

Abstract
We report a new model of chronic progressive renal failure in rats, produced by a single injection of microspheres (20 to 30 μm in diameter) into the left renal artery after right nephrectomy. Significant proteinuria appeared after 4 weeks, followed by hypoalbuminemia and hypercholesterolemia, in rats that received approximately 5 × 105 microspheres (0.8 mg). Renal function partially recovered by 4 weeks after nephrectomy and injection from postoperative dysfunction, but deteriorated again 12 weeks after operation. In the early stage, histologic examination showed tubules with cuff-like thickening of basement membranes scattered among apparently intact tubules. Many epithelial cells in the atrophic tubuli were immunoreactive for proliferating cell nuclear antigen (PCNA). Dilated tubules became apparent several weeks after development of tubular atrophy, most likely representing distal tubules. Dilated tubuli were mostly negative for the proliferation marker. These results showed similarity to findings in human chronic renal failure and strongly suggested that tubular atrophy and dilation in chronic tubulointerstitial lesions differ in pathogenesis. This new model of renal failure induced by microembolism should be useful for studying the interaction between normal and diseased tissue elements in histologically heterogenous lesions as well as the pathogenesis of interstitial fibrosis in disturbance of microcirculation.
Renal function is thought to deteriorate spontaneously and progressively after the number of functioning nephrons has decreased below a certain threshold. Thus, a common process appears to underlie functional deterioration in various renal diseases, irrespective of cause. The hyperfiltration theory 1 proposes nonimmunological mechanisms underlying renal functional deterioration and is supported by many observations in animal models in which nephron numbers are reduced by simple excision of tissue 2-4 or by ligation of specific branches of the renal artery. 5-7 In these models, the remaining kidney is histologically normal at the beginning of progressive impairment of renal function. However, in various severe human renal diseases, the decrease in the number of functioning nephrons is associated with marked tubulointerstitial changes, and relatively undamaged nephrons are admixed with those that are extensively damaged until the intact nephrons spontaneously deteriorate. These features are shared by both immunologically and nonimmunologically mediated kidney diseases.
Progressive chronic renal failure is characterized histologically by tubulointerstitial and vascular scarring as well as glomerular scarring. Renal dysfunction and outcome correlate better with tubulointerstitial scarring than with glomerular scarring. The extent of tubulointerstitial scarring sometimes exceeds that of glomerular sclerosis in rats with remnant kidneys, 8 in nephrotoxic serum nephritis, 9 and in adriamycin nephropathy. 10 Tubular cells in damaged kidneys are known to express or secrete various cytokines and growth factors. 11,12 Furthermore, tubular epithelial cells are capable of secreting interstitial collagens, 13 proteoglycans, and fibronectin. 14 Strutz et al 15 have shown in experimental models of renal disease that certain tubular cells expressed FSP1, a specific marker for fibroblasts, which might indicate some degree of transformation of tubular epithelial cells into fibroblasts. In addition, Nadasdy et al 16 have detected a high proliferation index in the atrophic tubules of human end-stage kidneys with interstitial fibrosis. Thus, the tubular cells in damaged kidneys may play a role in the progression of renal disease. Interactions between damaged and relatively undamaged nephrons has been neglected in studies of progression of end-stage renal disease, partly because of lack of an appropriate animal model.
We now present a model of nonimmunological progressive renal failure produced by a single injection of microspheres, in which relatively undamaged nephrons mingle with severely damaged ones beginning in the early stage of renal disease. This lesion distribution could overcome the drawbacks of conventional ablation models discussed above. In addition, the microembolization model should be useful in the study of mechanisms of progression of damage specifically related to disturbances of the renal microcirculation, such as arteriolosclerosis.
Methods
Renal failure was induced by arterial injection of microspheres into the remaining kidney of nephrectomized rats. Male Wistar rats 12 weeks of age were obtained from SLC (Hamamatsu, Japan) and were allowed free access to standard laboratory chow and water. Under anesthesia with sodium pentobarbital (40 mg/kg body weight, i.p.), the right kidney was removed and microspheres (acryl beads, 20 to 30 μm in diameter; kindly provided by Dr. Takabayashi, Hamamatsu College, University of Shizuoka) suspended in 0.5 ml of physiological saline were injected slowly into the aorta through a 27-gauge needle placed immediately caudal to the ostium of the left renal artery. During microsphere injection, the aorta caudal to the site of needle insertion as well as the anterior mesenteric and celiac arteries were clamped to ensure flow of microspheres into the left renal artery. After injection, the inserted needle was removed and the site of aortic puncture was gently compressed with a ball of cotton for approximately 2 minutes to stop bleeding. Blood flow through the left renal artery was maintained throughout this procedure.
Animals were grouped according to number of injected microspheres: group 1 received saline without microspheres (control); group 2 received 0.8 mg of microspheres (approximately 5 × 10 5 per rat); group 3 received 0.4 mg of microspheres; group 4 received 0.2 mg of microspheres; and group 5 received 0.1 mg of microspheres.
To evaluate the obstructed vascular volume resulting from microsphere injection, we measured renal blood flow before and shortly after injection in five rats in group 2 with an electromagnetic flowmeter (MFV-1200; Nihon Koden, Tokyo) connected to a flow probe (FJ-007TS, φ 0.7 mm; Nihon Koden) placed upon the left renal artery.
Body weight was measured before and 4, 8, and 12 weeks after surgery in all groups. The weight of the left kidney was determined after the rats were killed. Blood pressure was measured in conscious animals by the tail cuff method with a warmed restraining device (PS-300; Riken Kaihatsu, Tokyo) before and 4, 8, and 12 weeks after surgery in groups 1 and 2. Urinary excretion of albumin and serum concentrations of creatinine, albumin, and total cholesterol were determined at the same intervals for all groups. The animals were kept in metabolic cages while 24-hour urine specimens were collected. Urinary albumin was assayed by single radial immunodiffusion method using rabbit antiserum to rat albumin.
For planning the histological study, we made a preliminary examination of all groups for 12 weeks with a small number of animals. We found that the histological changes in group 2 were representative ones and in other groups, abnormal histological findings were scarce, especially in the early stage. Therefore, we focused our histological study on group 1 and 2. Five or six rats were killed at 2, 4, 8, and 12 weeks after surgery from each of groups 1 and 2, and at 12 weeks for groups 3, 4, and 5. Before killing, rats were anesthetized with pentobarbital (40 mg/kg body weight, i.p.) and the kidneys were perfused with 10 ml of cold saline through a 23 gauge needle placed in the abdominal aorta and connected to a bottle of saline placed 110 cm higher. The left kidney was removed, fixed in 10% buffeted formalin, embedded in paraffin, sliced into 2-μm-thick sections, and stained with periodic acid-Schiff (PAS) or Masson’s trichrome (MT) stain for light microscopic observation.
The point-counting method 17 was employed for morphometric evaluation of interstitial fibrosis, tubular basement membrane thickening, dilatation of tubular lumen, and cast formation. Twenty photographs with a final magnification of ×100 were analyzed from each kidney. Interstitial fibrosis was assessed by counting points in the cortical area stained green by the trichrome stain including the tubular basement membrane, except in the area immediately surrounding large vessels. Thickened basement membranes, which were discerned by their acellular homogeneous character and circular shape, were separately counted again. Areas of tubular lumens with and without casts were measured to assess dilation. Areas of casts were measured separately. All areas above were expressed as a percentage of total cortical area.
Another portion of the kidney was fixed in 4% paraformaldehyde in phosphate-buffeted saline and sections 4 μm thick were prepared for histochemical staining with a mouse monoclonal antibody reactive to proliferating-cell nuclear antigen (PCNA; Oncogene Science, Cambridge, MA), rabbit polyclonal antibody to cytokeratin (Dako, Japan), and sheep polyclonal antibody to human Tamm-Horsfall glycoprotein (THP; Chemicon International Inc., Temmecula, CA). Biotin-labeled peanut agglutinin (PNA; Biomeda, Foster City, CA) was used for detection of T antigen by the avidin-biotin-horseradish peroxidase method (Histofine SAB-PO kit; Nitirei, Tokyo).
Data are expressed as means ± SE. The statistical significance of differences was determined by analysis of variance (ANOVA) and either an unpaired or a paired t-test. A P value of <0.05 was considered statistically significant.
Results
After an initial decrease in the first 2 weeks after surgery (not shown), the body weight of rats in all groups gradually increased (Table 1) ▶ . The body weights of surviving rats in group 2 (receiving 0.8 mg of microspheres) did not increase after 8 weeks, possibly because of the onset of renal failure. The wet weight of the kidney remained virtually identical in groups 1 and 2 through 12 weeks after surgery, indicating no further increase in hypertrophy by microembolism in group 2 (Figure 1) ▶ .
Table 1.
Body Weight (g) of Animals by Group at Various Times after Surgery
| Group | Time | |||
|---|---|---|---|---|
| 0 weeks | 4 weeks | 8 weeks | 12 weeks | |
| 1 (control) | 271 ± 3.6 | 289 ± 5.7 | 311 ± 6.0d | 332 ± 7.0d |
| 2 (0.8 mg of microspheres) | 292 ± 3.7e | 299 ± 4.9d | 330 ± 6.0d | 324 ± 12.9d |
| 3 (0.4 mg of microspheres) | 287 ± 4.1b | 313 ± 3.6d,e | 335 ± 10.2b,d | 347 ± 14.8d |
| 4 (0.2 mg of microspheres) | 285 ± 4.4 | 308 ± 8.6b,d | 340 ± 13.3a | 366 ± 7.6b,c,d |
| 5 (0.1 mg of microspheres) | 282 ± 5.2 | 307 ± 4.9b,d | 345 ± 6.0d,e | 360 ± 3.8b,d |
a,b,c: P < 0.05 vs. 0 weeks, group 1, and group 3, respectively.
d,e: P < 0.01 vs. 0 weeks and group 1, respectively.
Figure 1.

The right kidney was weighed in group 1 (-Δ-, control) and group 2 (-•-, 0.8 mg of microspheres) at the indicated times after arterial microsphere injection following contralateral nephrectomy in rats.
Microsphere injection induced a significant increase in the blood pressure of rats in group 2 at 12 weeks (Table 2) ▶ . No significant change in blood pressure was apparent in control rats. Microsphere injection induced a dose-dependent increase in urinary excretion of albumin that was statistically significant by 4 weeks in group 2, by 8 weeks in group 3, and by 12 weeks in group 4 (Figure 2) ▶ . No significant change in the extent of albuminuria was apparent in control rats (group 1) over 12 weeks.
Table 2.
Systolic Blood Pressure (mm Hg) of Animals in Groups 1 and 2 at Various Times after Surgery
| Group | Time | |||
|---|---|---|---|---|
| 0 weeks | 4 weeks | 8 weeks | 12 weeks | |
| 1 (control) | 117 ± 4.0 | 115 ± 1.5 | 116 ± 2.7 | 114 ± 1.9 |
| 2 (0.8 mg of microspheres) | 118 ± 2.4 | 127 ± 3.8 | 132 ± 4.4 | 135 ± 3.7* |
*P < 0.01 vs. 0 weeks and group 1.
Figure 2.
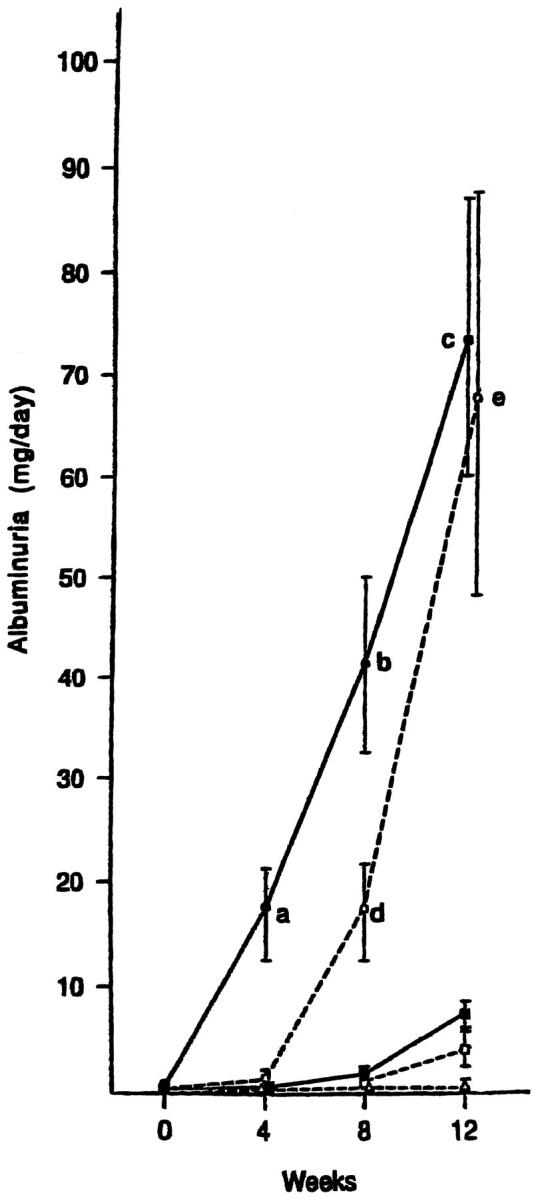
Urinary albumin excretion after microsphere injection. Groups of rats were as follows: (-Δ-) group 1, control, (-•-) group 2, 0.8 mg of microspheres; (-○-) group 3, 0.4 mg of microspheres; (-▪-) group 4, 0.2 mg of microspheres; (-□-) group 5, 0.1 mg of microspheres. a: P < 0.05 vs. the value before injection (0 weeks) and groups 1, 3, 4, and 5; b, P < 0.05 vs. group 3 and P < 0.01 vs. 0 weeks and groups 1, 4, and 5; c, P < 0.01 vs. groups 1, 4, and 5; d, P < 0.05 vs. 0 weeks and groups 1, 4, and 5; e, P < 0.05 vs. 0 weeks and groups 1 and 5.
Injection of microspheres induced a biphasic increase in serum creatinine concentration in groups 2 (Figure 3) ▶ . The first phase was apparent 2 weeks after injection. The second phase was more marked, with the serum creatinine concentration reaching 2.13 ± 0.46 mg/dL by 12 weeks. The serum concentration of creatinine had increased at 2 weeks in groups 3, 4, and 5 but subsequently demonstrated no further significant change over 12 weeks.
Figure 3.
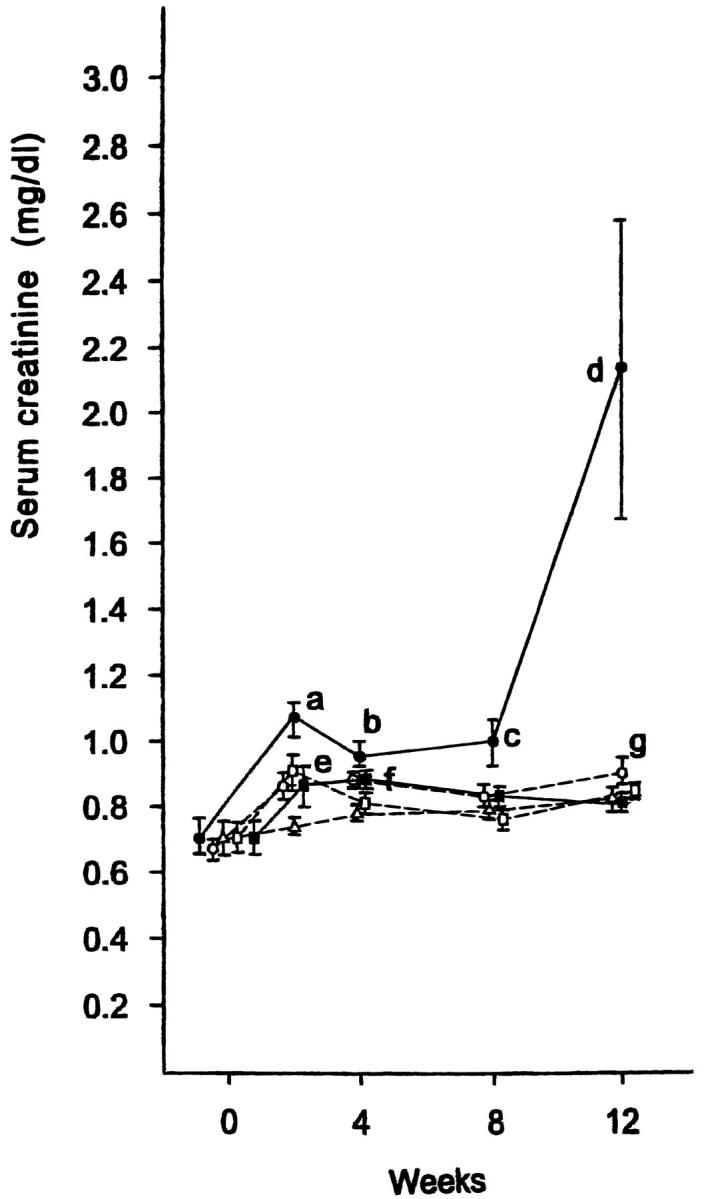
Serum creatinine concentration after microsphere injection. Groups of rats were: (-Δ-) group 1, control; (-•-) group 2, 0.8 mg of microspheres; (-○-) group 3, 0.4 mg of microspheres; (-▪-) group 4, 0.2 mg of microspheres; (-□-) group 5, 0.1 mg of microspheres. a, b, and f: P < 0.01 vs. the value before injection (0 weeks) and group 1; c: P < 0.05 vs. group 1, P < 0.01 vs. 0 weeks; d: P < 0.05 vs. groups 1, 3, 4, and 5, P < 0.01 vs. 0 weeks; e: P < 0.05 vs. 0 weeks; g: P < 0.01 vs. 0 weeks.
A significant decrease in serum albumin concentration was apparent in groups 2 and 3; the rate and extent of the decrease was dependent on the number of injected microspheres (Table 3) ▶ . The injection of microspheres also significantly increased the total serum cholesterol concentration in groups 2 and 3 compared to group 1. The time courses of the changes in serum concentrations of albumin and cholesterol were similar to that of the increase in urinary excretion of albumin (Figure 2 ▶ and Table 3 ▶ ).
Table 3.
Serum Concentrations of Albumin (g/dL) and Total Cholesterol (mg/dL) at Various Times after Surgery
| Group | Time (weeks) | |||
|---|---|---|---|---|
| 0 | 4 | 8 | 12 | |
| Albumin | ||||
| 1 | 4.53 ± 0.06 | 4.56 ± 0.05 | 4.61 ± 0.03 | 4.34 ± 0.06a |
| 2 | 4.49 ± 0.07 | 4.29 ± 0.08a | 4.00 ± 0.12f,g | 3.81 ± 0.15d,e,f,g |
| 3 | 4.48 ± 0.07 | 4.40 ± 0.05 | 4.31 ± 0.06a,g | 4.11 ± 0.02d,e,f,g |
| 4 | 4.58 ± 0.08 | 4.36 ± 0.08 | ND | 4.28 ± 0.07 |
| 5 | 4.44 ± 0.05 | 4.50 ± 0.03 | ND | 4.30 ± 0.10 |
| Total Cholesterol | ||||
| 1 | 61 ± 3 | 72 ± 3a | 70 ± 1a | 74 ± 3f |
| 2 | 63 ± 3 | 95 ± 5f,b,i,j | 150 ± 20f,g | 174 ± 19c,f,g,h,i,j |
| 3 | 60 ± 3 | 87 ± 4b,d,e,f | 90 ± 6b,f | 117 ± 12b,d,e,f |
| 4 | 59 ± 3 | 66 ± 3 | ND | 75 ± 3a |
| 5 | 64 ± 5 | 70 ± 3 | ND | 81 ± 5f |
a,b,c,d,e: P < 0.05 vs. 0 weeks, group 1, group 3, group 4, and group 5, respectively.
f,g,h,i,j: P < 0.01 vs. 0 weeks, group 1, group 3, group 4, and group 5, respectively.
Injection of 0.8 mg of microspheres in group 2 resulted in a rapid decrease in renal blood flow to approximately 19% of the preinjection value (3.46 ± 0.18 to 0.66 ± 0.15 mL/min); blood flow then gradually increased to 34% of the preinjection value (1.18 ± 0.07 mL/min) by 15 minutes and remained stable thereafter.
The most impressive histological abnormalities 2 to 4 weeks after injection of 0.8 mg of microspheres in group 2 were located predominantly in the interstitium (Figure 4A) ▶ . At this early stage, atrophic tubules and a small number of dilated tubules were evident, both admixed with normal tubules. The principal feature of atrophic tubules was a cuff-like, thickened basement membrane surrounding the tubular epithelial cells (Figure 4A) ▶ . This change was not apparent in dilated tubules. In the later stage, the kidney showed an increase in interstitial fibrosis and development of large dilated tubules with flattened epithelial cells and proteinaceous casts in their lumens (Figure 4B) ▶ .
Figure 4.
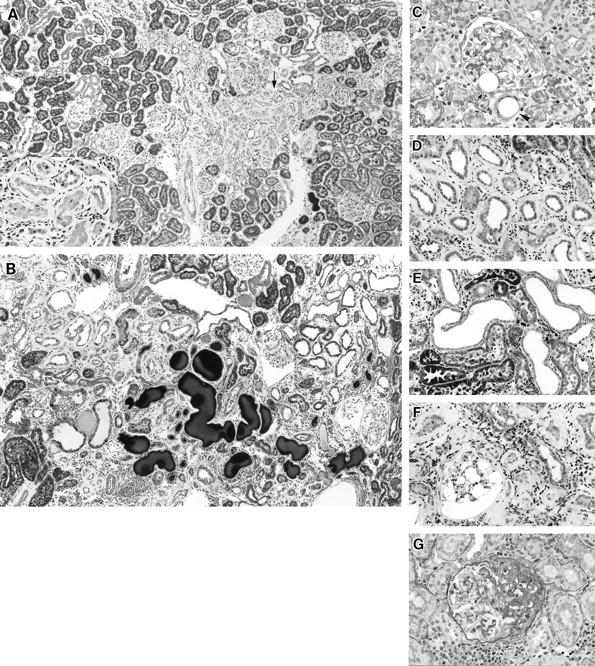
Light micrographs of kidneys 4 to 12 weeks after microsphere injection in group 2 (0.8 mg of microspheres). A: Four weeks after microsphere injection, a zone of atrophic tubules with surrounding fibrosis is evident between islands of apparently intact proximal tubules (arrow). Periodic acid-Schiff (PAS) stain; original magnification, ×60. Inset: A higher magnified micrograph of the indicated area showed atrophic tubules surrounded by thickened basement membrane. Original magnification, ×130. B: Twelve weeks after microsphere injection, large dilated tubules with or without cast formation are seen scattered among atrophic tubules. Masson trichrome stain; original magnification, ×60. C: Four weeks after microsphere injection. Transparent round microspheres are observed in the interstitium (arrowheads) and at the vascular pole of a glomerulus (arrow). The glomerulus containing the microsphere appears essentially normal. PAS stain; original magnification, ×260. D: Eight weeks after microsphere injection, a cuff-like, thickened basement membrane is more prominent, and infiltration of round cells is evident around atrophic tubules. Trichrome stain; original magnification, ×130. E: Eight weeks after microsphere injection, large dilated tubules consist of short, flattened epithelial cells without a thickened basement membrane. Trichrome stain; original magnification, ×130. F: Twelve weeks after microsphere injection, the thickness of the tubular basement membrane is increased. A glomerulus shows huge empty vacuolar change in glomerular epithelial cells. Trichrome stain; original magnification, ×130. G: Twelve weeks after microsphere injection, a glomerulus shows segmental glomerular sclerosis with hyalinosis and adhesion to Bowman’s capsule. Trichrome stain; original magnification, ×220.
Injected microspheres were trapped in small arterioles and in the glomerular capillary lumen near the vascular pole (Figure 4C) ▶ . Necrotic areas, however, were detected in only a small superficial portion of the cortex. The limited extent of the necrotic lesions in the cortex might have resulted from the disappearance of established ones before 4 weeks. More necrosis may have been evident sooner after the injection. For some glomeruli containing trapped microspheres, slight ischemic change was apparent in neighboring tufts. In the area without necrotic lesions, ischemic changes, such as wrinkling of capillary walls, were detected in a small percentage of glomeruli.
By 8 weeks after microsphere injection, the number of dilated tubules with relatively flattened epithelial cells had increased. The thickness of the cuff-like basement membrane of atrophic tubules had increased further, and some of these tubules were surrounded by infiltrating mononuclear cells (Figure 4, D and E) ▶ . Cytoplasmic vacuoles of glomerular epithelial cells also was conspicuous in some glomeruli.
At 12 weeks after injection, an increase in interstitial fibrosis and development of large dilated tubules and proteinaceous casts in their lumens were evident (Figure 4B) ▶ . However, almost none of the dilated tubules seen at 8 or 12 weeks were associated with thickening of the tubular basement membrane (Figure 4E) ▶ . The vacuoles in glomeruli had increased in number and extent (Figure 4F) ▶ , and some glomeruli had developed segmental sclerosis. The segmentally sclerotic glomeruli also showed an increased amount of PAS-positive mesangial matrix, collapsed capillaries, and PAS-positive inclusions in the glomerular epithelial cells (Figure 4G) ▶ . Totally obsolescent glomeruli were rarely observed.
Morphometric studies confirmed the above impression of progressive histological damage (Figure 5) ▶ . Interstitial fibrosis increased gradually after embolization in group 2. In contrast, no change in extent of fibrous tissue was apparent in control rats (group 1) during the experimental period. Consequently the extent of fibrous tissue in groups 1 and 2 became statistically significant 12 weeks after surgery. The difference in tubular basement membrane (TBM) thickening was more conspicuous than that of total fibrous tissue; as early as 4 weeks, thickening was evident in group 2. The areas of tubular lumens and casts occupying the lumen increased gradually, becoming significant at 8 weeks after microsphere injection. Although the morphometric score of tubular lumens does not necessarily mean the dilatation of each tubule, it seems to reflect the increase in number of large dilated tubules by light microscopic observation.
Figure 5.
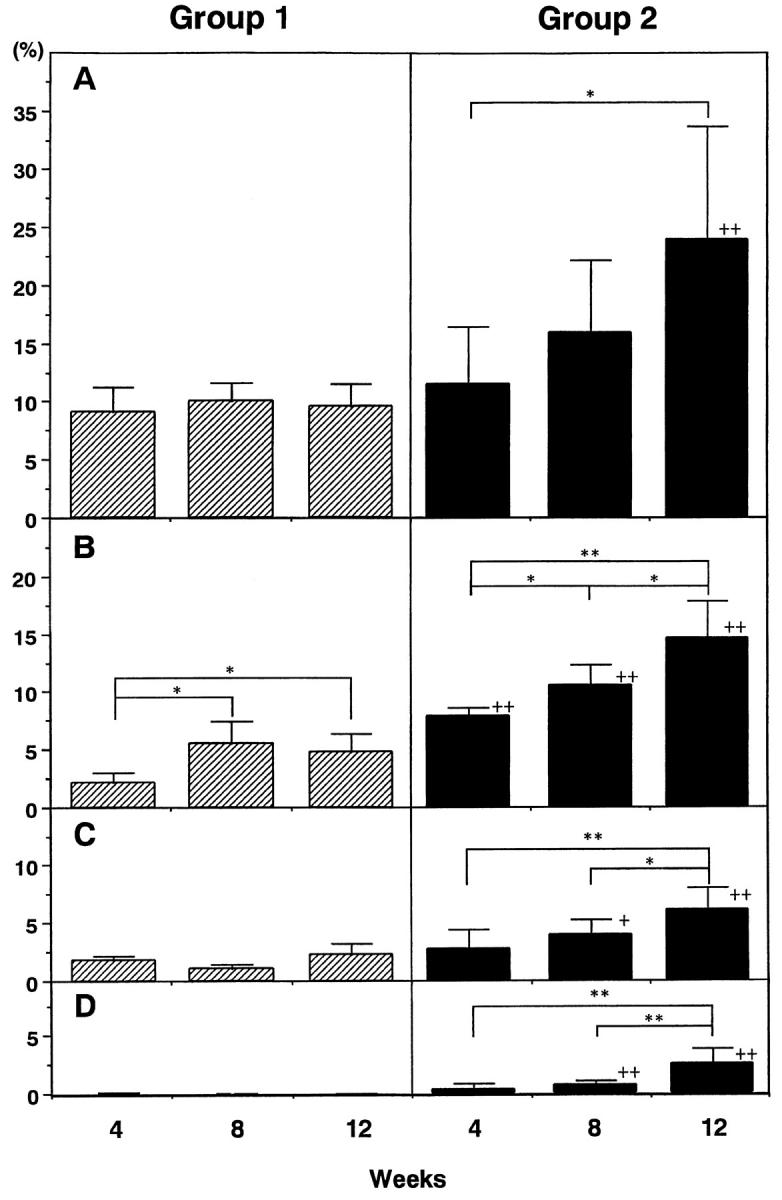
Morphometry of renal tubulointerstitial tissue of groups 1 (control) and 2 (0.8 mg of microspheres). Interstitial fibrous tissues (A), tubular basement membrane (B), tubular lumens (C), and casts within tubular lumens (D) were measured by a point-counting method and expressed as a percentage of total cortical area. * and **, P < 0.05 and 0.01, respectively. + and ++, P < 0.05 and 0.01 compared to group 1, respectively.
Atrophic tubules with basement membrane thickening did not show PNA or THP staining (Figure 6, A and C) ▶ . In contrast, the cytoplasm of dilated tubules were reactive with either PNA, antibody to THP, or both (Figure 6, B and D) ▶ which suggested that the dilated tubules were distal tubules. 16,18 PNA, which strongly stains the distal tubules, also weakly stains proximal tubular brush border. However, it is not difficult to distinguish them by their intracellular distribution and intensity (Figure 6, E and F) ▶ . Cytokeratin, which is demonstrable in connecting and collecting tubules, 19 was not immunostained in either atrophic or dilated tubules (data not shown). Numbers of PCNA-positive tubular epithelial cells were increased 2 weeks after microsphere injection, especially in the inner cortex and medulla, but had returned to basal values by 4 weeks (data not shown). During the experimental period, PCNA-positive cells were seen frequently in the atrophic tubules with cuff-like, thickened basement membranes, while only a few positive cells were apparent in intact or dilated tubules (Figure 6, G and H) ▶ . In control rats, numbers of PCNA-positive cells were slightly increased at 2 weeks, but the increase was not statistically significant.
Figure 6.
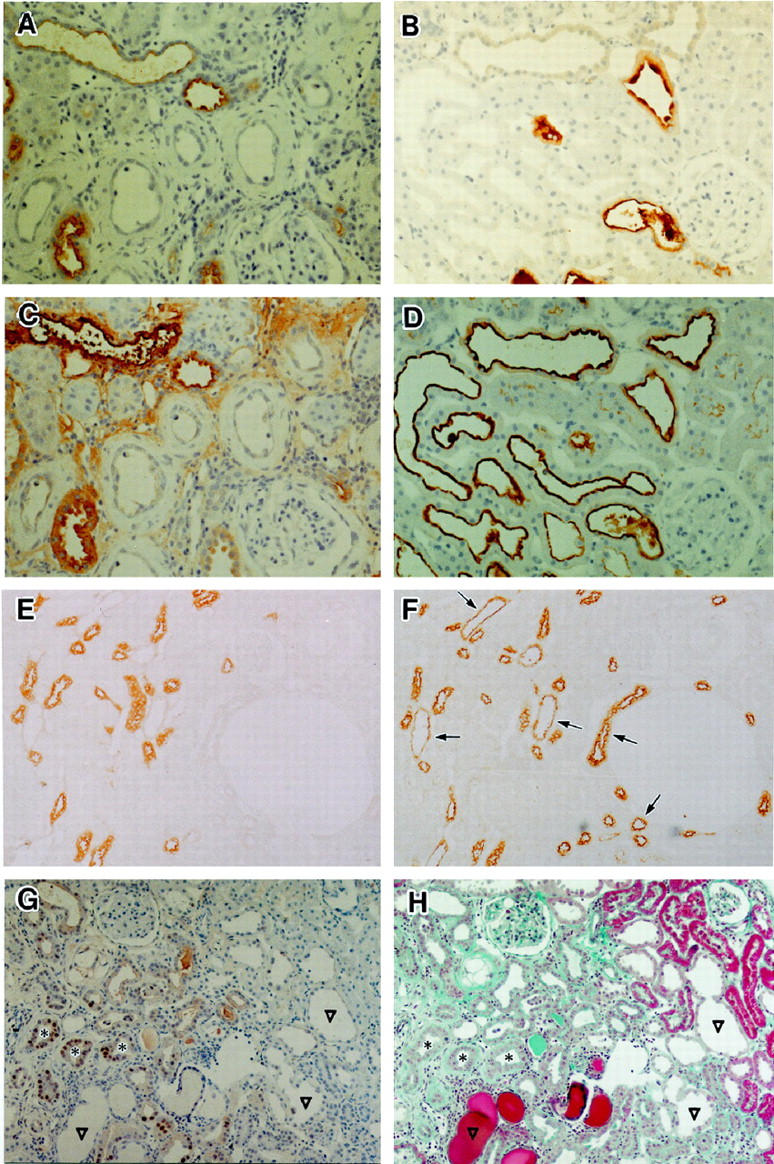
Histochemical staining with anti-THP antibodies (A, B, and E), biotin-labeled PNA (C, D, and F) and anti-PCNA antibodies (G) on kidneys from animals in group 2. Sections in A, B, and E are cut serially to ones in C, D and F, respectively. A: Atrophic tubular cells showed no staining for THP, 8 weeks after microsphere injection. Original magnification, ×90. B: Some dilated tubules stained for THP, but not all at 12 weeks. Original magnification, ×90. C: Atrophic tubular cells were not stained by PNA. Section cut serially to (A). Original magnification, ×90. D: Almost all dilated tubular cells were stained by PNA. Section cut serially to B. Original magnification, ×90. E and F: Serial sections of the deep cortex in the kidney from a rat before microsphere injection (normal control) were stained with antibodies to THP (E) or biotin-labeled PNA (F) PNA clearly stained many distal tubules (arrows) as well as THP-positive ones. Brush borders of proximal tubules were only faintly stained. Original magnification, ×90. G and H: Twelve weeks after microsphere injection, serial sections were stained with antibodies to PCNA (G) or Masson-Trichrome stain (H). PCNA-positive cells are evident only in tubules with a cuff-like, thickened basement membrane (*). Large dilated tubules (▿) did not include positive cells. Original magnification, ×60.
Discussion
We have described a new model of chronic renal failure induced by injection of microspheres of 20 to 30 μm in diameter. These microspheres occluded the preglomerular arterioles and intraglomerular capillaries near the vascular pole to induce a dose-dependent increase in urinary excretion of albumin and in the serum concentration of creatinine and total cholesterol with a reciprocal decrease in serum albumin. The creatinine concentration increased in group 2 (0.8 mg of microspheres) 12 weeks after embolization. Groups other than group 2 generally did not show significant increases in the creatinine concentration during the experimental period, although we found a significant increase in concentration of creatinine in group 3 (0.4 mg of microspheres) after 20 weeks (1.18 ± 0.11 mg/dl at 20 weeks and 1.64 ± 0.24 mg/dl at 24 weeks; unpublished data). Therefore this model, like the renal mass-reduction model, suggested that proteinuria preceded the increase in serum creatinine concentration. Blood pressure was mildly increased in rats injected with large amounts of microspheres.
These experiments are not the first attempt to induce chronic renal failure by injection of small particles. However, most reports published so far have focused on hypertension induced by renal infarction and did not analyze details of histological findings on progression. In addition, particles previously used have been larger than our microspheres or had widely varying sizes; as a consequence, the sizes of arteries affected were different than in the present study. 20-23 Apfelbach and Jensen 20 first reported producing renal failure by injection of charcoal particles into the dog renal artery in 1931. In 1961, Alexander et al 21 performed embolization of intrarenal arteries (arcuate or interlobular size) with several different particles to study the effect of embolism on renal parenchymal histology, blood pressure, and blood urea level. The typical resulting lesions, large wedge-shaped infarcts, differed from the present ones of small foci of atrophic tubules scattered among intact tubules. Koletsky and Rivera 22 infused plastic microspheres 37 to 74 μm in diameter, slightly larger than ours (20 to 30 μm), into the left renal arteries. Their studies focused on hypertensive nephrosclerosis, and their animals developed severe hypertension as early as 1 day after operation that reached up to 178 mm Hg at 1 week. In contrast, our rats developed hypertension only gradually; the mean systolic blood pressure at 12 weeks was only 135 mm Hg. Solez and Richter 23 also described the development of renal failure and hypertension after injection of Sephadex beads 20 to 80 μm in diameter into the rat aorta. This broad size range of emboli obstructed vessels ranging from arcuate arteries to small arterioles, producing considerable variation in histological change. Furthermore, these previous studies did not examine either the detailed time course of development of chronic renal failure or details of histological changes in affected kidneys.
The characteristic histological features of our present models, especially early findings of a scattered distribution of damaged and undamaged tubules, contrast sharply with the renal mass-ablation model where remaining tissue is normal at onset of renal damage. Mingling of damaged and relatively undamaged tubules is characteristic of many progressive kidney diseases in humans and may be important in disease pathogenesis, given that damaged tubular cells can secrete various cytokines and growth factors. 11,12,24 The nature of these atrophic tubules is not clearly established, but they did not express distal tubular markers such as THP or PNA-adhesive glycoprotein, and some of the cells showed apparent remnants of a brush border. Therefore, we believe at least some of these tubules to be proximal tubules. Proof will require detection of antigens specific to proximal tubular cells in the cells of atrophic tubules. The lack of staining for PNA or THP of cells in atrophied tubules does not necessarily mean that they are not distal tubules. They may no longer express those proteins as they are atrophic.
Another remarkable histological finding was that many dilated tubules became conspicuous as late as 8 weeks after injection. These were stained with either anti-THP antibody or labeled peanut agglutinin, which suggested a distal tubular origin. 18,19 Tubulointerstitial fibrosis with tubular atrophy, dilation, and cast formation collectively has been considered the hallmark of end-stage kidney disease. 25,26 Tubular atrophy, which can lead to tubulointerstitial scarring, has been suggested to result from maladaptive functional changes or increased metabolism in the hypertrophic (dilated) tubules, 26 although this hypothesis is not supported by conclusive evidence. The tubulointerstitial changes apparent in the present model included apparently independent development of atrophic tubules and large dilated tubules. Atrophic tubules were associated with a cuff-like, thickened basement membrane and PCNA-positive cells, whereas the dilated tubules appeared unrelated to them, with most originating from the distal tubule. Furthermore, atrophic tubules were numerous as early as 2 to 4 weeks after injection of microspheres, at which time only a small number of dilated tubules could be seen.
The mechanisms that underlie progressive renal dysfunction in the microsphere model are not clear from our present results. Development of massive proteinuria preceded an increase in the serum concentration of creatinine, and the amount of albuminuria at 8 weeks was correlated significantly with serum creatinine concentration at 12 weeks in groups 2 and 3 (data not shown). These observations resemble the clinical course of progressive human kidney diseases where massive proteinuria is a sign of poor renal functional prognosis. 25,26 Marked proteinuria per se may contribute directly to tubulointerstitial damage, as has been suggested for the renal mass-ablation model. 25 Alternatively, proteinuria might cause progressive deterioration of renal function through induction of hypercholesterolemia, which was observed as early as 4 weeks after microsphere injection. Direct toxicities of proteinuria 25 and hyperlipidemia, 27,28 both of which have been suggested as possible mechanisms for renal damage following partial renal ablation, 26 remain to be evaluated in the present model. Other factors, such as hyperfiltration or glomerular hypertrophy, also have been suggested to be important in the nonimmunological progression of renal disease. 1,5,29-32 We did not evaluate the glomerular filtration rate or glomerular size in the present study. The large epithelial vacuoles developed as time passed. The similar change of glomerular epithelial cells have been reported in adriamycin/daunomycin nephropathy 33 as well as some ablation models. 7 Although it is known that there is a close relationship between the large epithelial vacuolation and massive proteinuria due to hyperfiltration, resulting in glomerular sclerosis, the pathogenesis of vacuoles has not been elucidated in any case.
Hypertension also has been suggested as a cause of progressive renal failure in ablation models, 3,34,35 and it occurred in the present model. The extent of hypertension in rats in group 2, however, was less than that apparent in rats following a 5/6 nephrectomy. Recently we found that a low-protein diet effectively prevented deterioration in renal function induced by microsphere injection, even though rats developed hypertension as early as 4 weeks (unpublished data). Thus, we doubt that hypertension is the main cause for deterioration of renal function in our model.
In this model, we used uninephrectomy before microsphere injection. If we can induce progressive chronic renal failure by bilateral microembolization, that model would be more suitable for the study of the pathogenesis of human chronic renal diseases. However, we did not use such a method because we did not think that infusion of particles from aorta could guarantee the uniform distribution of particles in both kidneys. In addition, to induce progressive renal failure with two kidneys in our model, we would have to cause severe histological damage to one kidney, which would make it more difficult to get uniformly decreased renal function in a group of rats.
In summary, we describe a new model of progressive nonimmunological renal disease in rats in which atrophic and dilated tubules mingle with apparently normal tubules as in patients with various progressive renal diseases. We propose that atrophic tubules do not develop sequentially from dilated tubules and have a different pathogenesis, most likely interstitial ischemia. The present model should prove useful for characterizing pathophysiology and possibly for developing new therapies for progressive renal diseases.
Acknowledgments
We express special thanks to Ms. Fujishiro, Ms. Y. Ohta, Ms. M. Asano, and Ms. H. Takemura for their technical assistance.
Footnotes
Address reprint requests to M. Kimura, M.D., University of Shizuoka School of Nursing, 52-1 Yada, Shizuoka 422-8526, Japan. E-mail: kimura@u-shizuoka-ken.ac.jp.
Supported by a grant from the Ministry of Education of Japan.
References
- 1.Brenner BM, Meyer TW, Hostetter TH: Dietary protein intake and progressive nature of kidney disease: the role of hemodynamically mediated glomerular injury in the pathogenesis of progressive glomerular sclerosis in aging, renal ablation, and intrinsic renal disease. N Engl J Med 1982, 307:652-659 [DOI] [PubMed] [Google Scholar]
- 2.Chanutin A, Ferris EB: Experimental renal insufficiency produced by partial nephrectomy. Arch Intern Med 1932:767–787
- 3.Koletsky S, Goodsitt AM: Natural history and pathogenesis of renal ablation. AMA Arch Pathol 1960, 69:654-662 [PubMed] [Google Scholar]
- 4.Kaufman JM, DiMeola HJ, Siegel NJ, Lytton B, Kashgarian M, Hayslett J: Compensatory adaptation of structure and function following progressive renal ablation. Kidney Int 1974, 6:10-17 [DOI] [PubMed] [Google Scholar]
- 5.Pearce RM: The influence of kidney substance upon nitrogenous metabolism. J Exp Med 1908, 10:632-644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carrel A: Note on the production of kidney insufficiency by reduction of the arterial circulation of the kidney. Proc Soc Exper Biol Med 1909, 6:107-108 [Google Scholar]
- 7.Olson JL, Hostetter TH, Rennke HG, Brenner BM, Venkatachalam MA: Altered glomerular permselectivity and progressive sclerosis following extreme ablation of renal mass. Kidney Int 1982:112–126 [DOI] [PubMed]
- 8.Kleinknecht C, Laouari D: The influence of dietary components on experimental renal disease. Contemp Issues Nephrol 1986, 14:17-35 [Google Scholar]
- 9.El Nahas AM, Zoop SN, Evans DJ, Rees AJ: Chronic renal failure after nephrotoxic nephritis in rats: Contribution to progression. Kidney Int 1987, 36:173-180 [DOI] [PubMed] [Google Scholar]
- 10.Bertani T, Cutillo F, Zoja C, Broggini M, Remuzzi G: Tubulo-interstitial lesions mediate renal damage in adriamycin glomerulopathy. Kidney Int 1986, 30:488-496 [DOI] [PubMed] [Google Scholar]
- 11.Frank J, Enbler-Blum G, Rodemann HP, Müller GA: Human renal tubular cells as a cytokine source: PDGF-B, GM-CSF and IL-6 mRNA expression in vitro. Exp Nephrol 1993, 1:26-35 [PubMed] [Google Scholar]
- 12.Gröne H-J, Simon M, Gröne EF: Expression of vascular endothelial growth factor in renal vascular disease and renal allografts. J Pathol 1995, 177:259-267 [DOI] [PubMed] [Google Scholar]
- 13.Haverty TP, Kelly CJ, Hines WH, Amenta PS, Watanabe M, Harper RA, Kefalides NA, Neilson EG: Characterization of a renal tubular epithelial cell line which secretes the autologous target antigen of autoimmune experimental interstitial nephritis. J Cell Biol 1988, 107:1359-1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Humes HD, Nakamura T, Cieslinski DA, Miller D, Emmons RV, Border WA: Role of proteoglycans and cytoskeleton in the effects of TGF-β1 on renal proximal tubule cells. Kidney Int 1993, 43:575-584 [DOI] [PubMed] [Google Scholar]
- 15.Strutz F, Caron R, Tomaszewski J, Fumo P, Ziyadeh F, Neilson EG: Transdifferentiation: a new concept in renal fibrosis (abstr.). J Am Soc Nephrol 1994, 5:819 [Google Scholar]
- 16.Nadasdy T, Laszik Z, Blick KE, Johnson DL, Silva FG: Tubular atrophy in the end-stage kidney: A lectin and immunohistochemical study. Hum Pathol 1994, 25:22-28 [DOI] [PubMed] [Google Scholar]
- 17.Weibel ER: Principles and methods for the morphometric study of the lung and other organs. Lab Invest 1963, 12:131-155 [PubMed] [Google Scholar]
- 18.Ivanyi B, Olsen TS: Immunohistochemical identification of tubular segments in percutaneous renal biopsies. Histochemistry 1991, 95:351-356 [DOI] [PubMed] [Google Scholar]
- 19.Hemmi A, Mori Y: Immunohistochemical and scanning electron microscopic study of cytokeratin distribution in the collecting tubule of the rat kidney. Acta Pathol Jpn 1990, 40:307-313 [DOI] [PubMed] [Google Scholar]
- 20.Apfelbach CW, Jensen CR: Experimental renal insufficiency in dogs with special reference to arterial hypertension. J Clin Invest 1931, 10:162 [Google Scholar]
- 21.Alexander N, Heptinstall RH, Poickering GW: The effects of embolic obstruction of intrarenal arteries in the rabbit. J Pathol Bact 1961, 81:225-237 [DOI] [PubMed] [Google Scholar]
- 22.Koletsky S, Rivera-Velez JM: Renin-angiotensin system in microembolic renal hypertension. Arch Pathol 1968, 85:1-9 [PubMed] [Google Scholar]
- 23.Solez K, Richter GW: Microembolic renal disease in rats induced with sephadex. Am J Pathol 1972, 66:163-188 [PMC free article] [PubMed] [Google Scholar]
- 24.Grantham JJ: Mechanisms of progression in autosomal dominant polycystic kidney disease. Kidney Int 1997, 52(Suppl 63):S93-S97 [PubMed] [Google Scholar]
- 25.Remuzzi G, Buggenenti P, Benigni A: Understanding the nature of renal disease progression. Kidney Int 1997, 51:2-15 [DOI] [PubMed] [Google Scholar]
- 26.El Nahas AM: Mechanisms of experimental and clinical renal scarring. 2nd ed. Davison AM Cameron JS Grunfeld J-P Kerr DNS Ritz E Winearls CG eds. Oxford Textbook of Clinical Nephrology, 1998, :pp 1749-1788 Oxford Medical Publications, London [Google Scholar]
- 27.Kasiske BL, O’Donnell MP, Garvis WJ, Keane WF: Pharmacologic treatment of hyperlipidemia reduces glomerular injury in rat 5/6 nephrectomy model of chronic renal failure. Circ Res 1988, 62:367-374 [DOI] [PubMed] [Google Scholar]
- 28.Floege J, Gröne H-J: Progression of renal failure: what is the role of cytokines? Nephrol Dial Transplant 1995, 10:1575-1586 [PubMed] [Google Scholar]
- 29.Kaufman JM, DiMeola HJ, Siegel NJ, Lytton B, Kashgarian M, Hayslett J: Compensatory adaptation of structure and function following progressive renal ablation. Kidney Int 1974, 6:10-17 [DOI] [PubMed] [Google Scholar]
- 30.Yoshida Y, Fogo A, Ichikawa I: Glomerular hemodynamic changes vs. hypertrophy in experimental glomerular sclerosis. Kidney Int 1989, 35:654-660 [DOI] [PubMed] [Google Scholar]
- 31.Hostetter TH, Olson JL, Rennke HG, Venkatachalam MA, Brenner BM: Hyperfiltration in remnant nephrons: a potentially adverse response to renal ablation. Am J Physiol 1981, 241:F85-F93 [DOI] [PubMed] [Google Scholar]
- 32.Brenner BM: Nephron adaptation to renal injury or ablation. Am J Physiol 1985, 249:F324-F337 [DOI] [PubMed] [Google Scholar]
- 33.Fajardo LF, Eltringham JR, Stewart JR, Klauber MR: Adriamycin nephrotoxicity. Lab Invest 1980, 43:242-253 [PubMed] [Google Scholar]
- 34.Puerkerson ML, Hoffsten PE, Klahr S: Pathogenesis of the glomerulopathy associated with renal infarction in rats. Kidney Int 1976, 9:407-417 [DOI] [PubMed] [Google Scholar]
- 35.Bidani AK, Mithell KD, Schwartz MM, Navar LG, Lewis EJ: Absence of glomerular injury or nephron loss in a normotensive rat remnant kidney model. Kidney Int 1990, 38:28-38 [DOI] [PubMed] [Google Scholar]