

Abstract
Genetic changes underlying the tumorigenesis of sporadic adrenocortical tumors are poorly characterized. To search for characteristic genomic imbalances involved in adrenocortical tumors, we examined 41 adrenocortical lesions (12 carcinomas, 23 adenomas, and 6 hyperplasias) by comparative genomic hybridization. Our results show that genetic alterations are more frequent in malignant than in benign lesions and that they rarely occur in hyperplastic lesions. The most frequent DNA copy number changes in adrenocortical carcinomas included losses of 1p21–31, 2q, 3p, 3q, 6q, 9p, and 11q14-qter, as well as gains and amplifications of 5q12, 9q32-qter, 12q, and 20q. The genomic aberrations prevalently occurring in adrenocortical adenomas were gains of 17q, 17p, and 9q32-qter. Gains found in 2 of 6 adrenocortical hyperplastic lesions involved chromosome 17 or 17q only. These data indicate that oncogenes determining the early tumorigenesis of adrenocortical tumors may exist on chromosome 17 and that the number of genomic alterations is closely associated with tumor behavior in adrenocortical tumors.
Adrenocortical tumors consist of adenomas and carcinomas, which can either be endocrinologically silent or hormonally active. The vast majority of adrenocortical tumors are benign nonfunctioning adenomas, with a prevalence of 0.5 to 2% in people over 50 years of age. They are usually detected incidentally by imaging scans for various unrelated purposes. 1 Adrenocortical carcinomas are aggressive tumors with an annual incidence of two per million of population and accounts for 0.05 to 0.2% of all cancers. Diffuse or nodular hyperplasia of the adrenal cortex is generally considered a non-neoplastic lesion but can be a preneoplastic state. 2 Histopathological distinction of these adrenocortical lesions is often difficult, especially if they are small and well differentiated. 3 Thus, there is a need for markers of malignancy or progression at the molecular genetic level. Their availability would allow a better prediction of the biological and clinical behavior of adrenocortical lesions at the initial time of biopsy and, thereby, greatly favor therapeutic decisions.
Recent molecular genetics studies, especially on some rare hereditary syndromes with associated adrenocortical lesions, including the multiple endocrine neoplasia type 1 (MEN1), the Li-Fraumeni (LFS) and the Wiedemann-Beckwith syndromes (WBS), have given some insights into the genetic changes underlying tumorigenesis and development of adrenocortical tumors. The susceptibility gene for MEN1 that was mapped to 11q13 has been cloned. 4 LFS harbors germ line mutations in p53 located on 17p13 5 and the molecular basis of WBS is associated with structural abnormalities and allelic losses of 11p15. 6 These genetic aberrations have also been reported in some sporadic adrenocortical tumors. 7,8 Furthermore, other genetic alterations have occasionally been observed in sporadic adrenocortical tumors, such as loss of heterozygosity of 11p, 13q, and 17p 9 as well as mutations of gip2 10 and ACTH receptor genes. 11 The clinical significance of these genetic changes, however, remains to be clarified. More recently, comparative genomic hybridization (CGH), which allows a detection of all relative DNA sequence copy number alterations of the entire genome of a tumor in one examination, 12 was used to identify genomic alterations in adrenocortical tumors. 13,14 Because only small series of tumors were investigated in both of the reported studies, it is difficult to recognize characteristic genomic imbalances. In the present study, we used CGH to examine 41 adrenocortical lesions including 12 carcinomas, 23 adenomas, and 6 hyperplasias. In addition, we carried out fluorescence in situ hybridization (FISH) experiments to independently confirm some of the CGH results.
Materials and Methods
Patients and Samples
Clinical data of the patients examined are summarized in Table 1 ▶ . Adrenocortical lesions of 41 patients (39 frozen samples and 2 paraffin-embedded tumors) were analyzed. The samples included 12 adrenocortical carcinomas, 23 adenomas, and 6 hyperplasias. The average diameter of the adrenocortical adenomas and carcinomas was 4 (range, 2–9) and 8.4 (range, 4.5–13) cm, respectively. Sections of formaldehyde-fixed, paraffin-embedded samples from each tumor were stained with hematoxylin-eosin and used for histological assessment and classification according to previously published criteria. 3,15
Table 1.
Clinical Data and Genetic Findings in Patients with Adrenocortical Lesions
| No. | Age/sex | Diagnosis | Size (cm) | Clinical manifestation | Follow-up (yr)/findings | CGH data | FISH data | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Number of changes | Chromosomal changes | 11q13† | 17c‡ | 1c‡ | ||||||||||
| All* | Ampl | Gain | Loss | 11 | 17 | |||||||||
| 1 | 50 /F | Hyperplasia | 5 | Cushing | 6/ned | 1 | 0 | 1 | 0 | n | 17q+ | |||
| 2 | 69 /F | Hyperplasia | 4.5 | Cushing | 2/ned | 0 | 0 | 0 | 0 | n | n | |||
| 3 | 65 /F | Hyperplasia | 7 | Cushing | 2/ned | 0 | 0 | 0 | 0 | n | n | |||
| 4 | 35 /F | Hyperplasia | 3.5 | Cushing | 2/ned | 0 | 0 | 0 | 0 | n | n | |||
| 5 | 69 /F | Hyperplasia | nk | Cushing | nk | 2 | 0 | 2 | 0 | n | 17+ | |||
| 6 | 52 /F | Hyperplasia | 3 | Cushing | nk | 0 | 0 | 0 | 0 | n | n | |||
| 7 | 57 /F | Adenoma | 6 | Conn | 2/ned | 0 | 0 | 0 | 0 | n | n | |||
| 8 | 34 /M | Adenoma | 2 | Conn | 2/ned | 2 | 0 | 2 | 0 | n | 17+ | n | tris/tetras | tris/tetras |
| 9 | 63 /M | Adenoma | 9 | Conn | 1/ned | 0 | 0 | 0 | 0 | n | n | n | ||
| 10 | 42 /F | Adenoma | 2 | Conn | 1/ned | 0 | 0 | 0 | 0 | n | n | |||
| 11 | 42 /M | Adenoma | 4 | Conn | 1/ned | 0 | 0 | 0 | 0 | n | n | |||
| 12 | 61 /M | Adenoma | 5 | Conn | 1/ned | 2 | 0 | 1 | 1 | n | n | |||
| 13 | 62 /M | Adenoma | 2 | Conn | nk | 1 | 0 | 1 | 0 | n | 17q11-21+ | n | n | n |
| 14 | 44 /F | Adenoma | 3 | Conn | nk | 8 | 0 | 4 | 4 | n | n | n | ||
| 15 | 64 /M | Adenoma | 3.2 | Conn | nk | 2 | 0 | 1 | 1 | n | 17q+ | n | n | n |
| 16 | 29 /F | Adenoma | 5 | Cushing | 8/ned | 0 | 0 | 0 | 0 | n | n | |||
| 17 | 45 /M | Adenoma | 5 | Cushing | 5 | 0 | 0 | 0 | 0 | n | n | |||
| 18 | 37 /F | Adenoma | 2.5 | Cushing | 3/ned | 1 | 0 | 1 | 0 | n | 17q+ | |||
| 19 | 84 /F | Adenoma | 3 | Cushing | 2/ned | 2 | 0 | 2 | 0 | n | 17p+ | n | n | n |
| 20 | 37 /F | Adenoma | 3.5 | Cushing | 2/ned | 0 | 0 | 0 | 0 | n | n | |||
| 21 | 9 /M | Adenoma | 6.5 | Cushing | 4/ned | 3 | 0 | 2 | 1 | n | n | |||
| 22 | 61 /F | Adenoma | 4 | Cushing | nk | 1 | 0 | 1 | 0 | n | n | n | ||
| 23 | 47 /F | Adenoma | 3.5 | Cushing | nk | 4 | 0 | 1 | 3 | n | 17p+, 17q− | |||
| 24 | 48 /F | Adenoma | nk | nk | nk | 8 | 0 | 1 | 7 | L | 17q24-qter+ | |||
| 25 | 70 /F | Adenoma | 3.5 | nk | nk | 1 | 0 | 1 | 0 | n | n | |||
| 26 | 46 /F | Adenoma | 4 | nk | 3/ned | 0 | 0 | 0 | 0 | n | n | |||
| 27 | 56 /M | Adenoma | nk | nk | nk | 2 | 0 | 0 | 2 | n | n | n | ||
| 28 | 62 /F | Adenoma | 3.5 | nonfunctioning | 4/ned | 3 | 0 | 3 | 0 | n | 17+ | |||
| 29 | 16 /F | Adenoma | 4.6 | Virilising; hirsutism | nk | 12 | 0 | 8 | 4 | n | 17+ | tris | n | |
| 30 | 64 /F | Carcinoma | 10 | Cushing | 0.5/rec | 12 | 3 | 2 | 7 | n | n | |||
| 31 | 72 /M | Carcinoma | 13 | Cushing | 2 /rec | 34 | 3 | 14 | 18 | 11q− | 17+ | L | tris/tetras | n |
| 32 | 55 /F | Carcinoma | 5 | Cushing | 2/metas | 23 | 1 | 8 | 14 | L | 17−(1−) | L | monos | monos |
| 33 | 68 /F | Carcinoma | 7 | Cushing | 1 | 6 | 0 | 3 | 3 | n | 17+ | n | n | n |
| 34 | 54 /F | Carcinoma | 9 | Cushing | metas | 3 | 0 | 1 | 2 | n | n | n | ||
| 35 | 0.3 /M | Carcinoma | 4.5 | Cushing | nk | 4 | 0 | 4 | 0 | n | 17q+ | n | ||
| 36 | 43 /M | Carcinoma | 7.5 | nk | metas | 15 | 0 | 6 | 9 | L | n | L | ||
| 37 | 37 /F | Carcinoma | 7.5 | nk | rec | 14 | 2 | 5 | 9 | n | 17q23-qter− | n | ||
| 38 | 46 /M | Carcinoma | 11.5 | nk | 3/metas | 5 | 0 | 1 | 4 | n | n | L | ||
| 39 | 64 /F | Carcinoma | 13 | nk | nk | 29 | 0 | 16 | 13 | 11q− | n | n | ||
| 40 | 48 /M | Carcinoma | 9 | nk | nk | 4 | 0 | 2 | 2 | n | n | n | ||
| 41 | 52 /F | Carcinoma | 10 | nk | nk | 12 | 0 | 6 | 6 | 11q− | n |
*Total changes including losses, gains, and amplifications.
†MEN1 gene.
‡Centromeric probe.
M, male; F, female; Cushing, Cushing’s syndrome; Conn, Conn’s syndrome; nk, not known; n, no detectable changes; ned, no evidence of disease; L, loss; metas, metastasis; rec, recurrence; monos, monosomy; tris, trisomy; tetras, tetrasomy.
DNA Preparation for CGH
Isolation of genomic DNA from frozen tumor samples was performed using the D-5000 Puregene DNA Isolation Kit (Gentra Systems Inc., Minneapolis, MN). Approximately 2 mm 3 of frozen tumor material was homogenized and DNA extraction carried out according to the manufacturer’s recommendations. DNA extraction from paraffin-embedded tumors was performed as previously described. 16 Direct fluorescence labeling of DNA was performed by nick translation using a commercial kit (BioNick kit, Life Technologies, Gaithersburg, MD).
CGH Analysis
CGH was carried out as previously described. 16 The hybridization mixture consisted of 200 to 400 ng of Spectrum Green-labeled tumor DNA, 200 ng of Spectrum Red-labeled normal reference DNA, and 10 μg of unlabeled human Cot-1 DNA dissolved in 10 μl of hybridization buffer (50% formamide, 10% dextran sulfate, 2X SSC, pH 7.0). Hybridization took place over 3 days at 37°C to sex- matched normal metaphase spreads (Vysis, Downers Grove, IL). Digital images were collected from 6 to 7 metaphases using a Photometrics cooled CCD camera (Microimager 1400, Xillix Technologies, Vancouver, British Columbia, Canada). The VYSIS software program was used to calculate average green to red ratio profiles for each chromosome. At least four observations per autosome and two observations per sex chromosome were included in each analysis.
Thresholds used for definition of DNA sequence copy number gains and losses were based on the results of CGH analyses of normal tissues. A gain of DNA sequences was assumed at chromosomal regions where the hybridization resulted in a green to red ratio ≥1.20. Over-representations were considered amplifications when the fluorescence ratio values exceeded ≥1.5 in a subregion of a chromosome arm. A loss of DNA sequences was presumed at chromosomal regions where the tumor to normal ratio was ≤0.80. Since some false positive results were found in normal tissues at chromosomes 1p, 16p, 19, and 22, gains at these G-C-rich regions were excluded from all analyses.
FISH Analysis
Eight tumors (5 adenomas and 3 carcinomas) were analyzed for interphase cytogenetics by using a combination of two centromere probes specific for chromosomes 1 and 17, with the goal to independently confirm the CGH results of chromosome 17 abnormalities. Furthermore, eighteen tumors (8 adenomas and 10 carcinomas) were subjected to FISH analysis by an 11q13 probe (MEN1 gene), in combination with chromosome 11-specific centromeric probe, to compare deletions of this locus and the CGH findings of chromosome 11. Touch preparations from frozen tumor material were used. Centromere probes specific for chromosomes 1 and 17 were labeled using spectrum green-dUTP and spectrum red-dUTP (Vysis), respectively. The centromere probe specific for chromosome 11 was labeled using biotin (Boehringer Mannheim, Mannheim, Germany) and the 11q13 probe with spectrum green-dUTP. Hybridization, posthybridization washes, and detection of the hybridized signals were carried out as previously described. 17 At least 100 interphase nuclei with strong hybridization signals were scored for each tumor. Normal frozen adrenal or connective tissue in the vicinity of tumors served as control and exhibited two centromere and 11q13 signals in ≥95% of nuclei. An aneusomy was assumed if more than 30% of nuclei exhibited an abnormal number of centromere signals. It was considered a deletion when more than 30% of nuclei demonstrated only one 11q13 signal.
Statistics
Contingency table analysis and Student’s t-test were used to compare the number of aberrations and the frequency of individual changes between tumors of different types. Regression analysis was applied to compare the number of genetic changes and the tumor size.
Results
Genomic Alterations Detected by CGH
DNA copy number changes were observed in all carcinomas and 15 of 23 adenomas (Table 1) ▶ . The average number of alterations per carcinoma and adenoma was 14 ± 10.6 and 2 ± 3.2 (P = 0.0001), respectively. Of the 6 adrenocortical hyperplasias, one demonstrated a gain of chromosome 17 as only alteration and another a gain at 17q. The number of chromosomal alterations was strongly associated with malignancy. Comparison of the number of genetic changes and the tumor size by regression analysis showed only a weak correlation (r2 = 0.4) which, however, was statistically significant (P = 0.0001, Figure 1 ▶ ).
Figure 1.

Correlation between the number of genomic alterations and tumor size as analyzed by regression (r2 = 0.4, P = 0.0001).
Regions of Frequent Genomic Aberrations
The chromosomal regions with DNA copy number alterations (losses, gains, and amplifications) identified in all 41 tumors by CGH are illustrated in Figure 2 (A and B) ▶ . The most frequent DNA copy number changes in adrenocortical carcinomas included losses of 1p21–31 (67%), 9p (58%), 3p (50%), 2q, 3q, 6q, and 11q14-qter (42% each), 1q23–41, 2p21-pter, and 18q (33% each), as well as gains of 20q (50%), 5q12–13, 5q22-ter, 9q32-qter, 12q13–14, and 12q24 (42% each), and Xq13–21 (33%). Several amplifications were found at the regions of 4p16, 5p15, 5q13 (n = 2), 5q32-qter, 8q24, 9q32-qter, and 12q13–14 (n = 2) in 4 carcinomas. The genomic aberrations prevalently occurring in adrenocortical adenomas were gains at regions of 17q11.2–21 and 17q24–25 (35% each), 17p (26%), and 9q32-qter (22%). Gains occurring in 2 of 6 adrenocortical hyperplasias involved chromosome 17 or 17q only.
Figure 2.
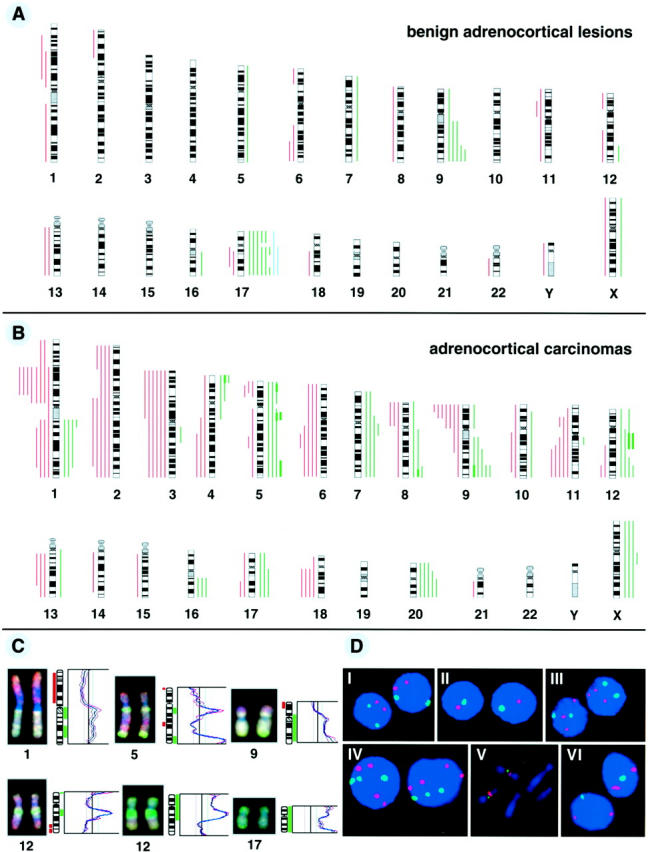
Summary of all DNA copy number alterations detected by CGH in 23 sporadic adrenocortical adenomas and 6 hyperplasias (A) and 12 carcinomas (B). The vertical green lines on the right side of the chromosome ideograms indicate gains, the red on the left losses of the corresponding chromosomal regions. The blue lines indicate gains detected in adrenocortical hyperplastic lesions. Amplifications are indicated as solid green bars. C: Representative examples of CGH digital images and profiles illustrating DNA copy number changes of chromosomes 1 (1p loss and 1q gain), 9 (loss of 9p21-pter and 9q gain), 5 and 12 (amplifications), and 17 (gain). Tumor DNA was labeled using green-dUTP and normal reference DNA with red-dUTP. The color ratio values 0.8, 1.2 and 1.5 were used as thresholds for chromosomal losses, gains and amplifications, respectively. D: Representative results of FISH analysis. I–IV: interphase touch preparation of sporadic adrenocortical tumors investigated using specific centromeric probes for chromosome 17 (red) and chromosome 1 (green). I, normal diploid; II, monosomy of both chromosomes 17 and 1 (tumor 32); III, trisomy of chromosome 17 (tumor 29); IV, tetrasomy of chromosome 17 and trisomy of chromosome 1 (tumor 8). V: MEN1 gene (green signal) located to 11q13 on metaphase chromosomes from normal human lymphocytes. Red signal, centromeric marker of chromosome 11. VI: allelic loss of one copy of the MEN1 gene (tumor 38; one green signal).
A comparison of the frequency of individual alterations among carcinomas, adenomas, and hyperplasias is shown in Table 2 ▶ . The majority of the detected genomic aberrations occurred in carcinomas as compared to adenomas or hyperplasias. This difference reached statistical significance for losses of 1p, 1q, 2p, 2q, 3p, 3q, 6p, 9p 11q, and 18q and for gains at 5q, 12q, 20q, and Xq. There was no significant difference for 9q+ between carcinomas and adenomas as well as for 17p+ and 17q+ among carcinomas, adenomas and hyperplasias. Additionally, an association between chromosomal losses at the distal part of 9p and gains of 9q32-qter in carcinomas was revealed.
Table 2.
Genomic Alterations in Sporadic Adrenocortical Lesions
| Locus of aberrations | Carcinoma % (n = 12) | Adenoma % (n = 23) | Hyperplasia % (n = 6) | P value* |
|---|---|---|---|---|
| 1p− | 67 | 9 | 0 | 0.0002 |
| 1q− | 33 | 4 | 0 | 0.0278 |
| 2p− | 33 | 4 | 0 | 0.0278 |
| 2q− | 42 | 0 | 0 | 0.0010 |
| 3p− | 50 | 0 | 0 | 0.0002 |
| 3q− | 42 | 0 | 0 | 0.0010 |
| 6q− | 42 | 9 | 0 | 0.0235 |
| 9p− | 58 | 0 | 0 | 0.0001 |
| 11q− | 42 | 4 | 0 | 0.0068 |
| 18q− | 33 | 4 | 0 | 0.0278 |
| 5q+ | 42 | 4 | 0 | 0.0068 |
| 9q+ | 42 | 22 | 0 | NS |
| 12q+ | 42 | 4 | 0 | 0.0068 |
| 17p+ | 17 | 26 | 17 | NS |
| 17q+ | 25 | 35 | 33 | NS |
| 20q+ | 50 | 0 | 0 | 0.0002 |
| Xq+ | 33 | 4 | 0 | 0.0278 |
*χ2 test.
NS, not significant.
Comparison of CGH and FISH
Eight tumors that showed alterations of chromosome 17 as identified by CGH, which included 4 gains and 1 loss of the whole chromosome 17, and 2 gains of 17q and 1 gain of 17p, were additionally analyzed by FISH using a combination of specific centromere probes for chromosomes 17 and 1. FISH analysis revealed trisomy and tetrasomy of chromosome 17 in 3 of the 4 tumors with a gain of this chromosome (Table 1) ▶ . The tumor showing loss of chromosome 17 exhibited a monosomy for this chromosome. The other 3 tumors, harboring 17q+ or 17p+, exhibited a diploidy pattern, which was as expected, since alterations involving only short or long arms of chromosomes cannot be detected by FISH using centromere probes. Two tumors, which showed a chromosome 17 gain (1 adenoma and 1 hyperplasia), could not be analyzed by FISH because of lack of tissues.
Among the 18 tumors (8 adenomas and 10 carcinomas) examined with an 11q13 probe (MEN1 gene), FISH revealed deletions of this locus in 4 carcinomas, in 3 of which CGH also detected losses of chromosome 11 or 11q (Table 1) ▶ . Both FISH and CGH did not show detectable changes in the 8 adenomas analyzed.
Collectively, these findings indicate that both CGH and FISH provided comparable results in 7 of the 8 tumors analyzed for chromosome 17, and in 16 of 18 tumors examined for the 11q13 locus. Representative examples of CGH images and corresponding profiles and interphase cytogenetics are shown in Figure 2 (C and D) ▶ .
Discussion
The present study, which represents a comprehensive survey of genomic imbalances involved in sporadic adrenocortical lesions, disclosed several novel genetic alterations and extended previous two limited CGH studies. 13,14 The genomic imbalances identified in the representative collection of sporadic adrenocortical lesions studied here provide new information, which could help to search for novel genes important for adrenocortical tumorigenesis and progression.
The prevalent genomic aberrations found in adrenocortical adenomas were chromosomal gains at 17q, 17p, and 9q. Interestingly, of the 6 patients with adrenocortical hyperplasia, 2 showed a DNA copy number gain involving 17q or the whole chromosome 17, which was at the same time the sole CGH finding in these lesions. This suggests that genes on chromosome 17 or 9q may be important during early tumorigenic events occurring in the adrenal cortex. It is known that the chromosomal area 17q11.2–21 harbors numerous putative candidate oncogenes such as erbB2, GAS, BRCA1, TOP2A, and NGFR. 18 Among them, erbB2 is the most promising candidate gene, because it appears to be overexpressed in a variety of human tumors. 19 However, we cannot exclude that other oncogenes possibly located on chromosomes 17q, 17p, or 9q may also participate in the early tumorigenesis of adrenocortical lesions. Recently, Figueiredo et al reported recurrent gains of 9q34 that were found in 8 of 9 adrenocortical tumors examined in their pilot CGH study. 14 Since the 9q34 locates at the telomeric region of chromosome 9, at which a false positive gain may appear, color ratio changes at this region should be cautiously interpreted.
The present study revealed extensive genomic alterations in adrenocortical carcinomas, including several high-level DNA copy number gains, which have not been reported previously. Most common sites with genomic deletions were 1p21–31, 1q23–41, 2p, 2q, 3p, 3q, 6q, 9p, 11q14-qter, and 18q, whereas the main regions with DNA copy number gains and/or amplifications were 5q, 9q32-qter, 12q, 20q, and Xq. Comparison of carcinomas and adenomas showed statistically significant differences in genomic changes at these regions, except for gains of 9q, 17p, and 17q (Table 2) ▶ . It can be speculated that such genomic imbalances may be important for tumor progression and malignant transformation leading to adrenocortical carcinomas. Studies are under way to gain a more detailed insight into these chromosomal changes.
Most of the chromosomal losses and gains observed in this study are also common to other human tumor types. Some changes, however, may be specific for adrenocortical tumors, including 1p21–31 deletions, co-occurrence of losses at the distal part of 9p and gains of 9q32-qter, and gains and amplifications (high-level gains) of chromosomes 5 and 12q. The prevalent losses of 1p21–31 found in 62% of adrenocortical carcinomas coincide with this region reported to undergo frequent loss of heterozygosity in other human tumors, such as breast cancer 20 and germ cell tumors. 21 This may imply that alterations of one or more tumor suppressor genes in this region may play a role in the development of adrenocortical tumors. The observed association between losses of the distal part of 9p and gains of 9q32-qter in adrenocortical carcinomas is noteworthy. These areas may harbor genes associated with each other in some specific pathways (eg, via rearrangements). The p15/p16 tumor suppressor genes are candidates on 9p21, whereas the ABL oncogene on 9q34 may be the overexpressed target at the region of 9q32-qter, although the oncogenic significance of the latter gene has only been established in hematopoietic cells thus far. The region of 12q13–14, where high-level gains were seen in 2 carcinomas, harbors several oncogenes such as MDM2, SAS, GLI, and CDK4, frequently amplified in different sarcoma types. 22,23 The biological significance of these genes in the development and progression of adrenocortical tumors, however, remains to be evaluated. Remarkably, the present study identified amplifications of three different loci on chromosome 5 (5p14, 5q13 and 5q32-qter), of which, to our knowledge, the two 5q areas have rarely been reported to be amplified in adrenocortical carcinomas as well as in other tumors. These amplification sites may harbor novel genes with a possible role in the progression of adrenocortical tumors.
Our FISH results showed aneusomies of chromosome 17 alone or together with aneusomies of chromosome 1 in 4 (2 adenomas and 2 carcinomas) out of 8 tumors examined, where DNA copy number gains of chromosome 17 were also detected by CGH. This implicates that aneusomies affecting only some chromosomes are involved in the development and progression of adrenocortical tumors, since CGH cannot detect aneusomies that simultaneously involve all chromosomes. The FISH analysis also revealed losses of 11q13 in 4 of 10 carcinomas examined, but not in adenomas, consistent with previous reports. 17 These data suggest that the MEN1 gene may not be a causative gene in adrenocortical tumorigenesis and that losses of this gene locus may represent only a late event in the development and progression of adrenocortical tumors. The good correlation between the results of FISH and CGH further supports the potential application of the CGH technique in screening for genomic alterations of tumors. In one adrenocortical carcinoma (tumor 38), CGH did not detect a deletion at the 11q13 region, whereas FISH revealed a loss of heterozygosity at this locus (Table 1) ▶ . This discrepancy can be explained by a limited sensitivity of CGH for alterations smaller than 5 to 10 megabases. This limitation might also explain why our CGH study revealed only few deletions involving 11p15 (2 tumors) and 17p (one tumor), in contrast to previous allelotyping studies. 9,24 The contradictory data between CGH and FISH observed in tumors 33 and 39 (Table 1) ▶ may be due to intratumoral heterogeneity.
CGH studies have demonstrated that an increased number of chromosomal alterations is generally associated with poor prognosis in different tumors types, such as renal cell carcinomas. 25 In agreement with this, we found a strong relationship between the average number of genomic alterations and tumor behavior. The size of adrenocortical tumors is used as a predictor of malignant potential. Our data exhibit that the number of genomic alterations is correlated, albeit not very strikingly, with tumor size, thus supporting the predictive value of tumor size. Surgical resection of an adrenocortical tumor is recommended if it exceeds 3 cm. However, we found genetic changes in 3 tumors smaller than 3 cm. This implicates that the biological behavior of adrenocortical tumors could be predicted earlier, based on the number of identified genetic changes.
In conclusion, frequent gains of 17q and 17p were found in sporadic adrenocortical adenomas and even in hyperplastic lesions of the adrenal cortex, indicating that genes important for early adrenocortical tumorigenesis may exist on this chromosome. The extensive genomic imbalances encountered in adrenocortical carcinomas indicate that the molecular pathogenesis of sporadic adrenocortical tumors is highly complex and that tumor progression and malignant transformation could be attributed to the accumulation of multiple genetic changes. Our data narrow down possible chromosomal loci frequently involved in sporadic adrenocortical lesions and thereby provide a basis for the search for novel genes playing a role in the initiation and progression of sporadic adrenocortical tumors.
Acknowledgments
We thank Dr. Guido Sauter and Dr. Holger Moch, University of Basel, Switzerland, for helpful discussions and supports, and Marlies Kasper and Prof. Michael J. Mihatsch, University of Basel, for providing frozen tissue samples. We also thank Norbert Wey for photographic and computer-assisted reproductions.
Footnotes
Address reprint requests to Dr. Paul Komminoth, Department of Pathology, University of Zurich, Schmelzbergstrasse 12, CH-8091 Zurich, Switzerland. E-mail: paul.komminoth@pty.usz.ch.
Supported by Swiss National Science Foundation Grant 31–53625.98 and the Hartmann Müller Foundation Grant 717.
References
- 1.Ross NS, Aron DC: Hormonal evaluation of the patient with an incidentally discovered adrenal mass. N Engl J Med 1990, 323:1401-1405 [DOI] [PubMed] [Google Scholar]
- 2.Barzilay JI, Pazianos AG: Adrenocortical carcinoma. Urol Clin North Am 1989, 16:457-468 [PubMed] [Google Scholar]
- 3.Schröder S, Komminoth P, Padberg B, Heitz PU: Morphological typing, evaluation of tumor dignity and prognosis and etiologic classification of adrenomedullary and adrenocortical neoplasias. Pathologe 1995, 16:307-314 [DOI] [PubMed] [Google Scholar]
- 4.Chandrasekharappa SC, Guru SC, Manickam P, Olufemi S, Collins FS, Emmert-Buck MR, Debelenko LV, Zhuang Z, Lubensky IA, Liotta LA, Crabtree JS, Wang Y, Roe BA, Weisemann J, Boguski MS, Agarwal SK, Kester MB, Kim YS, Heppner C, Dong Q, Spiegel AM, Burns AL, Marx SJ: Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science 1997, 276:404-406 [DOI] [PubMed] [Google Scholar]
- 5.Srivastava S, Zou ZQ, Pirollo K, Blattner W, Chang EH: Germ-line transmission of a mutated p53 gene in a cancer-prone family with Li-Fraumeni syndrome. Nature 1990, 348:747-749 [DOI] [PubMed] [Google Scholar]
- 6.Henry I, Bonaiti PC, Chehensse V, Beldjord C, Schwartz C, Utermann G, Junien C: Uniparental paternal disomy in a genetic cancer-predisposing syndrome. Nature 1991, 351:665-667 [DOI] [PubMed] [Google Scholar]
- 7.Ohgaki H, Kleihues P, Heitz PU: p53 mutations in sporadic adrenocortical tumors. Int J Cancer 1993, 54:408-410 [DOI] [PubMed] [Google Scholar]
- 8.Gicquel C, Raffin Sanson ML, Gaston V, Bertagna X, Plouin PF, Schlumberger M, Louvel A, Luton JP, LeBouc Y: Structural and functional abnormalities at 11p15 are associated with the malignant phenotype in sporadic adrenocortical tumors: study on a series of 82 tumors. J Clin Endocrinol Metab 1997, 82:2559-2565 [DOI] [PubMed] [Google Scholar]
- 9.Yano T, Linehan M, Anglard P, Lerman MI, Daniel LN, Stein CA, Robertson CN, LaRocca R, Zbar B: Genetic changes in human adrenocortical carcinomas. J Natl Cancer Inst 1989, 81:518-523 [DOI] [PubMed] [Google Scholar]
- 10.Lyons J, Landis CA, Harsh G, Vallar L, Grunewald K, Feichtinger H, Duh QY, Clark OH, Kawasaki E, Bourne HR, McCormick F: Two G protein oncogenes in human endocrine tumors. Science 1990, 249:655-659 [DOI] [PubMed] [Google Scholar]
- 11.Reincke M, Mora P, Beuschlein F, Arlt W, Chrousos GP, Allolio B: Deletion of the adrenocorticotropin receptor gene in human adrenocortical tumors: implications for tumorigenesis. J Clin Endocrinol Metab 1997, 82:3054-3058 [DOI] [PubMed] [Google Scholar]
- 12.Kallioniemi A, Kallioniemi OP, Sudar D, Rutovitz D, Gray JW, Waldman F, Pinkel D: Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science 1992, 258:818-821 [DOI] [PubMed] [Google Scholar]
- 13.Kjellman M, Kallioniemi OP, Karhu R, Hoog A, Farnebo LO, Auer G, Larsson C, Backdahl M: Genetic aberrations in adrenocortical tumors detected using comparative genomic hybridization correlate with tumor size and malignancy. Cancer Res 1996, 56:4219-4223 [PubMed] [Google Scholar]
- 14.Figeiredo BC, Stratakis CA, Sandrini R, DeLacerda L, Pianovsky MAD, Giatzakis C, Young HM, Haddad BR: Comparative genomic hybridization analysis of adrenocortical tumors of children. J Clin Endocrinol Metab 1999, 84:1116-1121 [DOI] [PubMed] [Google Scholar]
- 15.Komminoth P, Roth J, Saremaslani P, Schröder S, Heitz PU: Overlapping expression of immunohistochemical markers and synaptophysin mRNA in pheochromocytomas and adrenocortical carcinomas: implications for the differential diagnosis of adrenal gland tumors. Lab Invest 1995, 72:424-431 [PubMed] [Google Scholar]
- 16.Richter J, Jiang F, Gorog JP, Sartorius G, Egenter C, Gasser TC, Moch H, Mihatsch MJ, Sauter G: Marked genetic differences between stage pTa and stage pT1 papillary bladder cancer detected by comparative genomic hybridization. Cancer Res 1997, 57:2860-2864 [PubMed] [Google Scholar]
- 17.Görtz B, Roth J, Speel E-JM, Krähenmann A, de Krijger RR, Matias-Guiu X, Muletta-Feurer S, Rütimann K, Saremaslani P, Heitz PU, Komminoth P: MEN1 gene mutations in sporadic adrenocortical lesions. Int J Cancer 1999, 80:373-379 [DOI] [PubMed] [Google Scholar]
- 18.Anderson LA, Friedman L, Osborne LS, Lynch E, Weissenbach J, Bowcock A, King MC: High-density genetic map of the BRCA1 region of chromosome 17q12–q21. Genomics 1993, 17:618-623 [DOI] [PubMed] [Google Scholar]
- 19.Hynes NE, Stern DF: The biology of erbB-2/neu/HER-2 and its role in cancer. Biochim Biophys Acta 1994, 1198:165-184 [DOI] [PubMed] [Google Scholar]
- 20.Tsukamoto K, Ito N, Yoshimoto M, Kasumi F, Akiyama F, Sakamoto G, Nakamura Y, Emi M: Allelic loss on chromosome 1p is associated with progression and lymph node metastasis of primary breast carcinoma. Cancer 1998, 82:317-322 [PubMed] [Google Scholar]
- 21.Mathew S, Murty VV, Bosl GJ, Chaganti RS: Loss of heterozygosity identifies multiple sites of allelic deletions on chromosome 1 in human male germ cell tumors. Cancer Res 1994, 54:6265-6269 [PubMed] [Google Scholar]
- 22.Oliner JD, Kinzler KW, Meltzer PS, George DL, Vogelstein B: Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature 1992, 358:80-83 [DOI] [PubMed] [Google Scholar]
- 23.Khatib ZA, Matsushime H, Valentine M, Shapiro DN, Sherr CJ, Look AT: Coamplification of the CDK4 gene with MDM2 and GLI in human sarcomas. Cancer Res 1993, 53:5535-5541 [PubMed] [Google Scholar]
- 24.Gicquel C, Le BY: Molecular markers for malignancy in adrenocortical tumors. Horm Res 1997, 47:269-272 [DOI] [PubMed] [Google Scholar]
- 25.Moch H, Presti JJ, Sauter G, Buchholz N, Jordan P, Mihatsch MJ, Waldman FM: Genetic aberrations detected by comparative genomic hybridization are associated with clinical outcome in renal cell carcinoma. Cancer Res 1996, 56:27-30 [PubMed] [Google Scholar]