascular and involvement of white matter; +, perivascular; +/−, little white matter involvement.
Figure 3.
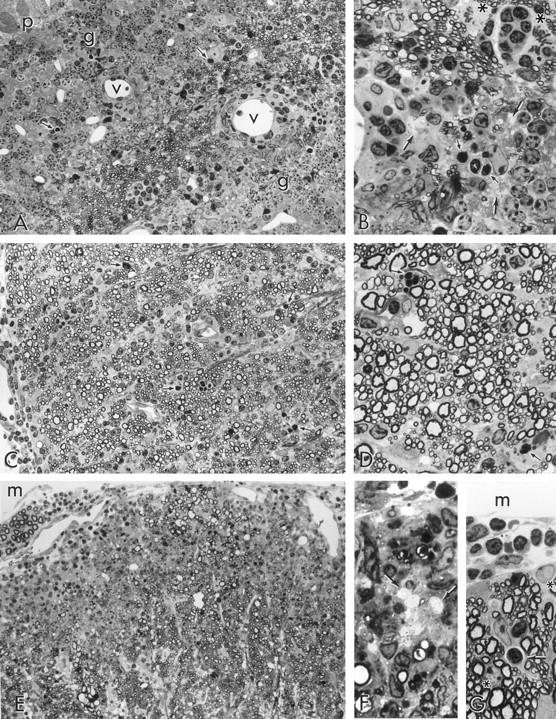
All figures are light micrographs taken from 1-μm epoxy sections stained with toluidine blue. A: Three-week-old recipient mouse; 7 dpt; cerebellum; no clinical signs. Note the presence of scattered infiltrating cells throughout the white matter of a cerebellar folium. The infiltrating cells are not organized into perivascular cuffs around the vessels (v) and many show dense clumping of the nuclear chromatin (arrows), typical of apoptosis. Granule cells (g) overlie white matter, and Purkinje cells (p) lie to the upper left. Original magnification, ×300. B: Detail from A showing infiltrating cells percolating white matter parenchyma. Some of the cells are apoptotic (small arrows). Note also clusters of demyelinated axons cut in cross section (large arrows) and two astrocytes undergoing mitosis (asterisks, upper right), flanking a pocket of small mononuclear cells. The cells to the lower right are granule cells (neurons), identified by round nuclei with clumped heterochromatin. Original magnification, ×750. C: Two-week-old recipient; 7 dpt; cervical spinal cord; no clinical signs. A lateral column which is still undergoing myelination (note wide variation in myelin sheath thickness), displays widespread infiltration by hematogenous cells throughout the parenchyma. Note that perivascular cuffing is not prominent and that many densely staining apoptotic profiles (eg, at arrows), are in evidence. Original magnification, ×300. D: Detail from C to show variation in myelin sheath thickness (ie, ongoing myelination) and leukocytes, some of which are apoptotic (arrows), between nerve fibers. Original magnification, ×750. E: Two-week-old recipient; 7 dpt; L6 spinal cord; no clinical signs. A large area of anterior column displays intense inflammation, extensive demyelination (naked axons are barely visible at this magnification), but little of the perivascular cuffing that is typical of EAE lesions in older age groups. Meningeal space (m) lies above and an anterior spinal nerve root can be seen to the upper left. Original magnification, ×300. F: Detail from E showing demyelinated axons in transverse section (arrows) and macrophages (above and below) containing myelin debris. Original magnification, ×750. G: Three-week-old recipient; 7 dpt.; L6 spinal cord; no clinical signs. A small area along the surface of the spinal cord shows numerous small lymphocytes within the meningeal space (m) and a lymphocyte undergoing apoptosis (arrow), located within a cytoplasmic chamber within an astrocyte, the nucleus of which lies above the apopoptic lymphocyte. The nucleus of a subpial astrocyte (asterisk, upper right) and of an oligodendrocyte (asterisk, lower left), can also be seen. Original magnification, ×750.
Long-term CNS changes in animals sensitized at 2 and 3 weeks were examined at 42 and 52 dpt, by which time younger animal groups had experienced at least one episode of clinical disease, and 69 dpt, by which time all animals had demonstrated a clinical relapse (Table 4) ▶ . CNS lesions in these animals were similar in distribution to those occurring in animals sensitized at a later age and displayed a wide gamut of activity from recent inflammation and demyelination (Figure 4A) ▶ to long-term demyelination and gliosis (Figure 4B) ▶ and chronic inflammation and remyelination (Figure 4, C and D) ▶ .
Table 4.
Histology of Chronic EAE in SJL Mice Sensitized at Different Ages
| Age (wks) at sensitization | Clinical course (no. of relapses) | Clinical grade | Day of sampling | Inflammation | Demyelination | Other |
|---|---|---|---|---|---|---|
| 2 | 2 | 2.0 | 69 | + | + | |
| 2 | 1 | 2.0 | 42 | ++ | + | |
| 3 | 1 | 2.0 | 42 | ++ | + | +RE |
| 4 | 1 | 2.0 | 42 | ++ | + | +RE,+WD |
| 4 | 1 | 2.0 | 42 | ++ | ++ | +RE |
| 8 | 1 | 1.0 | 42 | ++ | + | +RE |
| 8 | 3 | 3.0 | 42 | +++ | ++ | +RE |
| 12 | 3 | 3.0 | 69 | + | + | +RE,+WD |
Clinical grades: 1.0 = weak floppy tail; 2.0 = mild hind limb weakness; 3.0 = mild hind limb and forelimb weakness. For the complete range of grades, see Materials and Methods.
+RE, remyelination; +WD, Wallerian degeneration.
Figure 4.
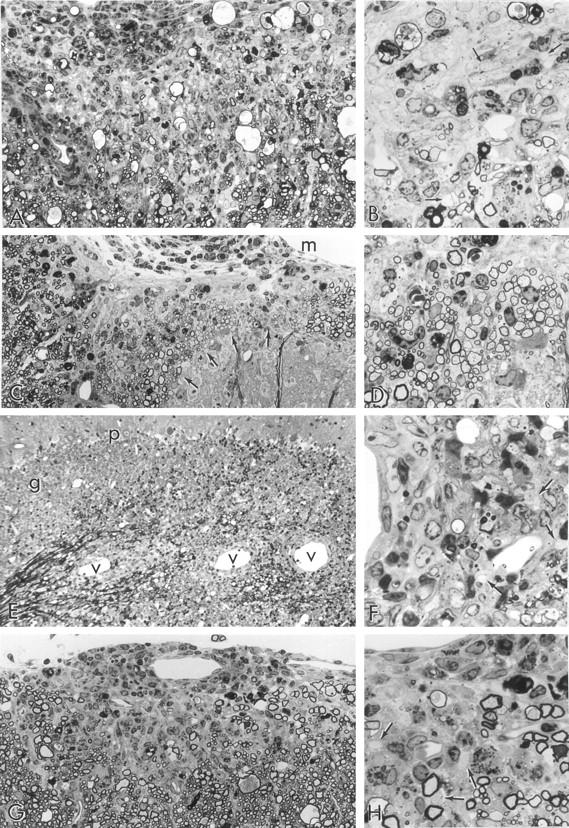
Light micrographs taken from 1-μm epoxy sections stained with toluidine blue. A: Two-week-old recipient; 52 dpt; L7 spinal cord; clinical signs 10 days. A large area of anterior column shows chronic demyelination, glial scarring, infiltrating cells (mostly within the meningeal space, upper left), and large myelin ovoids. The latter are the residue of fibers that have undergone Wallerian degeneration. Original magnification, ×300. B: Detail from A showing demyelinated axons (arrows), background fibrous astrogliosis, residual macrophage activity and a few thinly myelinated (remyelinated) axons. This appearance is fairly typical of a long-standing lesion and suggests that changes in this CNS preceded clinical signs by several weeks. Original magnification, ×750. C: Two-week-old recipient; 52 dpt; L6 spinal cord; clinical signs 10 days. An area of white matter overlying a dorsal horn (lower right) displays collections of nerve fibers (arrows), with disproportionately thin myelin sheaths, an appearance typical of remyelination. Original magnification, ×300. D: Detail of an area of remyelination shown at one of the arrows in C. Note the large group of uniformly thinly myelinated (remyelinated) fibers. m = meningeal surface. Original magnification, ×750. E: Eight-week-old recipient; 10 dpt; cerebellum; clinical signs 3 days. Note lesions typical of acute EAE visible as perivascular cuffs of infiltrating cells around blood vessels (v) along the white matter of this cerebellar folium. Granule (g) and Purkinje (p) cells are also seen. Original magnification, ×300. F: Detail of a lesion from E showing demyelinated axons (arrows) in relationship to a perivascular cuff (left). Some apoptotic nuclei are apparent. Original magnification, ×750. G: Eight-week-old recipient; 52 dpt; cervical spinal cord; clinical signs 42 days, recent relapse (3 days earlier). An intensely infiltrated, small demyelinating lesion is seen in the subpial region of spinal cord, overlaid by a perivascular cuff around a vessel in the leptomeninges. Such an acute lesion occurring in this chronically afflicated animal correlates with the recent relapse. Original magnification, ×300. H: Detail from G showing demyelinated axons (arrows) within an edematous, nonfibrous astrogliotic parenchyma. Macrophages containing myelin debris are present. Original magnification, ×750.
Lesion activity in animals sensitized at ≥6 weeks of age was identical to that detailed in previous works on this model, 18-20 with acute lesions occurring throughout the CNS white matter being centered on perivascular (Figure 4, E and F) ▶ and leptomeningeal infiltrates, correlating with clinical signs that developed between 6 and 8 dpt. In older-sensitized animals sampled during the chronic relapsing phase of the disease (eg, 52 dpt), lesions indicative of long-standing disease containing chronically demyelinated axons, fibrous astrogliosis, and remyelination were common, as described previously. 18 Also evident were recently active lesions displaying recent inflammation, polymorphonuclear cells (PMNs), demyelination, and macrophage activity occurring against an edematous parenchyma containing little evidence of glial scarring (Figure 4, G and H) ▶ .
TUNEL Staining and EM Show Prominent Apoptosis in Immature Groups
Staining by TdT labeling for DNA fragmentation in fixed sections of spinal cord sampled from animals at 7 dpt that had been sensitized at 2, 4, 8, 10, and 12 weeks of age revealed an appreciable number of TUNEL+ infiltrating cells in all animal groups. The number of TUNEL+ cells was greatest in the 2- and 4-week-old groups and lowest in the 12-week group (Table 5) ▶ . A single normal animal sampled at 2 weeks of age from which 30 sections of spinal cord were examined revealed a total of 25 TUNEL+ cells (cell density of 1.28 per cubic millimeter). This figure compares with the highest value of 1125 cells in the same number of sections from a 4-week-old recipient and a low value of 75 in the 12-week-old group. In heavily infiltrated spinal cord tissue from the 2-week group, TUNEL+ infiltrating cells were seen scattered among cells in perivascular cuffs (Figure 5, A and B) ▶ , and throughout the parenchyma (Figure 5C) ▶ . These apoptotic TUNEL+ cells were almost exclusively small lymphocytes. Glial cells, particularly oligodendroglial cells, were not affected. Indeed, chains of interfascicular oligodendrocytes could be readily distinguished by nuclear size from infiltrating cells and were never seen to contain apoptotic nuclei (eg, Figure 5B ▶ ). Apoptotic nuclear profiles were most common, as stated above, in the younger animal groups sensitized for EAE and sampled at 7 dpt.
Table 5.
Quantification of Labeled Apoptotic Cells
| Age (wks) at sensitization | Sampled (dpt)* | Number of TdT-labeled cells | |
|---|---|---|---|
| 30 slides/ spinal cord | Cells/mm3 | ||
| 2 | 7 | 848 | 39 |
| 4 | 7 | 1155 | 50 |
| 8 | 7 | 656 | 28.6 |
| 10 | 7 | 712 | 28.2 |
| 12 | 7 | 75 | 3.38 |
| 2 (normal) | n/a | 25 | 1.28 |
* Sampled at 7 dpt, at time of first clinical signs in 12-week-old animals.
Figure 5.
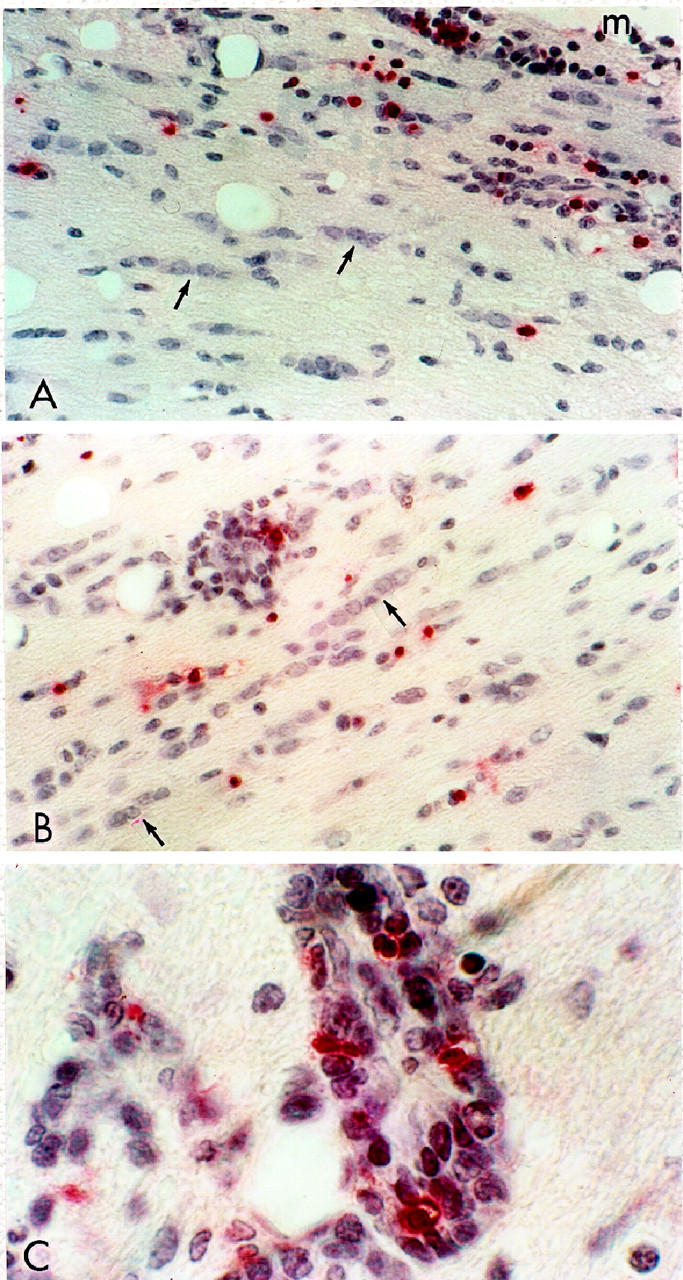
A: Two-week-old recipient; 7 dpt; lumbar spinal cord; no clinical signs. Numerous Fast Red staining, TUNEL+ infiltrating cells, indicative of DNA fragmentation (apoptosis), are seen in the subpial white matter, meninges (m), above. Chains of interfascicular oligodendrocytes (arrows), none of which show apoptosis, are also evident. Paraffin section; hematoxylin counterstain. Original magnification, ×300. B: Three-week-old recipient; 7 dpt; lumbar spinal cord; no signs. In this area of deep white matter, note the diffusely scattered TUNEL+ lymphocytes (some in a perivascular cuff) and the chains of normal-appearing nuclei of interfascicular oligodendrocytes (arrows). Original magnification, ×300. C: Same animal as B. Detail from a perivascular cuff shows numerous TUNEL+ infiltrating cells among others with no evidence of apoptosis. Original magnification, ×750.
By EM, CNS lesions in the 2- and 3-week-old groups sampled at 7 dpt (during the asymptomatic period) were typical of EAE in that they were inflammatory and demyelinating but atypical in that they were diffuse and rarely centered on perivascular infiltrates. Immediately apparent in these animals was the high incidence of apoptotic profiles. Very common was the presence of small lymphocytes at early stages of apoptosis (beginning clumping of nucleochromatin) encircled by astroglial cytoplasm. Sometimes apoptotic cells occurred in groups invested by the same cell (Figure 6A) ▶ . On the basis of cell shape, cytoplasmic content, and glycogen granules, the investing cells were identifiable as resident astroglia, as opposed to hematogenous macrophages or microglia. Indeed, microglial cells were not a prominent feature of lesions in the younger groups. The invested cells were usually small lymphocytes, although on occasion, cells with features of PMNs were also encountered undergoing apoptosis. Such appearances confirmed those encountered by light microscopy (see Figure 3, B, D, and G ▶ ). Oligodendrocytes were not seen to undergo apoptosis and sometimes lay adjacent to astrocytes containing lymphocytes undergoing apoptosis, apparently structurally normal, even to the extent of sharing membrane specializations (gap junctions) with the astrocyte (Figure 6, B and C) ▶ .
Figure 6.
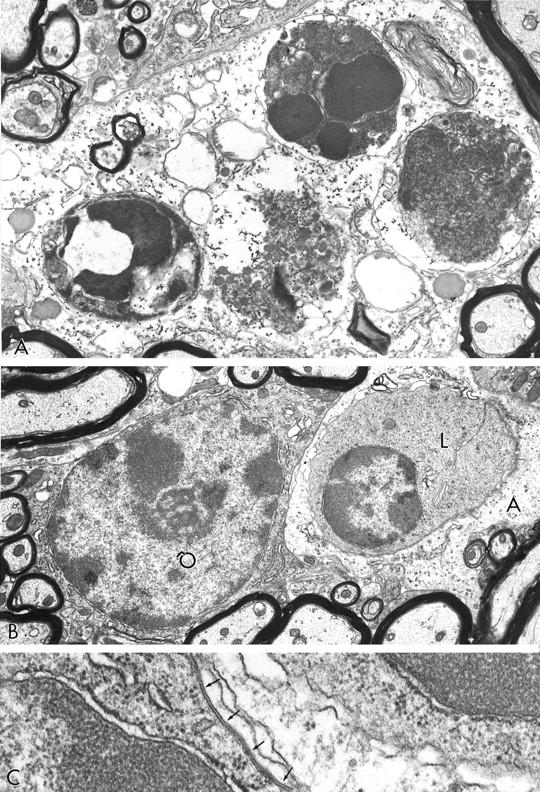
A: Two-week-old recipient; 7 dpt; cervical spinal cord; no clinical signs. A cluster of four leukocytes at advanced stages of apoptotic cell death are invested by astrocytic cytoplasm moderately rich in glycogen granules. Original magnification, ×13,500. B: Elsewhere in the same specimen, a portion of an astrocyte (A) investing a lymphocyte (L) in an early stage of apoptosis, lies next to an apparently healthy oligodendrocyte (O). Original magnification, ×11,000. C: Detail from B showing the presence of a gap junction (arrows) between the plasmalemma of the oligodendrocyte (nucleus lower left) and the astrocyte (right), containing the lymphocyte (nucleus, upper right). Original magnification, ×60,000.
APC Function and APC MHC Class II Expression of Recipient Animals Correlate with Age-Related Susceptibility of EAE
MHC Class II expression and antigen presentation by APC of CNS origin were of interest in this model, because we reasoned that age-related differences might be associated with the observed age-related resistance to clinical EAE in young mice. CNS-derived APC from 10- and 12-week-old animals, however, were markedly less viable in culture (without the addition of growth factors) than those from younger animals, making direct comparisons of CNS APC difficult. Cultures of spleen-derived APC from animals of different ages had similar viability and cellular make-up by FACS analysis at 72 hours, with similar proportions of CD4, CD8, and Mac-1-positive cells and were therefore used as an approach to study age-related differences in APC function and MHC expression and to give insight into possible age-related events in the CNS.
The cellular components, both infiltrating and resident, of CNS lesions from all age groups appeared similar despite the lack of clinical EAE in the younger age groups. To evaluate whether MHC Class II expression on APC might vary with age and correlate with clinical disease expression, APC from normal animals at different ages were studied using FACS analysis of splenocyte APC cultures in 6 separate experiments. Constitutive MHC Class II expression by APC, as measured by percentage of positive cells or mean fluorescence intensity at 6 through 72 hours in culture (Figure 7) ▶ , was found to increase with age. In addition, there was significantly less expression of Class II MHC by APC from 2-week-old animals compared with APC from 4-, 6- and 12-week-old groups. Constitutive expression by 2-week APC decreased over time in culture, in contrast to the older groups, which showed increasing constitutive Class II MHC. The increase in constitutive MHC Class II over time with increasing age paralleled the increased susceptibility to clinical EAE with age seen in this model.
Figure 7.

Constitutive MHC Class II expression at 6, 18, 24, 48, and 72 hours in culture was studied by FACS analysis of APC obtained from 2-, 4-, 6-, and 12-week-old animals. In a representative experiment, a clear difference with less MHC Class II expression in APC obtained from 2- week-old animals as compared with APC from older animals is seen. Two-week-old APC express decreased MHC class II at all time points, including the earliest time point after removal from donor animals. With increasing time in culture, APC from 2-week animals show no increase while APC from animals of increasing age show an increase in MHC class II expression. The MHC class II expression shows an increase at all time points as the age of the APC donor animals increases.
Antigen presentation by 2- and 10-week-old APC was measured by proliferation and 3H-thymidine incorporation of MBP-specific LNC obtained from animals immunized at 10 weeks of age. Antigen presentation by 2-week-old APC was less effective in stimulating proliferation when compared to 10-week-old APC. From a total of 4 experiments, the S.I. associated with 2-week-old APC was 10 to 40% of that seen with 10-week APC, suggesting an age-related difference in antigen presentation by APC in SJL mice (data not shown). On average, negative controls for 2- and 10-week APC produced, from triplicate samples, 2000 and 10,000 cpm 3H-methyl thymidine incorporation, respectively. This result has been previously reported under different experimental conditions. 32,33
Discussion
The present study on the induction of EAE in immature and adult mice has revealed the existence of clinical latency in younger age groups despite the development of CNS lesions. These observations have relevance to the clinical course of MS, which also shows evidence of both latency and age-related clinical expression. Clinical incidence of MS is rare below the age of 15 years and highest in the third decade of life. Epidemiological studies of various populations suggest that the risk of developing MS is established before the age of 12 and that a latent period ensues. The correlation with onset of pathological changes is unknown. However, MRI scanning at the time of (or occasionally before) diagnosis of MS frequently reveals evidence of accumulated, pre-existing white matter damage. 10
In contrast to MS, most models of EAE exhibit a predictable rapid onset of clinical signs associated with CNS lesions, usually within 7 to 10 days after transfer of CNS antigen-sensitized cells. Although previous studies on actively sensitized juvenile guinea pigs also developed subclinical disease, interpretation of the studies was complicated by the ongoing presence of injected antigenic emulsion. 16,17,34 The early subclinical phase, which may represent an early or age-related environmentally induced predisposition as well as an inherent genetic predisposition, was achieved here using juvenile mice from an EAE-susceptible strain that were sensitized for adoptive EAE at different ages. Our findings have shown that transfer of encephalitogenic cells into younger animals was typified by a latent period that was induced in animals up to 5 weeks of age and that preceded the expression of clinical signs and maximum clinical disease, the latter occurring during early adulthood. Prominent acute CNS lesions were present in all animals, including the animals that were youngest at time of transfer, despite the absence of overt clinical disease in young animals, whereas increased female clinical susceptibility was maintained at all ages. Regardless of the age at time of transfer and clinical disease expression, pathology was seen by 7 dpt in all age groups. This lack of correlation between clinical and pathological signs bespeaks a role for age-related compensatory mechanisms for CNS dysfunction resulting from autoimmune attack and is reminiscent of situations occasionally encountered in MS.
Although mice of all ages developed CNS pathology, some differences between young and adult animals were noted. These comprised the presence of higher numbers of apoptotic infiltrating cells, large reactive astrocytes (some investing multiple apoptotic lymphocytes), and normal myelination in the presence of ongoing demyelination, all evident in the younger but not the older groups of mice. The higher numbers of apoptotic cells were invariably associated with milder clinical disease in younger (2 to 4 weeks of age at time of transfer) animals. Whether or not widespread apoptosis of infiltrating effector cells was functionally linked to effective clearance of activated T cells from affected areas in the murine model has not been proven, but supportive evidence for this exists from other studies. 28,35-37 Studies in the rat model both support 38-42 and refute 41 this mechanism, though the use of the immunosuppressant cyclosporin-A in some studies 43 may complicate interpretation of some observations, especially in the absence of correlation with pathological EAE. In other experimental settings, defective Fas- and Fas ligand-associated decreases in target cell apoptosis have been linked to improved clinical course in gld and lpr mice. 44,45 Although the precise role of apoptosis of infiltrating cells in the present murine model remains to be defined, the abundance of apoptotic cells would support the presence of an intact mechanism for its induction. Developmental studies of cytokine gene expression in normal mouse CNS suggest the cytokine milieu may be supportive of apoptosis before 6 weeks of age and may change with maturation to a more resistant environment. Our preliminary work on CNS tissue from 2- to 12-week-old normal animals suggests that interleukin (IL)-4 is present during the first 6 weeks of development and absent in older animals. IL-2 and interferon (IFN)-γ are absent during the early weeks of life while IFN-γ is present for a short period in adult animals 46,47 (Smith ME, Scott DE, Ellen NE, McFarland HF, manuscript in preparation). The presence of IL-4 was noted in both normal SJL/J and SCID mouse strains during development without any experimental manipulation. This suggests a brain-derived rather than an infiltrating lymphocyte-derived origin for the cytokine message. IL-4 associated with an absence of IL-2 and IFN-γ in development has been previously described to be associated with delayed immune responses in neonatal lymphoid murine cells as well, but in lymphocytes this pattern changes after 5 days of age. 48 IL-4 has also been shown to both activate and deactivate macrophages and APCs, depending on the environment, and can enhance IFN-γ-induced (possibly of LNC origin) tumor necrosis factor (TNF)-α production, 49,50 which may be influential in the CNS and play a role in the age-related apoptosis and resistance to clinical expression of EAE shown here. The large number of cells not undergoing apoptosis in the EAE lesions, however, suggested that apoptosis formed but one component of the multifactorial resistance mechanism that an animal might mount against autoimmune CNS disease expression. In addition, chronic changes (42–69 dpt) in animals originally sensitized with transferred cells at early ages, when prominent acute CNS lesions were seen, consisted of fewer chronic lesions, thus providing supportive evidence that the developing CNS environment is more permissive to recovery from autoimmune inflammatory demyelination.
These results suggest that the maturation process within the CNS itself may hold part of the key to understanding susceptibility to clinical disease expression in MS. In the EAE model, it is conceivable that as the animal matures, a CNS environment less resistant to the consequences of autoimmunity and more conducive to the rapid expression of clinical EAE disease may develop. The immature CNS is an environment conducive to ongoing myelination that functions normally in both young animals and humans with incomplete myelin Together, perhaps, with the normal developmental changes in axon and myelin sheath integrity, eg, phenomena like reorganization of axonal sodium channels, 51,52 these features may contribute to the observed resistance to clinical signs. Interestingly, myelination in the CNS tapers off at approximately 6 weeks in mice and in the third decade in humans, and these time points correspond to the periods of increased susceptibility to clinically significant autoimmune demyelination.
Although sexual maturation was suspected to be related to increased EAE expression, in the present model this topic could not be readily approached because both females and males showed resistance to EAE both before and after sexual maturation. Despite this, it may be hypothesized that growth hormone as well as sex hormone levels during development provide an age-related natural protection from clinically relevant demyelination and TNF-α toxicity, 53,54 which has been shown to be associated with clinical events during CNS demyelination. 55,56 In the present model, transfer of encephalitogenic cells early in life ultimately produced clinical EAE, albeit in a delayed fashion, suggesting that a change in susceptibility to clinical EAE occurs during development as a process separate from predisposing events, such that two steps, immune predisposition and development of clinical susceptibility, are needed for complete manifestation of EAE.
The present work also explored selected immune parameters, focusing on either APC or T cell function and whether one facet of the immune process might correlate more closely with young animals’ resistant periods despite their genetic predisposition to EAE. The autoimmune basis of CNS lesions, including those in the youngest mice, was evidenced by lack of either clinical or pathological disease produced despite transferring large numbers of OVA-specific LNC and the requirement for CNS antigen-specific LNC. However, in comparison to the mature CNS, there was a disproportionately large number of lymphocytes within the immature CNS during EAE, and this may be related in part to the large number of MBP-specific cells transferred into very small animals. Of note, transfer of CNS antigen-specific LNC from either young or mature animals into susceptible 10-week-old mice consistently produced disease, showing that there was relatively little correlation between age-related delays in clinical EAE with age of LNC and young donor mice for the LNC. There was a similar ability to transfer disease with LNC from adult and young (2-week-old) mice, suggesting more mature T cell function with respect to CNS antigen-based autoimmunity, even at younger ages, and fewer grounds to implicate age-related differences in EAE expression. Moreover, the ability to transfer disease with LNC stimulated by whole-molecule MBP provides evidence of comparable antigen processing between the different age groups.
Age-related differences in APC, large scale effector cell apoptosis, and the close relationship of astrocytes with apoptotic cells in the immature CNS environment where ongoing myelination and resistance to chronic lesion accumulation were seen, seemed to correlate more closely with age-associated delays in expression of clinical EAE. MHC Class II expression in vitro by APC was most decreased in 2-week-old animals, and a trend of increased constitutive expression with increasing age was noted. An attempt was made to confirm this finding in vivo by immunocytochemistry on the CNS of very young animals, but the results were difficult to interpret because the CNS tissue was extremely delicate and did not withstand processing. Functionally, APC from the youngest animals presented antigen less well to MBP-specific LNC and baseline proliferation was decreased as compared to 10-week-old animals. The ages at which MHC Class II expression and antigen presentation approximated that of adult animals paralleled the increase in clinical expression of EAE as animals matured. This suggested that a similar situation might also occur in the CNS, where both infiltrating APC and resident CNS APC may contribute to the transient protection from clinical EAE seen in the younger animals. However, successful direct study of specific interactions of the APC from young animals was somewhat limited. In addition, attempts to study the effects of young APC by maintaining MBP-specific LNC beyond the usual culture period after removal from donor animals using 2-week-old APC were unsuccessful in producing longer term survival of the LNC.
Low levels of Class II MHC expression by peripheral APC from young mice and poor function of APC during development observed here and elsewhere 57 may reflect a similar situation in the CNS of young mice. Class II expression by keratinocytes has been found to parallel similar expression on CNS astrocytes in vitro, 58 and thus, poor function or decreased MHC expression by peripheral APC in these young age groups may correlate with immature function of astrocytes, microglia, or endothelial cells. The latter CNS cell types are known to possess the ability to express a number of immune system molecules in vivo and in vitro 59-65 and to interact with lymphocytes in the adult animal. 66-69 Astrocytes appeared actively involved in immunomodulation in CNS lesions, especially when cells undergoing apoptosis were in high concentration. Moreover, astrocytes in the youngest animal groups appeared most active in this role compared to the older groups. Oligodendroglia and microglia appeared to be less involved in these effector cell interactions.
The present developmental model of immune-mediated CNS demyelination affords yet another approach for the study of a clinically significant CNS demyelinating syndrome with adult onset, despite the induction of risk conferred by the transfer of CNS antigen-sensitized LNC during the process of maturation. These observations may have relevance to MS, for which a large number of immunosuppressant treatments is used in its management and its pathological expression and which is believed to have an immune-mediated pathogenesis. 70,71 In MS, it is surmised that onset of immune dysfunction probably precedes clinical expression. Understanding the former event may uncover important predictive information of relevance to the therapeutic modulation of this perplexing human disease.
Acknowledgments
This paper is dedicated to the memory of Dale E. McFarlin, M.D., who was instrumental in its early support. M. E. S. appreciates the early instruction by Kenneth Swaiman, M.D. and Gary Birnbaum, M.D., who turned her interest toward the maturing nervous system’s age effects during autoimmunity. We thank Everett Swanson, Miriam Pakingan, and Howard Finch for technical assistance and Patricia Cobban-Bond for preparation of the manuscript.
Footnotes
Address reprint requests to Dr. Mary E. Smith, Division of Acquired Immunodeficiency Syndrome, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Building 6700B, Room 5105, Rockledge Drive, Bethesda, MD 20892. E-mail: BS161v@nih.gov.
Supported in part by U. S. Department of Health and Human Services grants NS 08952, NS 11920, and NS 07098, by National Multiple Sclerosis grant RG 1001-I-9 to C. S. R., by the Sol Goldman Charitable Trust, and by the Wollowick Family Foundation (to C. S. R.). M. K. R. is a Harry Weaver Neuroscience Scholar of the National Multiple Sclerosis Society and the Young Investigator in Multiple Sclerosis of the American Academy of Neurology Education and Research Foundation.
References
- 1.Whitaker JN, Mitchell G: Clinical features of multiple sclerosis. Raine CS McFarland HF Tourtellotte WW eds. Multiple Sclerosis: Clinical and Pathogenetic Basis. 1997, :pp 3-20 Chapman and Hall, London [Google Scholar]
- 2.Raine CS: The neuropathology of multiple sclerosis. Raine CS McFarland HF Tourtellotte WW eds. Multiple Sclerosis: Clinical and Pathogenetic Basis. 1997, :pp 151-171 Chapman and Hall, London [Google Scholar]
- 3.Kurtzke JF: Epidemiological evidence for MS as an infection. Clin Microbiol Rev 1993, 6:382-427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poskanzer D, Schapira K, Miller H: MS and poliomyelitis. Lancet 1963, 11:917-921 [DOI] [PubMed] [Google Scholar]
- 5.Kurtzke J, Hyllested K: Multiple sclerosis in the Faroe Islands. I. Clinical and epidemiological features. Ann Neurol 1979, 5:6-21 [DOI] [PubMed] [Google Scholar]
- 6.Norman J, Kurtzke J, Beebe G: Epidemiology of multiple sclerosis in U.S. veterans: 2. Latitude, climate and the risk of multiple sclerosis. J Chron Dis 1983, 36:551-559 [DOI] [PubMed] [Google Scholar]
- 7.Xu XX, McFarlin DE: Oligoclonal bands in CSF of twins with multiple sclerosis. Neurology 1984, 34:769-774 [DOI] [PubMed] [Google Scholar]
- 8.McFarland HF, Patronas NJ, McFarlin DE, Mandler RN, Beall SS, Cross AH, Goodman A, Krebs H: Studies of multiple sclerosis in twins using MRI (abstract). Neurology 1985, 35:137 [Google Scholar]
- 9.Kurtzke J, Hyllested K: Multiple sclerosis in the Faroe Islands. II. Clinical update, transmission, and the nature of multiple sclerosis. Neurology 1986, 36:307-328 [DOI] [PubMed] [Google Scholar]
- 10.Uitdehaag BM, Polman CH, Valk J, Koetsier JC, Lucas CJ: Magnetic resonance imaging studies in multiple sclerosis twins. J Neurol Neurosurg Psychiat 1989, 52:1417-1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mumford CJ, Wood NW, Kellar-Wood H, Thorpe JW, Miller DH, Compston DA: The British Isles survey of multiple sclerosis in twins. Neurology 1994, 44:11-15 [DOI] [PubMed] [Google Scholar]
- 12.Thorpe JW, Mumford CJ, Compston DA, Kendall BE, MacManus DG, McDonald WI, Miller DH: British Isles survey of MS in twins: MRI. J Neurol Neurosurg Psychiat 1994, 57:491-496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferraro A, Cazzullo CL: Chronic experimental allergic encephalomyelitis in monkeys. J Neuropathol Exp Neurol 1948, 7:235-245 [DOI] [PubMed] [Google Scholar]
- 14.McFarlin DE, Blank SE, Kibler RF: Recurrent experimental allergic encephalomyelitis in the Lewis rat. J Immunol 1974, 113:712-715 [PubMed] [Google Scholar]
- 15.Stone SH, Lerner EM: Chronic disseminated allergic encephalomyelitis in guinea pigs. Ann NY Acad Sci 1965, 122:227-241 [DOI] [PubMed] [Google Scholar]
- 16.Raine CS, Snyder DH, Valsamis MP, Stone SH: Chronic experimental allergic encephalomyelitis in inbred guinea pigs: an ultrastructural study. Lab Invest 1974, 31:369-380 [PubMed] [Google Scholar]
- 17.Brown A, McFarlin DE, Raine CS: Chronologic neuropathology of relapsing experimental allergic encephalomyelitis in the mouse. Lab Invest 1982, 46:171-185 [PubMed] [Google Scholar]
- 18.Raine CS, Mokhtarian F, McFarlin DE: Adoptively transferred chronic relapsing EAE in the mouse: neuropathologic analysis. Lab Invest 1984, 51:534-546 [PubMed] [Google Scholar]
- 19.Raine CS: Biology of Disease. The analysis of autoimmune demyelination: its impact upon multiple sclerosis. Lab Invest 1984, 50:608-635 [PubMed] [Google Scholar]
- 20.Mokhtarian F, McFarlin DE, Raine CS: Adoptive transfer of myelin basic protein sensitized cells produces chronic relapsing demyelinating disease in mice. Nature 1984, 309:356-358 [DOI] [PubMed] [Google Scholar]
- 21.Riikonen R, Ketonen L, Sipponen J: Magnetic resonance imaging, evoked responses and CSF findings in a follow-up study of children with optic neuritis. Acta Neurol Scand 1988, 77:44-49 [DOI] [PubMed] [Google Scholar]
- 22.Riikonen R, Donner M, Erkkila H: Optic neuritis in children and its relationship to multiple sclerosis: a clinical study of 21 children. Dev Med Child Neurol 1988, 30:349-359 [DOI] [PubMed] [Google Scholar]
- 23.Wolfson C, Wolfson DB: The latent period of multiple sclerosis: a critical review. Epidemiology 1993, 4:464-470 [DOI] [PubMed] [Google Scholar]
- 24.Deibler GE, Martenson RE, Kies MW: Large scale preparation of myelin basis protein from central nervous tissue of several mammalian species. Prep Biochem 1972, 2:139-165 [DOI] [PubMed] [Google Scholar]
- 25.Racke MK, Critchfield JM, Quigley L, Cannella B, Raine CS, McFarland HF, Lenardo MJ: Intravenous antigen therapy as therapy for autoimmune demyelinating disease. Ann Neurol 1996, 39:46-56 [DOI] [PubMed] [Google Scholar]
- 26.Smith ME, McFarlin DE, Dhib-Jalbut S: Differential effect of interleukin-1β on Ia expression in astrocytes and microglia. J Neuroimmunol 1993, 46:97-104 [DOI] [PubMed] [Google Scholar]
- 27.Gavrieli Y, Sherman Y, Ben-Sasson SA: Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol 1992, 119:493-503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gold R, Schmied M, Giegerich G, Breitschopf H, Hartung HP, Toyka KV, Lassmann H: Differentiation between cellular apoptosis and necrosis by the combined use of in situ and tailing and nick translation techniques. Lab Invest 1994, 71:219-225 [PubMed] [Google Scholar]
- 29.Bonetti B, Raine CS: Multiple sclerosis: oligodendrocytes display cell death-related molecules in situ but do not undergo apoptosis. Ann Neurol 1997, 42:74-84 [DOI] [PubMed] [Google Scholar]
- 30.Cua D, Hinton D, Stohlman S: Self-antigen induced Th2 responses in experimental allergic encephalomyelitis (EAE)-resistant mice. J Immunol 1995, 155:4052-4059 [PubMed] [Google Scholar]
- 31.Voskuhl RR, Pitchekian-Halabi H, MacKenzie-Graham A, McFarland HF, Raine CS: Gender differences in autoimmune demyelination in the mouse: implications for multiple sclerosis. Ann Neurol 1996, 39:724-733 [DOI] [PubMed] [Google Scholar]
- 32.Mosier DE, Tigelaar RE, Cohen PL: Ontogeny of in vitro correlates of graft versus host reactions. Transplant Proc 1976, 8:371-374 [PubMed] [Google Scholar]
- 33.Mosier DE, Cohen PL: Ontogeny of mouse T-lymphocyte function. Fed Proc 1975, 34:18-24 [PubMed] [Google Scholar]
- 34.Raine CS, Stone SH: Animal model for multiple sclerosis: chronic experimental allergic encephalomyelitis in inbred guinea pigs. NY State J Med 1977, 77:1693-1696 [PubMed] [Google Scholar]
- 35.Critchfield JM, Racke MK, Zuniga-Pflucker JC, Cannella B, Raine CS, Goverman J, Lenardo MJ: T cell deletion in high antigen dose therapy of autoimmune encephalomyelitis. Science 1994, 263:1139-1143 [DOI] [PubMed] [Google Scholar]
- 36.Critchfield JM, Lenardo MJ: Antigen-induced programmed T cell death as a new approach to immune therapy. Clin Immunol Immunopathol 1995, 75:13-19 [DOI] [PubMed] [Google Scholar]
- 37.Bonetti B, Pohl J, Gao YL, Raine CS: Cell death during autoimmune demyelination: effector but not target cells are eliminated by apoptosis. J Immunol 1997, 159:5733-5741 [PubMed] [Google Scholar]
- 38.Pender MP, McCombe PA, Yoong G, Nguyen KB: Apoptosis of α-beta T lymphocytes in the nervous system in experimental autoimmune encephalomyelitis: its possible implications for recovery and acquired tolerance. J Autoimmun 1992, 5:401-410 [DOI] [PubMed] [Google Scholar]
- 39.Schmied M, Breitschopf H, Gold R, Zischler H, Wekerle H, Lassman H: Apoptosis of T lymphocytes in experimental autoimmune encephalomyelitis: evidence for programmed cell death as a mechanism to control inflammation in the brain. Am J Pathol 1993, 143:446-455 [PMC free article] [PubMed] [Google Scholar]
- 40.Tabi Z, McCombe PA, Pender MP: Apoptotic elimination of Vβ8.2+ cells from the central nervous system during recovery from experimental autoimmune encephalomyelitis induced by the passive transfer of Vβ8.2+ encephalitogenic T cells. Eur J Immunol 1994, 24:2609-2617 [DOI] [PubMed] [Google Scholar]
- 41.Tabi Z, McCombe PA, Pender MP: Antigen-specific down-regulation of myelin basic protein-reactive T cells during spontaneous recovery from EAE: further evidence of apoptotic deletion of autoreactive T cells in the central nervous system. Int Immunol 1995, 7:967-974 [DOI] [PubMed] [Google Scholar]
- 42.McCombe PA, Nickson I, Tabi Z, Pender MP: Apoptosis of Vβ8.2+ T lymphocytes in the spinal cord during recovery from experimental autoimmune encephalomyelitis induced in Lewis rats by inoculation with myelin basic protein. J Neurol Sci 1996, 139:1-9 [PubMed] [Google Scholar]
- 43.Kohji T, Tanuma N, Aikawa Y, Kawazoe Y, Suzuki Y, Kohyama K, Matsumoto Y: Interaction between apoptotic cells and reactive brain cells in the central nervous system of rats with autoimmune encephalitis. J Neuroimmunol 1998, 82:168-174 [DOI] [PubMed] [Google Scholar]
- 44.Sabelko KA, Kelly KA, Nohm MH, Cross AH, Russell JH: FAS and FAS ligand enhance the pathogenesis of experimental allergic encephalomyelitis but are not essential for immune privilege in the central nervous system. J Immunol 1997, 159:3096-3099 [PubMed] [Google Scholar]
- 45.Waldner H, Sobel RA, Howard E, Kuchroo VK: Fas and FasL-deficient mice are resistant to induction of autoimmune encephalomyelitis. J Immunol 1997, 159:3100-3103 [PubMed] [Google Scholar]
- 46.Smith ME, Raine CS, McFarland HF: An EAE model which allows examination of developmental changes associated with clinically relevant CNS demyelination (abstract). Trans FASEB, 1995, Atlanta, GA
- 47.Smith ME, Raine CS, McFarland HF, Scott DS: Resistance to clinical EAE correlates with Th2 cytokine gene expression. FASEB 9th International Congress of Immunology, July 1985, San Francisco
- 48.Adkins B, Ghanei A, Hamilton K: Developmental regulation of IL-4, IL-2 and IFN-γ production by murine peripheral T lymphocytes. J Immunol 1991, 151:6617-6626 [PubMed] [Google Scholar]
- 49.Bogdan C, Vodovotz Y, Paik J, Xie Q, Natham C: Mechanism of suppression of nitric oxide synthase expression by IL-4 in primary mouse macrophages. J Leukoc Biol 1994, 55:227-233 [DOI] [PubMed] [Google Scholar]
- 50.Stenger S, Solbach W, Rollinghoff M, Bogdan C: Cytokine interactions in experimental cutaneous leishmaniasis. II. Endogenous tumor necrosis factor-α production by macrophages is induced by the synergistic action of IFN-γ and IL-4 and accounts for the antiparastic effect mediated by IFN-γ and IL-4. Eur J Immunol 1991, 21:1669-1675 [DOI] [PubMed] [Google Scholar]
- 51.Foster RE, Connors BW, Waxman SG: Rat optic nerve: electrophysiological, pharmacological and anatomical studies during development. Brain Res 1982, 255:371-386 [DOI] [PubMed] [Google Scholar]
- 52.Waxman SG, Kocsis JD, Black JA: Type III sodium channel mRNA is expressed in embryonic but not adult spinal sensory neurons, and is re-expressed following axotomy. J Neurophysiol 1994, 72:466-470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Selmaj KW, Raine CS, Cross AH: Anti-tumor necrosis factor therapy abrogates autoimmune demyelination. Ann Neurol 1991, 30:694-700 [DOI] [PubMed] [Google Scholar]
- 54.Selmaj KW, Raine CS: EAE: immunotherapy with anti-tumor necrosis factor antibodies and soluble tumor necrosis factor receptors. Neurology 1995, 45:S44-S49 [DOI] [PubMed] [Google Scholar]
- 55.Sharief MK, Hentges R: Association between tumor necrosis factor-α and disease progression in chronic progressive multiple sclerosis. N Engl J Med 1991, 325:467-472 [DOI] [PubMed] [Google Scholar]
- 56.Rieckmann P, Albrecht M, Kitze B, Weber T, Tumani H, Broocks A, Luer W, Helwig A, Poser S: Tumor necrosis factor-α messenger RNA expression in patients with relapsing-remitting multiple sclerosis is associated with disease activity. Ann Neurol 1995, 37:82-88 [DOI] [PubMed] [Google Scholar]
- 57.Stohlman SA, Matsushima GK, Casteel N, Frelinger JA: The defect in delayed-type hypersensitivity of young adult SJL mice is due to a lack of functional antigen-presenting cells. Eur J Immunol 1985, 15:913-916 [DOI] [PubMed] [Google Scholar]
- 58.Massa PT, ter Meulen V: Inducibility of Ia molecules on keratinocytes reflects genetic control on astrocytes in the brain. J Neuroimmunol 1988, 19:69-75 [DOI] [PubMed] [Google Scholar]
- 59.Hirsh MR, Wietzerbin J, Pierres M, Goridis C: Expression of Ia antigens by cultured astrocytes treated with γ-interferon. Neurosci Lett 1983, 41:199-204 [DOI] [PubMed] [Google Scholar]
- 60.Sobel RA, Blanchette BW, Bhan AK, Colvin RB: The immunopathology of experimental allergic encephalomyelitis. II. Endothelial cell Ia increases prior to inflammatory cell infiltration. J Immunol 1989, 25:2402-2407 [PubMed] [Google Scholar]
- 61.Rose LM, Peterson R, Mehra R, Alvord EC, Jr: Endothelial cell Ia increases before inflammatory cell infiltration in EAE induced in long-tailed macaques. Ann NY Acad Sci 1988, 540:315-318 [DOI] [PubMed] [Google Scholar]
- 62.Sasaki A, Levison SW, Ting JP-Y: Comparison and quantitation of Ia antigen expression on cultured macroglia and ameboid microglia from Lewis rat cerebral cortex: analysis and implications. J Neuroimmunol 1989, 25:63-74 [DOI] [PubMed] [Google Scholar]
- 63.McCarron RM, Wang L, Cowan EP, Spatz M: Class II MHC antigen expression by cultured human cerebral vascular endothelial cells. Brain Res 1991, 566:325-328 [DOI] [PubMed] [Google Scholar]
- 64.McCarron RM, Wang L, Stanimirovic DB, Spatz M: Endothelin induction of adhesion molecule expression on human brain microvascular endothelial cells. Neurosci Lett 1993, 156:31-34 [DOI] [PubMed] [Google Scholar]
- 65.Fontana A, Fierz W, Wekerle H: Astrocytes present myelin basic protein to encephalitogenic T-cell lines. Nature 1984, 307:273-276 [DOI] [PubMed] [Google Scholar]
- 66.McCarron RM, Kempski O, Spatz M, McFarlin DE: Presentation of myelin basic protein by murine cerebral vascular endothelial cells. J Immunol 1985, 134:3100-3103 [PubMed] [Google Scholar]
- 67.McCarron RM, Spatz M, Kempski O, Hogan RN, Muehl L, McFarlin DE: Interaction between myelin basic protein-sensitized T lymphocytes and murine cerebral vascular endothelial cells. J Immunol 1986, 137:3428-3435 [PubMed] [Google Scholar]
- 68.Frei K, Siepl C, Groscurth P, Bodmer S, Schwerdel C, Fontana A: Antigen presentation and tumor cytotoxicity by interferon-γ-treated microglial cells. Eur J Immunol 1987, 17:1271-1278 [DOI] [PubMed] [Google Scholar]
- 69.McCarron RM, Wang L, Racke MK, McFarlin DE, Spatz M: Cytokine-regulated adhesion between encephalitogenic T lymphocytes and cerebrovascular endothelial cells. J Neuroimmunol 1993, 43:23-30 [DOI] [PubMed] [Google Scholar]
- 70.Martin R, McFarland HF: Immunology of multiple sclerosis and experimental allergic encephalomyelitis. Raine CS McFarland HF Tourtellotte WW eds. Multiple Sclerosis: Clinical and Pathogenetic Basis. 1997, :pp 221-242 Chapman and Hall, London [Google Scholar]
- 71.Raine CS: The lesion in multiple sclerosis and chronic relapsing encephalomyelitis: a structural comparison. Multiple Sclerosis: Clinical and Pathogenetic Basis. Edited by CS Raine, HF McFarland, WW Tourtellotte. 1997:pp 243-286 Chapman and Hall, London