

Abstract
Alzheimer’s Disease (AD) is a progressive neurodegenerative disorder involving select neurons of the hippocampus, neocortex, and other regions of the brain. Markers of end stage disease include fibrillar lesions, which accumulate hyperphosphorylated tau protein polymerized into filaments, and granulovacuolar lesions, which appear primarily within the hippocampus. The mechanism by which only select populations of neurons develop these lesions as well as the relationship between them is unknown. To address these questions, we have turned to AD tissue to search for enzymes specifically involved in tau hyperphosphorylation. Recently, we showed that the principal phosphotransferases associated with AD brain-derived tau filaments are members of the casein kinase-1 (CK1) family of protein kinases. Here we report the distribution of three CK1 isoforms (Ckiα, Ckiδ, and Ckiε) in AD and control brains using immunohistochemistry and Western analysis. In addition to colocalizing with elements of the fibrillar pathology, CK1 is found within the matrix of granulovacuolar degeneration bodies. Furthermore, levels of all CK1 isoforms are elevated in the CA1 region of AD hippocampus relative to controls, with one isoform, Ckiδ, being elevated >30-fold. We propose that overexpression of this protein kinase family plays a key role in the hyperphosphorylation of tau and in the formation of AD-related pathology.
Alzheimer’s Disease (AD) is characterized pathologically by the appearance of two principal intracellular lesions. The first, termed granulovacuolar degeneration (GVD), involves the cytoplasmic accumulation of abnormally large (≤5 μm diameter) vacuoles containing a dense-cored granule. 1 The molecular composition of GVD bodies, which appear primarily within the cell bodies of affected hippocampal pyramidal neurons, is unknown. The second lesion, characterized by the accumulation of fibrils or filaments within neuronal cell bodies (neurofibrillary tangles; NFT), neuronal processes (neuropil threads), and within dystrophic neurites associated with amyloid plaques (neuritic plaques; NP), comprises the neurofibrillary or fibrillar pathology. Each manifestation of fibrillar pathology accumulates filaments comprised of the microtubule-associated protein tau. 2 As a result, affected brain regions contain nearly an order of magnitude more tau than normal controls. 3 In addition to these quantitative differences, the quality of tau differs as well, having fold higher stoichiometries of covalently bound phosphate than normal tau. 4 Hyperphosphorylation affects tau function 5 and is a sensitive marker of disease. 6
Multiple strategies have been used to identify the phosphotransferases mediating tau hyperphosphorylation in AD. First, in vitro approaches have proved that tau is an efficient substrate for most protein kinases, many of which are capable of filling known phosphorylation sites on filamentous tau. 7 These studies have shown that the number of phosphotransferases involved in tau hyperphosphorylation is potentially large. Cell-based approaches confirm that elevation of selected protein kinases in situ can increase occupancy of sites found on filamentous tau, 8 but again it is not clear which of these enzymes may actually be involved in disease pathogenesis. A third approach has focused on authentic AD tissue to identify phosphotransferases that are tightly associated with elements of AD pathology or that change levels or specific activity in disease. Applying the last strategy, we showed that the principal phosphotransferases associated with AD brain-derived tau filaments are members of the casein kinase-1 (CK1) family of protein kinases. 9 Once considered a single entity, human CK1 is now known to consist of multiple isoforms encoded by distinct genes (Ckiα, γ1, γ2, γ3, δ, ε). At least one of these isoforms, Ckiα, was shown to be a major constituent of purified tau filaments, comprising as much as 0.5% of the preparation by weight, suggesting it is localized appropriately to play a role in tau hyperphosphorylation. 9
Here we tested the hypothesis that CK1 isoforms correlate with AD pathogenesis by returning to authentic AD tissue and examining the distribution of isoforms Ckiα, δ, and ε by immunohistochemistry and Western analysis. The results confirm that CK1 isoforms associate with elements of AD pathology in tissue, with large increases in levels accompanying the formation of AD pathology in hippocampus. Furthermore, the data presented here establish CK1 isoforms as unambiguous markers for GVD in AD tissue, suggesting a regulatory nexus between GVD and the fibrillar pathologies.
Materials and Methods
Primary Antibodies
Monoclonal antibodies specific for Ckiα (IC94.1), 9 Ckiδ (IC128A), 9 class IIIβ isoform of tubulin (Tuj1), 10 and filamentous tau (Tau2) 11 were purified and handled as described. 12 Purified monoclonal antibody against Ckiε came from a commercial source (#40520; Transduction Laboratories, Lexington, KY).
Human Subjects
AD cases had a clinical diagnosis of probable AD that was confirmed on neuropathological evaluation in which the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) age-adjusted criteria were met. Control cases were nondemented clinically and failed to fulfill the age-adjusted neuropathological criteria for AD. 13
Tissue Preparation
Postmortem interval for all specimens averaged 8.8 ± 5.6 hours (range, 4–20 hours). For all immunohistochemical analyses described below, brains from four AD patients (one female and three males, mean age 81.8 ± 2.6 years; range, 78–84 years) and three nondemented controls (one female and two males, mean age 66.0 ± 2.0 years; range, 64–68 years) were harvested, sliced into 1-cm-thick coronal slabs, and fixed in chilled 4% paraformaldehyde for 30 hours as described. 14 Fixed slabs containing the hippocampus and adjacent temporal cortices were cut into 40-μm-thick sections on a freezing sliding microtome and stored in cryoprotectant until processed.
For preparation of homogenates, hippocampi were dissected from five AD patients (three female and two males, mean age 67.0 ± 4.9 years; range, 63–75 years) and four age-matched controls (two female and two males, mean age 65.3 ± 2.9 years; range, 63–69 years), snap-frozen in liquid nitrogen, and stored at −80°C until processed. All samples were coded and analyzed by an investigator blinded to the code.
Immunohistochemistry
Free-floating, 40-μm-thick tissue sections were processed for immunohistochemistry as described, 14 except that 1% nonfat dry milk replaced horse serum in all incubations involving antibodies. Sections were immunostained with monoclonal antibodies IC94.1 (10 μg/ml), IC128A (2 μg/ml), 40520 (1 μg/ml), or Tau2 (2 μg/ml) overnight at room temperature. Biotinylated goat anti-mouse antibody (6.8 μg/ml;Vector Laboratories, Burlingame, CA) was used as secondary antibody. Stained sections were labeled for 1 h in an avidin-biotin complex (Elite kit; Vector Laboratories; diluted 1:275), developed with 3,3′-diaminobenzidine (Metal enhanced DAB Substrate Kit, Pierce, Rockford, IL), mounted on gelatin-coated slides, dehydrated through graded alcohols (70, 90, 100%), cleared in xylene, and coverslipped with Permount (Fisher Scientific, Pittsburgh, PA). Selected sections were counterstained with cresyl violet (0.11% in H2O) or Thioflavin S (0.1% in H2O) before dehydration. Paraffin-embedded sections (6 μm) were treated similarly after dewaxing, except that they were heat treated (120°C for 7 minutes in 10 mmol/L Tris-NaOH, pH 10.0 at 25°C) before immunostaining. 15 To clarify whether monoclonal antibodies recognized phosphoepitopes, selected sections were dephosphorylated with calf intestine alkaline phosphatase (Type VII-L, Sigma, St. Louis, MO) before immunohistochemistry as described previously. 11,16
Immuno-Electron Microscopy
Coronal sections of hippocampus (40 μm) were subjected to immunohistochemistry as described above, except that Triton X-100 was omitted from all solutions. Immunostaining was detected with either 3,3′-diaminobenzidine or silver-enhanced colloidal gold. 17 After immunolabeling, tissue for electron microscopy was osmicated, dehydrated, embedded in plastic, sectioned, and counterstained. 18 Light microscopy was used to select immunolabeled pyramidal cells from the CA1 region of the embedded section. These were then sectioned thinly and examined in a JEOL 100CX electron microscope.
Quantitation of Lesions
The number of lesions was quantified from images captured at ×200 magnification using a Nikon Eclipse 800 light microscope coupled to a Photometrics SenSys digital camera. In hippocampus, three fields (0.153 mm2/field) from each region of Ammon’s horn (CA1–4) were counted using the Metamorph Imaging System. In neocortical samples, three fields per case were counted in each of Layers III and V. After each field was captured, the stage was moved manually to a new field using fiduciary landmarks to ensure a completely nonredundant evaluation. The location of each field was further verified by maintaining a record of stage coordinates for each field examined.
Tissue Homogenates
Aliquots of fresh-frozen hippocampus were thawed, dissected to yield the CA1 sector, and snap-frozen in dry ice/ethanol for storage before homogenization. Samples of CA1 (30–200 mg) were suspended in 10 volumes (v/w) of 50 mmol/L Tris, pH 7.4, 50 mmol/L NaF, 1 mmol/L phenylmethylsulfonyl fluoride, 5 μg/ml leupeptin, and 1 mmol/L dithiothreitol and homogenized with 10 passes in a motor-driven glass-Teflon homogenizer. Aliquots of homogenates were immediately subjected to boiling in sodium dodecyl sulfate-polyacrylamide gel electrophoresis sample buffer.
Analytical Methods
The protein content of monoclonal antibodies was estimated spectrophotometrically, whereas brain homogenates were estimated by the method of Bradford. 9 Levels of CK1 isoforms in tissue homogenates were estimated by Western analysis as described previously. 9 Statistical errors are reported as ± one SD.
Results
Monoclonal Antibodies
Three monoclonal antibodies were used to characterize the distribution of CK1 isoforms in human brain: IC94.1, raised against Ckiα; IC128A, raised against Ckiδ; and 40250, raised against Ckiε. 9,19 The specificities of IC94.1 and IC128A were shown previously, 9 whereas that of 40250 is shown in Figure 1 ▶ . These data confirm that 40250 reacts on Western blots with Ckiε but not with Ckiα, γ3, or δ. In homogenates of human neocortex, cerebellum, and brain stem, Ckiε immunoreactivity appeared as a single species migrating close to its calculated molecular mass (47.3 kd). 19 This is in contrast to Ckiα, which appears as three distinct species derived from alternative splicing of a single gene. On the basis of quantitative Western analysis, the levels of Ckiε as a percentage of total protein (mean ± range) averaged 0.0032 ± 0.0004% (w/w). Thus Ckiε, like other CK1 homologues examined to date, is widely distributed within human brain at concentrations similar to those of Ckiδ. 9
Figure 1.

Monoclonal antibody 40250 is a specific immunological probe for Ckiε. Recombinant CK1 standards (∼4 ng each) and extracts of selected human brain regions were subjected to Western analysis with anti-Ckiε monoclonal antibody 40250. Lane 1, mixture of Ckiα1, α2, and α3; Lane 2, Ckiγ3; Lane 3, Ckiδ; Lane 4, Ckiε; Lane 5, cerebellum extract (∼63 μg); Lane 6, cerebral cortex extract (∼63 μg); Lane 7, brain stem extract (∼63 μg). The positions of protein molecular mass standards are shown in kilodaltons on left. Under these conditions, monoclonal antibody 40250 does not label Ckiα, Ckiγ3, Ckiδ (Lanes 1–3), or other proteins in Eschericia coli extracts, but reacts with glutathione-S-transferase-Ckiε, (a recombinant fusion protein, Lane 4). Monoclonal antibody 40250 is also specific for Ckiε in crude brain extracts (Lanes 5–7).
Distribution of CK1 Isoforms in Normal Brain
To examine the cellular distribution of CK1 isoforms in human brain, free-floating sections of temporal lobe were subjected to immunohistochemical analysis with each of the three anti-CK1 antibodies described above. First, the distribution in aged, nondemented control temporal lobe was established. The results revealed that Ckiδ had the most selective localization of the three CK1 isoforms examined, being highly enriched in the bodies of pyramidal neurons (Figure 2A) ▶ . Thus, although the total brain concentration of Ckiδ is only threefold greater than that of Ckiα, 9 the neuronal concentration is probably much higher, owing to neuron-selective distribution. In contrast, Ckiε staining was widespread in gray matter, with immunoreactivity appearing in both cell bodies and the neuropil (data not shown). Thus, despite being nearly identical to Ckiδ in primary structure within the protein kinase catalytic domain, 19 these two CK1 isoforms appear differentially distributed in human brain. The final isoform, Ckiα, showed the broadest distribution of the three, being found in all areas of the section. No selective localization was apparent (data not shown).
Figure 2.
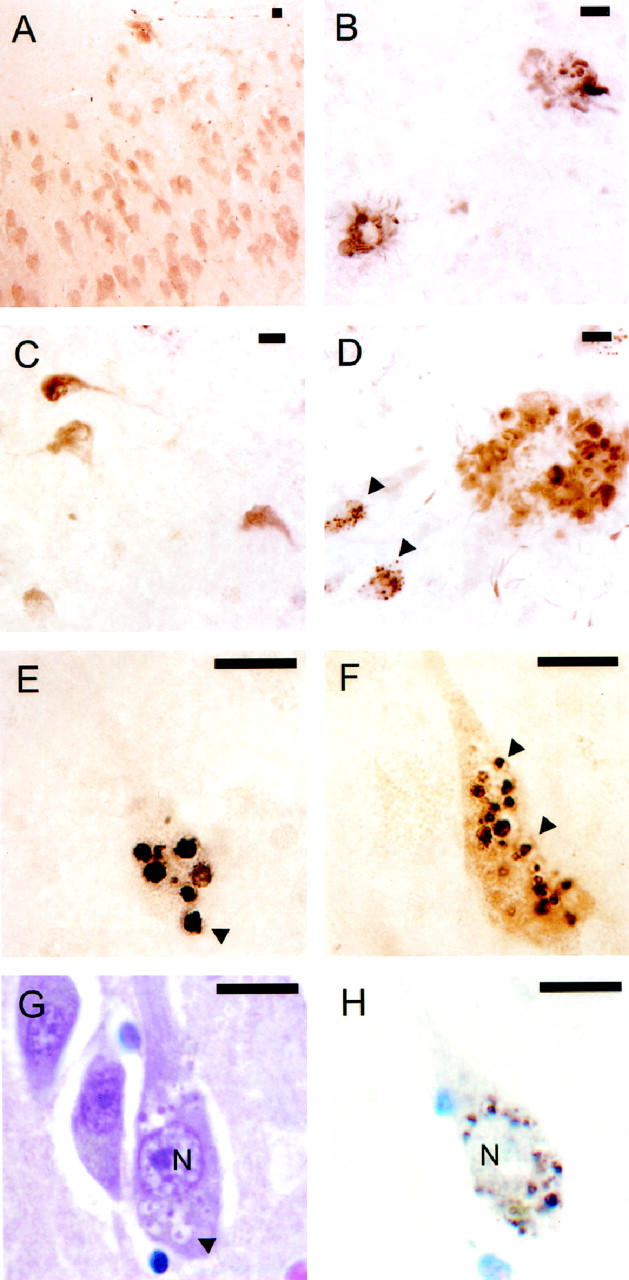
Differential distribution of CK1 isoforms in AD and normal brain. Sections of temporal lobe (free-floating) prepared from control (A) and AD (B-H) brains were stained with monoclonal antibodies against CK1 isoforms and examined by light microscopy. In control hippocampus, Ckiδ immunoreactivity appears enriched in pyramidal neurons of Ammon’s horn (A). In AD superior temporal gyrus, Ckiδ colocalizes with both NPs (B) and NFTs (C). In AD hippocampus, Ckiε colocalizes with both fibrillar and granular lesions (D, arrowheads point to two neurons containing granules). This pattern is seen only in severe, late stage disease. Typically, as shown in AD hippocampus for Ckiα (E) and Ckiδ (F), immunostaining is more intense in granules than in NFTs or NPs. The granules, which can be up to 4 μm in diameter, lie within vacuoles (arrowheads point to selected examples). Paraffin-embedded 6-μm-thick sections of hippocampus stained with hematoxylin and eosin (G) or cresyl violet and monoclonal antibody against Ckiδ (H) reveal that the intracellular distribution and morphology of Ckiδ-immunoreactive bodies corresponds to those of authentic GVD bodies in an adjacent neuron (arrowhead points to one of several examples within an affected neuron). N, nucleus. All scale bars, 10 μm.
CK1 Isoforms Colocalize with Neurofibrillary Lesions
To determine whether CK1 isoforms colocalized with fibrillar pathology in AD brain, as predicted by their copurification with tau filaments, 9 sections of neuropathologically confirmed AD temporal lobe were examined immunohistochemically. We focused initially on neocortical regions (eg, superior temporal gyrus) because these develop extensive fibrillar pathology in AD but not in aged, nondiseased controls. 20 The results showed that all three CK1 isoforms examined colocalized with fibrillar pathology in this region. Staining was most intense in layers III and V, with NPs predominating in the former and NFTs in the latter (shown for Ckiδ only; Figure 2, B and C ▶ ). Approximately 63% of thioflavin-S-positive NPs and 90% of thioflavin-S-positive NFTs contained anti-CK1 immunoreactivity in these layers (shown for Ckiδ only; Figure 3 ▶ ). Typically, NPs stained more intensely than either neuropil threads or NFTs. The latter gave the greatest variability, with some NFTs being strongly immunostained while others stained only weakly. Overall, the staining pattern observed for each of the anti-CK1 monoclonal antibodies closely resembled that of Tau2, a monoclonal antibody that selectively recognizes filamentous tau. 11,12
Figure 3.

Ckiδ colocalizes with NFTs and NPs. Serial sections (free-floating) of AD superior temporal gyrus were stained separately with anti-Ckiδ monoclonal antibody IC128 (dark bars) or thioflavin S (light bars). Numbers of stained NFTs and NPs were then counted as described in Materials and Methods. Approximately 63% of thioflavin-S-positive NPs and 90% of thioflavin-S-positive NFTs contained anti-Ckiδ immunoreactivity. Bars reflect mean ± SD (n = 9 observations from 3 cases).
Staining of fibrillar lesions was seen in coronal sections of hippocampus as well. Yet here the intensity of fibrillar staining was dwarfed by strongly immunoreactive granules within the pyramidal cell layer. Similar staining intensity for both fibrillar and granular lesions was seen only in late stage cases containing florid fibrillar pathology in the hippocampus (shown for Ckiε; Figure 2D ▶ ). Immunoreactive granules were large (up to 4 μm in diameter) and reacted with all three anti-CK1 monoclonal antibodies described above but not with Tau2. On closer inspection, the granules appeared to lie within cytoplasmic vacuoles (shown for Ckiα and Ckiδ; Figure 2, E and F ▶ ). The general appearance of these lesions resembled that of granulovacuolar degeneration (GVD), a feature of AD pathology characterized by the accumulation of granule-bearing vacuoles of characteristic size, morphology, and intrahippocampal distribution. 1
CK1 Isoforms Colocalize with GVD Bodies
On the basis of four criteria, CK1 isoforms are biochemical markers for GVD. First, the intracellular distribution of authentic GVD bodies, as detected by the classic procedure of hematoxylin and eosin histology, correlate with CK1 immunostaining in serial, 6-μm-thick paraffin-embedded coronal sections of hippocampus (shown for Ckiδ; Figure 2, G and H ▶ ). Both methods detect granule-bearing vacuoles of characteristic size and perinuclear distribution. Second, the intrahippocampal distribution of CK1-positive bodies follows the pattern established for GVD, 21,22 with the greatest number of involved neurons lying within the CA1 region of Ammon’s horn, followed by CA2 and CA3/CA4 (shown for Ckiδ and Ckiε in Figure 4 ▶ ). Although present in aged, nondemented controls, these lesions are encountered far less frequently than in the AD group (Figure 4) ▶ . Third, CK1-positive granules retain the ultrastructure of GVD bodies, 1 consisting of coarse, electron-dense granules in the center of large cytoplasmic vacuoles (shown for Ckiδ; Figure 5A ▶ ). Labeling of these structures with colloidal gold confirmed that Ckiδ is localized primarily within the matrix of the vacuole (Figure 5B) ▶ . Finally, CK1-positive bodies, like those observed in authentic GVD, are large (see above; Figures 2 and 5 ▶ ▶ ), having three to five orders of magnitude larger volume than normal intracellular bodies such as endosomes or lysosomes. 23
Figure 4.

The regional distribution of CK1-positive granules in AD hippocampus parallels that of granulovacuolar degeneration. Sections (free-floating) of AD (dark bars) and control (light bars) hippocampi were stained separately with anti-Ckiδ monoclonal antibody IC128 or anti-Ckiε monoclonal antibody 40250 and examined by light microscopy. The number of pyramidal neurons accumulating GVD-like granules that were positive for Ckiδ (A) or Ckiε (B) in each region of Ammon’s horn (CA1–4) was then quantified as described in Materials and Methods. The frequency of neurons containing Ckiδ- and Ckiε-positive granules parallels that established for GVD. Bars reflect mean ± SD (n = 9 observations from 3 cases).
Figure 5.
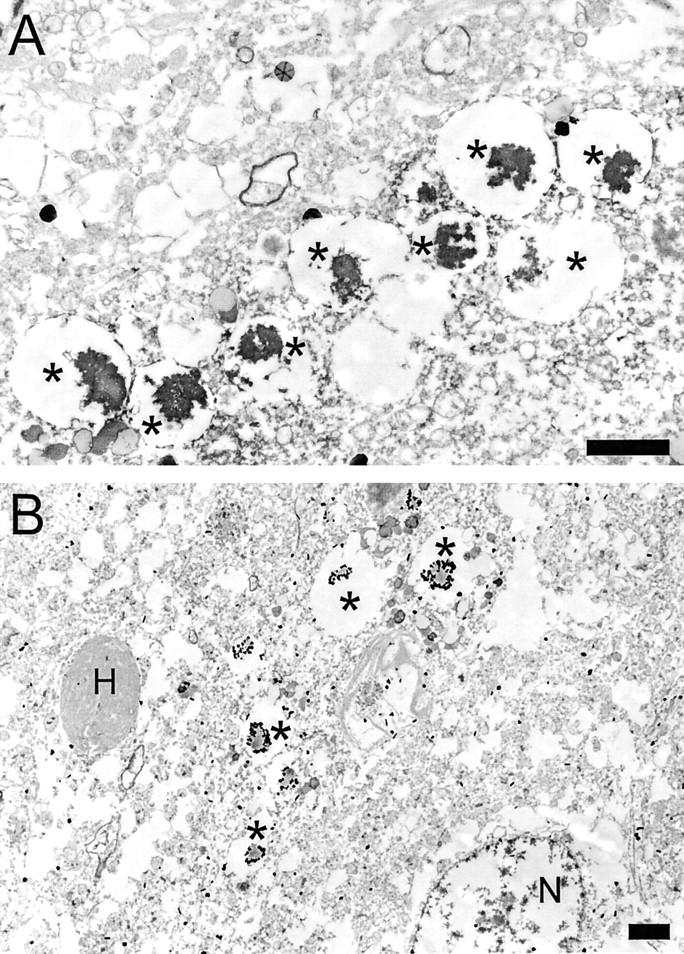
Ultrastructure of granules labeled with anti-Ckiδ monoclonal antibody. Vacuole-bearing lesions identified immunohistochemically with anti-Ckiδ monoclonal antibody IC128 were processed for electron microscopy as described in Materials and Methods. A: 3,3′-diaminobenzidine staining reveals structures consist of coarse, irregular, electron-dense cores (*) lying within hollow vacuoles. The eight cored vacuoles (*) shown in this field average 2.48 ± 0.51 μm in diameter. B: A single vacuole-bearing lesion identified as above but stained with colloidal gold-linked secondary antibody shows the presence of gold particles within the matrix of the vacuole (*). Monoclonal antibody against Ckiδ does not label Hirano bodies (H) or cell nuclei (N) under these conditions. Scale bars, 2 μm.
Anti-CK1 Immunoreactivity Is Not Phosphatase-Sensitive
The pattern of immunoreactivity described above for Ckiα, δ, and ε suggests these enzymes are selectively enriched in both fibrillar and granulovacuolar lesions of AD. Yet these same lesions are rich in phosphoepitopes and cross-react strongly with monoclonal antibodies that recognize phosphate moieties as part of their epitope selectivity. 24,25 Although it has been shown that the monoclonal antibodies used to detect Ckiα, δ, and ε are monospecific for each protein kinase isoform and do not cross-react with other phosphoproteins on Western blots of crude brain extracts 9 (see Figure 1 ▶ ), it is still possible that phosphate moieties mediate the association of the monoclonal antibodies with each CK1 isoform studied. Therefore, the differences in CK1 immunoreactivity observed in AD and control tissue could reflect protein kinase phosphorylation (eg, arising from changes in trans- or autophosphorylation) rather than changes in the levels of protein kinase polypeptides. To examine this possibility, tissue sections were preincubated with or without alkaline phosphatase (an enzyme that hydrolyzes phosphoepitopes found in NFT) 16,26 before immunostaining with anti-CK1 monoclonal antibodies. The efficacy of the treatment is shown in Figure 6 ▶ . In the absence of phosphatase pretreatment, tissue sections derived from AD brain stained poorly with monoclonal antibody Tau1 (Figure 6A) ▶ . Pretreatment with alkaline phosphatase, however, removed covalently bound phosphate and unmasked the Tau1 epitope (Figure 6B) ▶ . 16 In contrast, alkaline phosphatase pretreatment did not attenuate the ability of anti-CK1 antibodies to react with either fibrillar or granulovacuolar lesions (shown for Ckiδ only in Figure 6, C and D ▶ ). Combined with the monospecificity illustrated in Figure 1 ▶ , these data suggest that the monoclonal antibodies used in this study do not bind CK1 isoforms through phosphoepitopes and that the large increases in anti-CK1 immunoreactivity seen in AD tissue reflect increased levels of CK1 polypeptides in disease.
Figure 6.
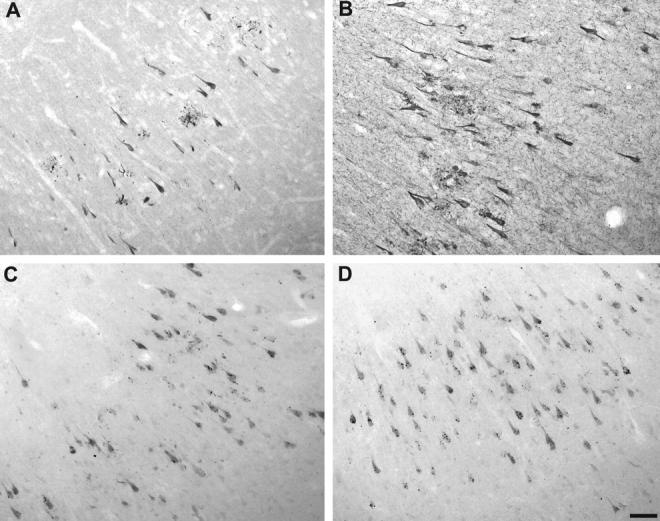
Ckiδ immunostaining is not alkaline phosphatase-sensitive. Serial sections (free-floating) of AD hippocampus pretreated without (A and C) or with (B and D) alkaline phosphatase were stained separately with the phosphorylation-sensitive, anti-tau monoclonal antibody Tau1 (A and B) or the anti-Ckiδ monoclonal antibody IC128 (C and D) as described in Materials and Methods. Although alkaline phosphatase treatment markedly augments immunostaining with monoclonal antibody Tau1, immunostaining with monoclonal antibody IC128 is unattenuated.
CK1 Isoforms Are Elevated in AD Tissue
To confirm that levels of Ckiα, δ, and ε increase in AD, each isoform was quantified by Western analysis of tissue homogenates prepared as described in Materials and Methods. The CA1 region of hippocampus was chosen for analysis because it is a rich source of both GVD and fibrillar pathologies. 27 As shown in Figure 7, A–C ▶ , the results confirm that the strong immunohistochemical staining of individual CK1 isoforms in AD hippocampus results from elevations in the amounts of these protein kinases, not from cross-reactivity with other cell consituents. Quantitation of Western blots revealed elevations in CK1 levels ranging from ∼2.4-fold for Ckiα to about ninefold for Ckiε to ∼33-fold for Ckiδ (Figure 7E) ▶ . In contrast, levels of type IIIβ tubulin, a neuron-specific form of tubulin, 10 did not differ between the AD and age-matched control groups (Figure 7, D and E) ▶ . Together, these data suggest that the CK1 family of enzymes, in particular the Ckiδ isoform, is greatly elevated in AD and associated with neurofibrillary and granulovacuolar lesions.
Figure 7.

Protein kinase levels are elevated in AD hippocampus relative to nondemented controls. Equal volumes of crude homogenates prepared from fresh-frozen AD and control CA1 regions of hippocampus were subjected to Western analysis with monoclonal antibodies against Ckiα (A), Ckiδ (B), Ckiε (C), and ClassIIIβ tubulin (D). AD cases: Lanes 1, 4, 5, 6, and 7. Control cases: Lanes 2, 3, 8, and 9. E: Normalization of the data (A-D) for protein content reveals elevation in levels of Ckiα (2.4-fold), Ckiδ (33-fold), and Ckiε (9-fold) relative to age-matched control hippocampus, but tubulin levels are not altered. Bars represent mean ± SD of duplicate determinations from five AD and four control homogenates prepared as described in Materials and Methods.
Discussion
These data establish a new molecular link between the fibrillar and granulovacuolar pathologies of AD: both intracellular lesions accumulate CK1 protein kinases. On the basis of immunostaining, levels of Ckiα, δ, and ε are greatest in GVD bodies, followed by NPs, followed by NFTs. In the former lesion, CK1 isoforms are sequestered from the neuronal cytoplasm within the matrix of vacuoles. In neurofibrillary lesions, CK1 isoforms are positioned appropriately to mediate tau hyperphosphorylation, a reaction they catalyze in vitro. 28 The association between CK1 and tau filaments appears to be physical, as it is maintained through stringent purification methods. 9 As a result, at least one CK1 isoform, Ckiα, is a major co-isolate of purified tau filaments, comprising as much as 0.5% (w/w) of immunopurified preparations.
Normally, Ckiα, δ, and ε are relatively low-abundance enzymes, differentially distributed in brain and ranging from 0.002–0.003% (w/w) of total cellular protein. 9 With the appearance of AD pathology, however, CK1 levels increase markedly, with one isoform, Ckiδ, rising >30-fold in the CA1 sector of Ammon’s Horn. This is the first demonstration of a phosphotransferase being elevated by an order of magnitude in authentic AD tissue. It will be important to assess whether these increases result from changes in transcriptional regulation or from changes in protein turnover and whether they are accompanied by increases in CK1 activity. On the basis of immunohistochemical localization, the increase in protein kinase levels parallels the appearance of AD-related lesions.
Other links between GVD bodies and tau filaments have been proposed previously. Chief among these are tau and neurofilament proteins. 24,29 Yet despite clear evidence for tau protein in neurofibrillary lesions, its presence in GVD bodies is controversial. Although some monoclonal antibodies raised against PHF-tau label GVD bodies strongly, 24,25 it has not been possible to identify tau or neurofilament peptides directly in these lesions. 30 On the contrary, it appears that these antibodies are capable of cross-reacting with phosphoepitopes found within unrelated phosphoproteins, 16 and that neither tau- nor neurofilament-derived polypeptides are abundant in GVD bodies. 30 In contrast, we have shown that CK1 immunoreactivity is not mediated by phosphoepitopes and that the appearance of GVD and neurofibrillary lesions is accompanied by major increases in the amounts of these protein kinases. Because of their location, the CK1 isoforms identified here may play a role in generating the abundant phosphoepitopes found in both GVD and neurofibrillary lesions.
Another protein found consistently in NFTs and NPs, 31 and frequently in GVD, 30 is ubiquitin. The physiological importance of the latter observation is unclear, as ubiquitin is a key component of proteosome-mediated proteolysis 32 unrelated to the autophagic lysosomal pathway thought to underlie GVD body formation. 33 Autophagy is distinguished from other degradative processes in that it involves cytoplasmic sequestration and employs a modification cascade distinct from the ubiquitin pathway. 34 Yet because it functions by a relatively nonselective volume uptake mechanism, most organelles and macromolecules are subject to vacuolar isolation in proportion to their cytoplasmic abundance. This may account for the inconsistent presence of ubiquitin in GVD bodies.
Similarly, the association of CK1 isoforms with GVD bodies and tau filaments reported here may reflect their abundance within degenerating neurons or nonspecific trapping within proteinaceous aggregates. Yet the high levels of CK1 found in these lesions, coupled with the emerging biological functions of CK1, suggests both associations may be of regulatory significance. First, the budding yeast homologue of Ckiδ, the CK1 isoform most upregulated in AD hippocampus, functions as a negative regulator of vesicle budding from the endoplasmic reticulum. 35 Thus, Ckiδ is positioned to participate in the pathological formation of GVD bodies, which are enclosed by membranes morphologically related to and potentially derived from the endoplasmic reticulum. Second, overexpression of CK1 isoforms in budding yeast can suppress trafficking defects arising from null mutants of guanine nucleotide-binding proteins or their effectors. 36 Although particulate forms of CK1 are normally essential for budding yeast viability, their loss can be complemented by null mutations in adapter proteins associated with clathrin-mediated transport. 37 Together these data implicate roles for CK1 homologues in the modulation of intracellular trafficking and participation in transport-dependent cellular processes such as autophagy, secretion, and phagocytosis. Other phenotypes associated with CK1 in lower eukaryotes, including control of DNA repair, 38 cell morphology, 39 and circadian rhythm, 40,41 may result indirectly from their role in intracellular transport. For example, DNA repair defects can result from impaired protein turnover. 42
Although the precise molecular mechanism by which CK1 isoforms modulate trafficking is unknown, the intracellular distribution of CK1 isoforms investigated to date suggests they exert their effects at the level of the cytoskeleton. First, in cycling cells, Ckiα colocalizes with both the mitotic spindle and the centrosome, revealing that this isoform has access to at least a subpopulation of microtubules before filament formation. 43 Second, Ckiα copurifies with neurofilament proteins, suggesting a direct interaction with these cytoskeletal proteins in situ. 44,45 Third, deletion of Ckiδ homologues in fission yeast results in enhanced sensitivity to benomyl, a microtubule depolymerization agent. 46 These data suggest that interaction with the cytoskeleton is a normal function of some CK1 isoforms.
Members of the CK1 family of protein kinases share a common structure and enzymology, suggesting they serve a similar function in different contexts. 47,48 Each of the three isoforms investigated here colocalizes with intracellular AD pathology and retains the unique phosphate-directed substrate selectivity characteristic of family members. 9 Yet the data presented here suggest that Ckiδ is the isoform most closely linked with the appearance of AD pathology. First, its levels are dramatically elevated in AD versus normal control tissue, rising over 30-fold. In contrast, increases in Ckiα levels averaged only 2.4-fold. Second, Ckiδ is selectively localized to neuronal populations in normal tissue, unlike Ckiα and Ckiε, which appear widely distributed. Third, its function as currently defined in model systems is associated with trafficking events occurring at the endoplasmic reticulum. Although the gene encoding Ckiδ maps to 17q25, 49 in the vicinity of the locus for frontotemporal dementia with parkinsonism, chromosome 17 type (17q21–22), 50 no familial frontal lobar atrophies have been linked to Ckiδ.
A hypothetical model for a role of CK1 in AD pathogenesis is presented in Figure 8 ▶ . We propose that breakdown of the microtubule cytoskeleton 51 beginning in early stage AD leads to disruption of motor-driven intracellular transport 52 and results in loss of functionality of intracellular organelles such as endoplasmic reticulum and the Golgi apparatus. 53,54 Increased CK1 levels, particularly of Ckiδ, may be a physiological response of neurons to suppress these trafficking defects in AD, much as they do in lower eukaryotes. 35 Elevated levels of CK1 could then hyperphosphorylate substrates at their point of action, the cytoskeleton. Because hyperphosphorylation of tau leads to microtubule dissolution, 51 which in turn can produce Golgi fragmentation and other trafficking defects, 52,54 elements of a pathological positive feedback loop are in place. The population of neurons affected may dictate whether the product of the loop is manifested primarily as neurofibrillary or granulovacuolar degeneration.
Figure 8.

A hypothetical model for the role of CK1 isoforms in AD pathogenesis. See text for details.
CK1 enzymes are important regulators of normal cell biology, and the discovery that members of this protein kinase family are highly elevated in AD suggests they play an important role in pathology as well. Yet these protein kinases are also valuable as markers. A robust biochemical marker for GVD bodies will facilitate their purification, assessment of their molecular composition, precise mapping of their distribution in neurodegenerative diseases, and insight into their origin.
Acknowledgments
We thank Prof. E. J. Mufson, Dr. B. Quinn, A. Guillozet, and M. King for guidance in hippocampal dissection and immunostaining and Prof. A. Frankfurter for monoclonal antibody Tuj1. Generous access to AD tissue was provided by the Northwestern (AG13854) and Rush (AG10161) Alzheimer’s Disease Centers.
Footnotes
Address reprint requests to Jeff Kuret, Ph.D., Ohio State University College of Medicine, Department of Medical Biochemistry, 1060 Carmack Road, Columbus, OH 43210. E-mail: kuret.3@osu.edu.
Supported by grants from the National Institutes of Health (AG09466, AG14452, GM56292) and the Alzheimer’s Association (RG2–29-076).
References
- 1.Okamoto K, Hirai S, Iizuka T, Yanagisawa T, Watanabe M: Reexamination of granulovacuolar degeneration. Acta Neuropathol 1991, 82:340-345 [DOI] [PubMed] [Google Scholar]
- 2.Goedert M: Tau protein and the neurofibrillary pathology of Alzheimer’s disease. Trends Neurosci 1993, 16:460-465 [DOI] [PubMed] [Google Scholar]
- 3.Khatoon S, Grundke-Iqbal I, Iqbal I: Brain levels of microtubule-associated protein tau are elevated in Alzheimer’s disease: a radioimmuno-slot-blot assay for nanograms of the protein. J Neurochem 1992, 59:750-753 [DOI] [PubMed] [Google Scholar]
- 4.Ksiezak-Reding H, Liu WK, Yen SH: Phosphate analysis and dephosphorylation of modified tau associated with paired helical filaments. Brain Res 1992, 597:209-219 [DOI] [PubMed] [Google Scholar]
- 5.Lindwall G, Cole RD: Phosphorylation affects the ability of tau protein to promote microtubule assembly. J Biol Chem 1984, 255:5301-5305 [PubMed] [Google Scholar]
- 6.Braak E, Braak H, Mandelkow E-M: A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol 1994, 87:554-567 [DOI] [PubMed] [Google Scholar]
- 7.Hanger DP, Betts JC, Loviny TLF, Blackstock WP, Anderton BH: New phosphorylation sites identified in hyperphosphorylated tau (paired helical filament-tau) from Alzheimer’s disease brain using nanoelectrospray mass spectrometry. J Neurochem 1998, 71:2465-2476 [DOI] [PubMed] [Google Scholar]
- 8.Sperber BR, Leight S, Goedert M, Lee VMY: Glycogen synthase kinase-3 β phosphorylates tau protein at multiple sites in intact cells. Neurosci Lett 1995, 197:149-153 [DOI] [PubMed] [Google Scholar]
- 9.Kuret J, Johnson GS, Cha D, Christenson ER, DeMaggio AJ, Hoekstra MF: Casein kinase 1 is tightly associated with paired helical filaments isolated from Alzheimer’s disease brain. J Neurochem 1997, 69:2506-2515 [DOI] [PubMed] [Google Scholar]
- 10.Lee MK, Tuttle JB, Rebhun LI, Cleveland DW, Frankfurter A: The expression and post-translational modification of a neuron-specific β-tubulin isotype during chick embryogenesis. Cell Motil Cytoskeleton 1990, 17:118-132 [DOI] [PubMed] [Google Scholar]
- 11.Papasozomenos SC, Binder LI: Phosphorylation determines two distinct species of Tau in the central nervous system. Cell Motil Cytoskeleton 1987, 8:210-226 [DOI] [PubMed] [Google Scholar]
- 12.Carmel G, Mager EM, Binder LI, Kuret J: The structural basis of monoclonal antibody Alz50’s selectivity for Alzheimer’s disease pathology. J Biol Chem 1996, 271:32789-32795 [DOI] [PubMed] [Google Scholar]
- 13.Mirra SS, Hart MN, Terry RD: Making the diagnosis of Alzheimer’s disease: a primer for practicing pathologists. Arch Pathol Lab Med 1993, 117:132-144 [PubMed] [Google Scholar]
- 14.Mufson EJ, Lavine N, Jaffar S, Kordower JH, Quirion R, Saragovi HU: Reduction in p140-TrkA receptor protein within the nucleus basalis and cortex in Alzheimer’s disease. Exp Neurol 1997, 146:91-103 [DOI] [PubMed] [Google Scholar]
- 15.Evers P, Uylings HBM, Suurmeijer AJH: Antigen retrieval in formaldehyde-fixed human brain tissue. Methods 1998, 15:133-140 [DOI] [PubMed] [Google Scholar]
- 16.Grundke-Iqbal I, Iqbal K, Tuna Y-C, Quinlan M, Wisniewski HM, Binder LI: Abnormal phosphorylation of the microtubule-associated protein, tau in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA 1986, 83:4913-4917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chan J, Aoki C, Pickel VM: Optimization of differential immunogold-silver and peroxidase labeling with maintenance of ultrastructure in brain sections before plastic embedding. J Neurosci Methods 1990, 33:113-127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smiley JF, Goldman-Rakic PS: Silver-enhanced diaminobenzidine-sulfide (SEDS): a technique for high-resolution immunoelectron microscopy demonstrated with monoamine immunoreactivity in monkey cerebral cortex and caudate. J Histochem Cytochem 1993, 41:1393-1404 [DOI] [PubMed] [Google Scholar]
- 19.Fish KJ, Cegielska A, Getman ME, Landes GM, Virshup DM: Isolation and characterization of human casein kinase I epsilon (CKI), a novel member of the CKI gene family. J Biol Chem 1995, 270:14875-14883 [DOI] [PubMed] [Google Scholar]
- 20.Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT: Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 1992, 42:631-639 [DOI] [PubMed] [Google Scholar]
- 21.Ball MJ: Topographic distribution of neurofibrillary tangles and granulovacuolar degeneration in hippocampal cortex of aging and demented patients. Acta Neuropathol 1978, 42:73-80 [DOI] [PubMed] [Google Scholar]
- 22.Xu M, Shibayama H, Kobayashi H, Yamada K, Ishihara R, Zhao P, Takeuchi T, Yoshida K, Inagaki T, Nokura K: Granulovacuolar degeneration in the hippocampal cortex of aging and demented patients: a quantitative study. Acta Neuropathol 1992, 85:1-9 [DOI] [PubMed] [Google Scholar]
- 23.Nixon RA, Cataldo AM: The endosomal-lysosomal system of neurons: new roles. Trends Neurosci 1995, 18:489-496 [DOI] [PubMed] [Google Scholar]
- 24.Dickson DW, Liu Y, Kress Y, Ku J, DeJesus O, Yen S-HC: Phosphorylated tau immunoreactivity of granulovacuolar bodies (GVB) of Alzheimer’s disease: localization of two amino terminal tau epitopes in GVB. Acta Neuropathol 1993, 85:463-470 [DOI] [PubMed] [Google Scholar]
- 25.Vincent I, Zheng J-H, Dickson DW, Kress Y, Davies P: Mitotic phosphoepitopes precede paired helical filaments in Alzheimer’s disease. Neurobiol Aging 1998, 19:287-296 [DOI] [PubMed] [Google Scholar]
- 26.Vincent I, Rosado M, Davies P: Mitotic mechanisms in Alzheimer’s disease? J Cell Biol 1996, 132:413-425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ball MJ, Nuttall K: Topography of neurofibrillary tangles and granulovacuoles in hippocampi of patients with Down’s syndrome: quantitative comparison with normal ageing and Alzheimer’s disease. Neuropathol Appl Neurobiol 1981, 7:13-20 [DOI] [PubMed] [Google Scholar]
- 28.Singh TJ, Grundke-Iqbal I, Iqbal K: Phosphorylation of tau protein by casein kinase-1 converts it to an abnormal Alzheimer-like state. J Neurochem 1995, 64:1420-1423 [DOI] [PubMed] [Google Scholar]
- 29.Dickson DW, Ksiezak-Reding H, Davies P, Yen S-H: A monoclonal antibody that recognizes a phosphorylated epitope in Alzheimer neurofibrillary tangles, neurofilaments and tau proteins immunostains granulovacuolar degeneration. Acta Neuropathol 1987, 73:254-258 [DOI] [PubMed] [Google Scholar]
- 30.Lübke U, Mercken M, Vandermeeren M, Ceuterick-de-Groote C, Vanmechelen E, Martin J-J, Cras P: Comparative study of granulovacuolar degeneration in neurodegenerative diseases. Adv Biosci 1993, 87:149-150 [Google Scholar]
- 31.Ii K, Ito H, Tanaka K, Hirano A: Immunocytochemical co-localization of the proteasome in ubiquintinated structures in neurodegenerative diseases and the elderly. J Neuropathol Exp Neurol 1997, 56:125-131 [DOI] [PubMed] [Google Scholar]
- 32.Haas AL, Sipemann T: Pathways of ubiquitin conjugation. FASEB J 1997, 11:1257-1268 [DOI] [PubMed] [Google Scholar]
- 33.Mortimore GE, Miotto G, Venerando R, Kadowaki M: Autophagy. Subcell Biochem 1996, 27:93-135 [DOI] [PubMed] [Google Scholar]
- 34.Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Kionsky DJ, Ohsumi M, Oshumi Y: A protein conjugation system essential for autophagy. Nature 1998, 395:395-398 [DOI] [PubMed] [Google Scholar]
- 35.Murakami A, Kimura K, Nakano A: The inactive form of a yeast casein kinase I suppresses the secretory defect of the sec12 mutant. J Biol Chem 1998, 274:3804-3810 [DOI] [PubMed] [Google Scholar]
- 36.Wang X, Hoekstra MF, DeMaggio AJ, Dhillon N, Vancura A, Kuret J, Johnston GC, Singer RA: Prenylated isoforms of yeast casein kinase I, including that encoded by the novel YCK3 gene, suppress the gcs1 blockage of cell proliferation from stationary phase. Mol Cell Biol 1996, 16:5375-5385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Panek HR, Stepp JD, Engle HM, Marks KM, Tan PK, Lemmon SK, Robinson LC: Suppressors of YCK-encoded yeast casein kinase 1 deficiency define the four subunits of a novel clathrin AP-like complex. EMBO J 1996, 16:4194-4204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoekstra MF, Liskay RM, Ou AC, Demaggio AJ, Burbee DG, Heffron F: HRR25, a putative protein kinase from budding yeast: association with repair of damaged DNA. Science 1991, 253:1031-1034 [DOI] [PubMed] [Google Scholar]
- 39.Robinson LC, Menold MM, Garrett S, Culbertson MR: Casein kinase I-like protein kinases encoded by YCK1 and YCK2 are required for yeast morphogenesis. Mol Cell Biol 1993, 13:2870-2881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Price JL, Blau J, Rothenfluh A, Abodeely M, Kloss B, Young MW: Double-time is a novel Drosophila clock gene that regulates PERIOD protein accumulation. Cell 1998, 84:83-95 [DOI] [PubMed] [Google Scholar]
- 41.Kloss B, Price JL, Saez L, Blau J, Rothenfluh A, Wesley CS, Young MW: The Drosophila cock gene double-time encodes a protein closely related to human casein kinase 1. Cell 1998, 84:97-107 [DOI] [PubMed] [Google Scholar]
- 42.Schauber C, Chen L, Tongaonkar P, Vega I, Lambertson D, Potts W, Madura K: Rad23 links DNA repair to the ubiquitin/proteasome pathway. Nature 1998, 391:715-718 [DOI] [PubMed] [Google Scholar]
- 43.Brockman JL, Gross SD, Sussman MR, Anderson RA: Cell cycle-dependent localization of casein kinase I to mitotic spindles. Proc Natl Acad Sci USA 1992, 89:9454-9458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Floyd CC, Grant P, Gallant PE, Pant HC: Principal neurofilament-associated protein kinase in squid axoplasm is related to casein kinase I. J Biol Chem 1991, 266:4987-4994 [PubMed] [Google Scholar]
- 45.Hollander BA, Bennett GS, Shaw G: Localization of sites in the tail domain of the middle molecular mass neurofilament subunit phosphorylated by a neurofilament-associated kinase and by casein kinase I. J Neurochem 1996, 66:412-420 [DOI] [PubMed] [Google Scholar]
- 46.Dhillon N, Hoekstra MF: Characterization of two protein kinases from Schizosaccharomyces pombe involved in the regulation of DNA repair. EMBO J 1994, 13:2777-2787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu R, Carmel G, Sweet RM, Kuret J, Cheng X: Crystal structure of casein kinase-1, a phosphate-directed protein kinase. EMBO J 1995, 14:1015-1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hoekstra M, Dhillon N, Carmel G, DeMaggio AJ, Lindberg RA, Hunter T, Kuret J: Budding and fission yeast casein kinase I isoforms have dual-specificity protein kinase activity. Mol Biol Cell 1994, 5:877-886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kusuda J, Hidari N, Hirai M, Hashimoto K: Sequence analysis of the cDNA for the human casein kianse 1 δ (CSNK1D) gene and its chromosomal localization. Genomics 1996, 32:140-143 [DOI] [PubMed] [Google Scholar]
- 50.Bird TD, Wijsman EM, Nochlin D, Leehey M, Sumi SM, Payami H, Poorkaj P, Nemens E, Rafkind M, Schellenberg GD: Chromosome 17 and hereditary dementia: linkage studies in three non-Alzheimer families and kindreds with late-onset FAD. Neurology 1997, 48:949-954 [DOI] [PubMed] [Google Scholar]
- 51.Alonso AC, Grundke-Iqbal I, Iqbal K: Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med 1996, 2:783-787 [DOI] [PubMed] [Google Scholar]
- 52.Ebneth A, Godemann R, Stamer K, Illenberger A, Trinczek B, Mandelkow E-M, Mandelkow E: Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer’s disease. J Cell Biol 1998, 143:777-794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stieber A, Mourelatos Z, Gonatas NK: In Alzheimer’s disease the Golgi apparatus of a population of neurons without neurofibrillary tangles is fragmented and atrophic. Am J Pathol 1996, 148:415-426 [PMC free article] [PubMed] [Google Scholar]
- 54.Yang W, Storrie B: Scattered Golgi elements during microtubule disruption are initially enriched in trans-Golgi proteins. Mol Biol Cell 1998, 9:191-207 [DOI] [PMC free article] [PubMed] [Google Scholar]