

Abstract
Glial cytoplasmic inclusions (GCI) are the hallmark of multiple system atrophy (MSA), a rare movement disorder frequently associated with autonomic dysfunction. In this study of 21 cases of MSA, GCI were consistently immunoreactive for α-synuclein and double-immunostained for ubiquitin and oligodendroglial markers, but not glial fibrillary acidic protein. No statistically significant difference was found in the density of GCI in various brain regions in the two forms of MSA, striatonigral degeneration (SND) and olivopontocerebellar atrophy (OPCA). Postmortem brain samples from 9 cases of MSA were fractionated according to solubility in buffer, Triton-X 100, sodium dodecyl sulfate (SDS), and formic acid, and α-synuclein immunoreactivity was measured in Western blots. Total α-synuclein immunoreactivity was increased in MSA compared to controls, with no statistically significant difference between SND and OPCA. Most of the increase was due to α-synuclein in SDS fractions. In controls this fraction had little or no immunoreactivity. In 7 cases and 4 controls correlations were investigated between quantitative neuropathology and biochemical properties of α-synuclein. Surprisingly, the amount of SDS-soluble α-synuclein correlated poorly with the number of GCI in adjacent sections. Furthermore, areas with few or no GCI unexpectedly had abundant SDS-soluble α-synuclein. These findings provide evidence that modifications of α-synuclein in MSA may be more widespread than obvious histopathology. Moreover, these alterations may constitute a biochemical signature for the synucleinopathies.
Graham and Oppenheimer coined the term multiple system atrophy (MSA) for a nonheritable neurodegenerative disease characterized by Parkinsonism, cerebellar ataxia, and idiopathic orthostatic hypotension. 1 The concept of MSA unifies three separate entities, namely, olivopontocerebellar atrophy (OPCA), Shy-Drager syndrome, and striatonigral degeneration (SND). MSA has an average age of onset between 30 and 50 years of age and a disease duration of decades. 2 There are no known genetic risk factors or genetic mutations in MSA. 3 The predominant signs and symptoms depend on neuronal systems predominantly affected; cerebellar ataxia is prominent in OPCA, whereas Parkinsonism and orthostatic hypotension are major features of SND. In epidemiological studies about 3 to 7% of Parkinsonism is due to MSA. 4
In addition to variable atrophy of pons, inferior olive, and cerebellum, softening and discoloration of the posterolateral putamen, and loss of pigment in the ventrolateral substantia nigra, white matter pathology is increasingly recognized in MSA. The brunt of the white matter pathology is in fiber tracts in affected areas; however, recent studies suggest that white matter pathology may be more widespread than previously suspected. 5 Given the consistent presence of white matter pathology in MSA, it is not entirely surprising that oligodendroglial changes are detected. Lantos and coworkers first described oligodendroglial inclusions in MSA and coined the term glial cytoplasmic inclusions (GCI) 6 for argyrophilic and ubiquitin-immunoreactive flame- or sickle-shaped inclusions in oligodendrocytes. GCI are variably immunoreactive for tubulin, αB-crystallin, and tau, 7 in addition to ubiquitin. At the ultrastructural level, GCI are non-membrane-bound cytoplasmic inclusions composed of filaments (20–40 nm) and granular material. 6,8-10 They are distinctly different from filamentous oligodendroglial inclusions, called coiled bodies, found in other neurodegenerative diseases, including progressive supranuclear palsy, corticobasal degeneration, and Braak’s argyrophilic grain disease. 11-14 Although tau antibodies readily stain the latter inclusions, GCI are negative for tau. Recent studies suggest that if GCI contain tau, it is largely nonphosphorylated. 15 More recently, GCI have been shown to have immunoreactivity for α-synuclein. 7 We independently made this observation 16 and, meanwhile, have explored the biochemical basis for immunoreactivity of GCI for α-synuclein. 17-22 In a previous report, 20 a preliminary biochemical analysis of α-synuclein was reported in MSA. The present study represents a significant extension of biochemical studies of α-synuclein in MSA.
α-synuclein, also referred to as precursor of the non-amyloid component of plaques (NACP), is a 140-amino acid protein that is normally present in presynaptic terminals in the human brain. 23 It is similar to microtubule-associated protein tau in terms of thermal stability and being a natively unfolded protein. 24 The exact subcellular distribution of α-synuclein is not known. While non-neuronal cells are known to express α-synuclein, immunostaining of brains shows almost exclusive localization to gray matter. It remains unknown to what extent normal oligodendrocytes express α-synuclein and what function it may play in non-neuronal cells. In normal brain extracts α-synuclein distributes almost entirely in a soluble cytosolic fraction, 25 but it is also detected in membrane fractions, including those rich in vesicles and synaptic membranes. 23 The present study reports biochemical analyses of α-synuclein in postmortem brain tissue in MSA and correlates biochemical findings with quantitative regional neuropathology. The major findings suggest that MSA has widespread modifications of α-synuclein and that these biochemical changes correlate imperfectly with neuropathology.
Materials and Methods
Case Material
The cases used for the various analyses are listed in Table 1 ▶ . Cases are subdivided according to whether the predominant clinical and pathological findings were those of OPCA or SND. A total of 21 cases of MSA were studied, including 13 men and 8 women with an average age of 64.9 ± 9.0 years. Fifteen cases had SND and 6 had OPCA. Frozen tissue was available from 9 cases for fractionation and Western blot analysis. The average postmortem interval for the MSA cases used for biochemical studies was 8.4 ± 4.4 hours. Control cases were slightly older, 76.4 ± 7.3 (t-test, P < 0.05), but the postmortem delay, 10.0 ± 5.1 hours, was not significantly different between cases and controls. For quantitative Western blot analyses a subset of 7 cases of MSA were compared to 4 controls matched for age and postmortem delay. The quantitative analyses were repeated at least twice for each case and averaged; comparison of cases and controls was based on the average of these averages. Fixed tissue was available for immunocytochemistry on 20 cases, including the 7 cases used for quantitative Western blot studies.
Table 1.
MSA and Controls
| MSA type | Source | Age | Sex | PMI (hr) | Fixed & paraffin | Frozen |
|---|---|---|---|---|---|---|
| SND | UMDNJ-RWJMS | 70 | M | + | ||
| SND | UMDNJ-RWJMS | 73 | F | + | ||
| SND | UMDNJ-RWJMS | 66 | M | + | ||
| SND | UMDNJ-RWJMS | 63 | F | + | ||
| SND | UMDNJ-RWJMS | 60 | M | + | ||
| SND | Mayo Clinic | 73 | M | + | ||
| SND | Mayo Clinic | 68 | F | + | ||
| SND | Mayo Clinic | 71 | F | + | ||
| SND | Mayo Clinic | 54 | M | + | ||
| SND | Mayo Clinic | 83 | M | + | ||
| SND | Mayo Clinic | 68 | M | + | ||
| OPCA | Univ. Virginia | 49 | M | + | ||
| OPCA | Mayo Clinic | 58 | F | 16 | + | + |
| OPCA | Mayo Clinic | 68 | M | 2 | + | + |
| OPCA | Univ. Michigan | 63 | M | 13 | + | + |
| OPCA | Univ. Michigan | 65 | F | 9 | + | + |
| OPCA | Univ. Michigan | 77 | F | 5 | + | + |
| SND | Univ. Michigan | 62 | F | 6 | + | + |
| SND | Univ. Michigan | 52 | M | 6 | + | + |
| SND | Univ. Michigan | 72 | M | 5 | + | + |
| SND | Univ. Michigan | 49 | M | 14 | + | |
| Summary | 64.9± 9.0 | 8 F & 13 M | 8.4± 4.4 | 20 | 9 |
| Controls for biochemical studies | ||||
|---|---|---|---|---|
| Path. dx | Source | Age | Sex | PMI (hr) |
| N | A Einstein Coll Med | 82 | F | 14 |
| AD | A Einstein Coll Med | 71 | M | 10 |
| N | A Einstein Coll Med | 88 | F | 4 |
| N | A Einstein Coll Med | 69 | M | 16 |
| PND | Mayo Clinic | 72 | M | 6 |
| Summary | 76.4± 7.3 | 2 F, 3 M | 10± 5.1 |
PMI, postmortem interval; SND, striatonigral degeneration; OPCA, olivopontocerebellar atrophy; N, normal; AD, Alzheimer’s disease; PND, pallidonigral degeneration.
α-Synuclein Antibodies
A 19-amino acid peptide, DQLGKNEEGAPQEGILED-C, with a cysteine residue at its C-terminus and corresponding to α-synuclein amino acids 98–115 (NACP98), and a 10-amino acid peptide, C-GILEDMPVD, with a cysteine residue at its N-terminus and corresponding to α-synuclein amino acids 111–119 (NACP111), were used for antibody production. The peptides were coupled to an equal amount of maleimide-activated keyhole limpet hemocyanin (KLH; Pierce Chemicals, Rockford, IL) as recommended by the manufacturer. Female New Zealand White rabbits were immunized. For most of the Western blot studies and all of the quantitative immunoanalyses, NACP98 was used as affinity-purified antibody, which was prepared by an affinity column with NACP98 peptide immobilized to sulfolink gel (Pierce). Both NACP98 and NACP111 were used for immunocytochemistry as diluted antisera.
The specificity of the antibodies was tested with immunocytochemistry and Western blots. Two previously characterized monoclonal antibodies specific for α-synuclein were also used to confirm the results with the polyclonal antibodies. Dr. T. Iwatsubo of the University of Tokyo provided a previously characterized monoclonal antibody (LB509 26 ; Zymed, Inc., South San Francisco, CA), and the other was purchased from Transduction Laboratories (Lexington, KY). The latter antibody was raised to a synthetic peptide spanning amino acids 15–123 of α-synuclein.
Tissue Processing
Tissue processed in a variety of ways was used for immunocytochemical studies. Paraffin-embedded tissue was available from cases from Mayo Clinic Jacksonville (MCJ), Mayo Clinic Rochester (MCR), the Robert Wood Johnson School of Medicine (RWJSM), and the University of Michigan. Fixed tissue was available on another subset of cases, including one case from the University of Virginia (Charlottesville, VA) that was fixed briefly at the time of autopsy in Bouin’s fixative. This case and two cases fixed in 4% paraformaldehyde were used for staining of free-floating vibratome sections.
Immunocytochemistry
Sections of brain were stained with single and double immunocytochemical methods. Sections were double stained with rabbit antisera to α-synuclein and several different mouse monoclonal antibodies, including glial fibrillary acidic protein (GFAP; BioGenex, San Ramon, CA), myelin basic protein (MBP; Boehringer-Mannheim, Indianapolis, IN), Leu7 (Becton-Dickinson, San Jose, CA); C4d (Biogenesis, Poole, UK), ubiquitin (5–25 and 3–39, Senentek, St. Louis, MO), phospho-tau (PHF-1; Peter Davies, Albert Einstein College of Medicine, New York, NY) and synaptophysin (EP10; Peter Davies).
For paraffin sections, 5-μm-thick sections were deparaffinized in xylene and alcohols and incubated in 3% H2O2 for 30 minutes to block endogenous peroxidase and then in 5% normal goat serum for 10 minutes to block nonspecific antibody binding. The primary antibodies were incubated overnight at 20°C. Antibody binding was detected with the avidin biotin complex method (Vector Labs, Burlingame, CA). The chromogen was 3,3′diaminobenzidine (Sigma Chemical, St. Louis, MO). The sections were lightly counterstained with hematoxylin and dehydrated, and coverslips were mounted with Permount (Fisher Scientific, Pittsburgh, PA).
For double labeling experiments with light microscopy, sections were incubated simultaneously with both antibodies. After extensive washing in buffer, the antibodies were detected with peroxidase- and alkaline phosphatase-labeled isotype-specific secondary antibodies (Southern Biotechnology, Birmingham, AL). The chromogens 3,3′diaminobenzidine and 5-bromo-4-chloro-3-indolyl-phosphate (BCIP Nitro blue tetrazolium, Sigma) were developed sequentially.
Laser confocal microscopy was performed on 40-μm-thick vibratome sections of formaldehyde- or paraformaldehyde-fixed tissue. The sections were incubated in 0.4% Triton X-100 and 5% normal goat serum before overnight incubation with rabbit antisera to α-synuclein and mouse antibodies (ubiquitin, tau, GFAP, or C4d) antibodies. After several buffer washes the antibodies were detected with rhodamine- and fluorescein-conjugated isotype-specific goat secondary antibodies. The sections were mounted on glass slides and coverslips mounted with Aqua-Mount (Lerner Labs., Pittsburgh, PA). The slides were viewed with an Olympus Fluoview BX50 confocal microscope.
Distribution of α-Synuclein in Brain Fractions
Brain proteins were partitioned into four fractions according to their solubility in phosphate buffered saline (PBS), Triton X-100, sodium dodecyl sulfate (SDS), or formic acid. The posterolateral putamen was dissected from frozen brain slabs of 7 cases of MSA, as well as 4 control brains, including 3 normal elderly and a case of Alzheimer’s disease. The brain tissue samples were homogenized in 10 volumes (gram weight per volume) of PBS+, a solution of PBS, pH 7.4, containing a mixture of protease inhibitors and phosphatase inhibitors (25 mmol/L NaF, 25 mmol/L β-glycerophosphate, 0.1 mmol/L Na-vanadate, 1 mmol/L EDTA, 1 mmol/L EGTA, 1 mmol/L phenylmethylsulfonyl fluoride, 5μg/ml leupeptin). The homogenate was subsequently centrifuged at 1000 × g for 10 minutes. The supernatant (fraction S1) contained all of the α-synuclein immunoreactivity and was used for serial extractions. Before further extraction, the samples were normalized based on the amount of protein. Each S1 solution was diluted to a final protein concentration of 1 mg/ml based on protein determination by the Lowry method. 27 Equal amounts of S1 were then centrifuged at 100,000 × g for 1 hour to separate the soluble cytosolic fraction (S2) and insoluble pellet (P2). The P2 pellet was washed twice with PBS+ to remove S2 contaminants and then extracted with PBS+ containing 1% Triton X-100 with 0.5% deoxycholate and 0.1% SDS. After centrifugation at 100,000 × g for 1 hour, the supernatant fraction, S3, and the pellet, P3, were collected. The P3 pellet was washed twice with PBS+ to remove S3 contaminants. The P3 pellet was then extracted with PBS+ containing 2% SDS. After another centrifugation at 100,000 × g for 1 hour, the SDS-soluble fraction, S4, and the pellet, P4, were collected. The P4 pellet was washed with PBS+, and extracted with 70% formic acid. The formic acid extract was centrifuged at 100,000 × g for 1 hour. At this point, most samples had minimal or no pellet after centrifugation. The formic acid extract was dried in a SpeedVac, and the residue was dissolved in PBS+ as fraction S5.
For regional studies, 10 brain regions of a single MSA brain and an age- and postmortem interval-matched normal brain were used for comparison. The dissections included regions with few GCI (hippocampus, amygdala, and several cortical regions) as well as regions with many GCI (thalamus, globus pallidus, and corpus striatum). Because of the limited amount of starting material in the microdissections, 1% Triton X-100, 0.5% deoxycholate, and 0.1% SDS were added directly to S1 before centrifugation, and the resulting supernatant fraction is equivalent to S2 and S3.
For immunoblotting, each fraction was separated on precast 10 to 20% SDS gradient gels (BioRad, Hercules, CA) and transblotted onto nitrocellulose membrane. 28 The blots were probed first with primary antibodies and then with anti-IgG secondary antibody conjugated with horseradish peroxidase (Boehringer Mannheim, Indianapolis, IN). Select blots were reprobed with monoclonal antibodies to synaptophysin (EP10) or ubiquitin. 5-25 The immunoreactivity was detected by the Enhanced Chemiluminescence Plus system (ECL+, Amersham Life Science, Piscataway, NJ). The immunoreactive images of the blot were scanned and saved on the Storm 860 system (Molecular Dynamics Co., Sunnyvale, CA), and the densities of the immunoreactive bands were quantified using ImageQuant software (Molecular Dynamics).
Statistical analyses were performed with Microsoft Excel (t-tests and linear regression) and Jandel Scientific SigmaStat (Spearman correlation analysis, analysis of variance on ranks, and Wilcoxon signed rank test). All t-tests were two-tailed and a P value <0.05 was required for significance.
Results
Characterization of Polyclonal Antibodies to α-Synuclein
The two antibodies to α-synuclein, NACP98 and NACP111, gave consistent and reproducible staining of Lewy bodies and Lewy neurites (not shown). In cryostat sections fixed briefly in acetone the antibodies were specific at dilutions up to 1:20,000. For paraffin sections the antibodies were used at 1:1000–1:5000 dilution. Oligodendroglial staining was not observed in normal brains with any means of fixation and processing. Synaptic neuropil staining and staining of abnormal structures were completely abolished in immunoabsorption experiments. The antibodies worked very well on vibratome sections of tissue that had been fixed briefly in 4% paraformaldehyde or in Bouin’s fixative with prominent labeling of dystrophic neurites in Lewy body disease (LBD) in the latter fixative.
Double labeling experiments were performed with antibodies to tau, PHF-tau, and ubiquitin (not shown). As in other published studies, 29 there was virtually no overlap between α-synuclein staining and either tau or PHF-tau immunoreactive structures in brains that had both types of lesions. In contrast to tau, ubiquitin antibodies colabeled virtually all of the pathological structures that were stained with α-synuclein. There was no ubiquitin staining of synaptic structures.
α-Synuclein Immunoreactivity in MSA
The α-synuclein polyclonal antibodies stained inclusion bodies in neurons and glia in MSA (Figure 1) ▶ . All 20 cases of MSA that were available for immunocytochemistry had α-synuclein-immunoreactive GCI. Only a few of the cases had α-synuclein-immunoreactive neuronal inclusions, and neuronal inclusions were most often found in the pontine base. Such cases also often had a few α-synuclein-immunoreactive cell processes (Figure 1) ▶ .
Figure 1.
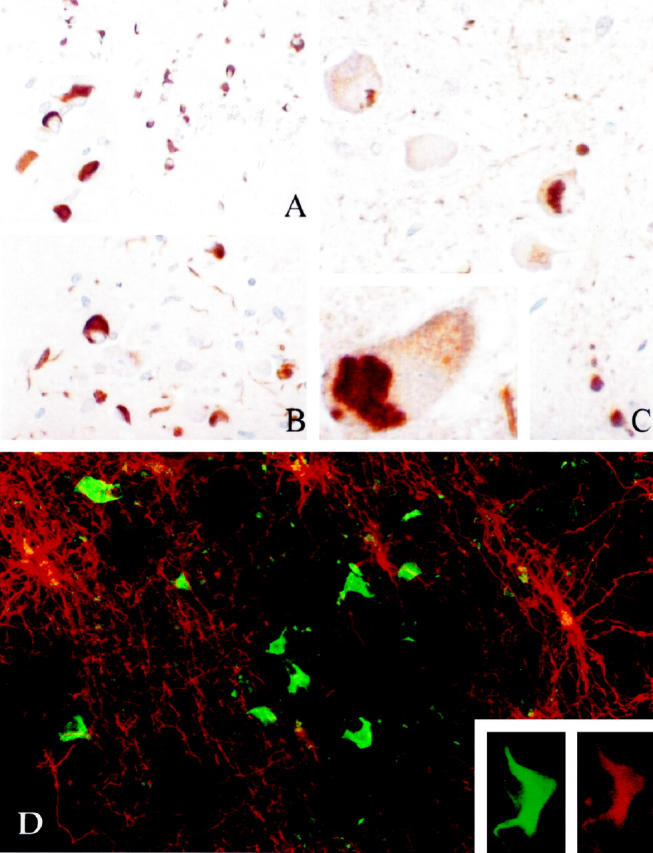
Immunostaining for α-synuclein in paraffin sections (A, B, and C) in MSA. Note many GCI in white matter in basal ganglia (A and inset). In the pons (B and C) was a mixture of glial and neuronal inclusions. The neuronal inclusions were cytosolic and pleomorphic (C and inset). D: Double immunostaining of Vibratome sections with GFAP (rhodamine filter, red) and α-synuclein (fluorescein filter, green). Note the absence of colocalization of the two signals. Inset shows a GCI that is double labeled with α-synuclein (fluorescein filter, green) and C4d (rhodamine filter, red).
Double labeling immunocytochemistry showed that GCI were positive for ubiquitin but negative for tau and GFAP. The several markers used to label oligodendroglia, including MBP, C4d, and Leu7, gave weak immunoreactivity of some of the inclusions (Figure 1) ▶ , due in part to the well-recognized lack of good cellular markers for mature oligodendrocytes.
Distribution of α-Synuclein-Immunoreactive GCI
The neuroanatomical distribution of the density of GCI (Figure 2) ▶ was determined by manual counting of immunostained sections of 7 cases (3 men and 4 women; 3 OPCA and 4 SND; average age 62.1 ± 9.4 years) by averaging the counts at 400× magnification in three representative fields. These same cases were used for quantitative immunoblot analysis (see below). There was a trend for OPCA cases to have fewer inclusions than SND cases, especially in the brain stem regions, but there was no statistically significant difference between the average GCI counts in SND and OPCA. The greatest density of GCI was in the basal ganglia in both OPCA and SND. Not only the putamen, but also the pallidum, had many inclusions. For this reason basal ganglia was the region chosen for more detailed biochemical studies.
Figure 2.

The number of GCI were counted in multiple neuroanatomical regions in the 4 cases of SND (gray bars) and 3 cases of OPCA (black bars) used for quantitative immunochemical studies (see Figures 4 and 5 ▶ ▶ ). Note that GCI were detected in all regions in all cases, but that the numbers varied widely and in no clear pattern with respect to clinical subtype.
The density of inclusions was to some extent a function of the density of myelinated fibers in the region. Most of the inclusions in the putamen were in the pencil fibers, whereas those in the globus pallidus were in the rich plexus of myelinated fibers in this nucleus. No neuronal inclusions were present in the basal ganglia. The inclusions in the pontine base were more abundant in transverse fibers than pontine nuclei. In SND there were GCI in oligodendroglia in areas with no obvious gross pathology, most notably the cerebellar white matter. Cerebral cortical white matter had very few inclusions, as in previous studies. 30
Immunoblots of α-Synuclein in MSA and Control Brains
Western blots of brain extracts from basal ganglia in 9 cases demonstrated significant differences in the physicochemical properties of α-synuclein in MSA compared to controls. The fractions from MSA brains had more intense α-synuclein immunoreactivity than corresponding fractions from normal brains (Figure 3) ▶ . The S2 (buffer-extractable) and S3 (Triton-soluble) fractions from AD brains sometimes had slightly higher α-synuclein immunoreactivity than normal controls (Figure 3B) ▶ , but the increase was not comparable to that observed in MSA. For this reason Alzheimer’s disease cases were included as controls in quantitative analyses. The α-synuclein antibody labeled proteins of molecular weight 19 kd more intensely than those of 29–36 and 45–55 kd. Similar immunoblotting patterns were detected with both polyclonal antibodies (NACP98 and NACP111) as well as both monoclonal antibodies to α-synuclein (data not shown), indicating that these proteins were α-synuclein or related proteins. Some MSA cases had detectable α-synuclein immunoreactivity in S5 fraction (formic acid-soluble), but α-synuclein immunoreactivity in S5 was usually completely absent in controls. Significantly more α-synuclein was detected in S4 in MSA compared to controls, and this fraction also had heterogeneous high molecular weight species. Some of the high molecular weight species were resolved as sharp bands around 30 to 40 kd and others appeared as smears. The 19-kd band in these fractions represents intact α-synuclein, whereas the higher molecular weight species most likely represent aggregated, posttranslationally modified, or complexed forms of α-synuclein. These findings were reproducible in 9 cases of MSA.
Figure 3.

Western blots of MSA (C) and 2 controls (A, normal; B, Alzheimer’s disease) with respect to brain fractions. Note the overall increase in α-synuclein immunoreactivity in MSA in both the 19-kd band, corresponding to α-synuclein, and higher molecular weight species, especially in S4.
Quantitative Analysis of α-Synuclein Immunoreactivity
Quantitative immunoblot analysis was performed on dissections of the posterior putamen of 7 cases of MSA and 4 controls, which were selected based on availability of tissue for immunocytochemistry. The cases and controls did not differ in terms of age (MSA 62.1 ± 9.4 years versus controls 70.7 ± 1.2 years, P = 0.20) or postmortem delay (MSA 8.7 ± 4.2 hours versus controls 11 ± 5, P = 0.58). The amount of α-synuclein per wet weight (units/g) was determined by dividing the immunoreactivity readout (after subtraction of background) by the sample volume loaded on the gel, and the total amount of α-synuclein in the given fraction was estimated by multiplying this immunoreactivity value by the volume of the fraction.
There was a trend for total α-synuclein immunoreactivity to be increased in MSA compared to controls (154 ± 41 versus 89 ± 47, P = 0.056), and this was statistically significant when the Alzheimer’s disease case was excluded from the controls (154 ± 41 versus 62 ± 6.1, P < 0.01). In MSA the α-synuclein immunoreactivity was almost equally distributed (about 45%) between the Triton- (S2 + S3) and SDS- (S4) extractable fractions. This was markedly different from controls, where almost 80% of the α-synuclein was in the Triton-extractable fraction (S2 + S3; Figure 4a ▶ ). Only a minor amount of α-synuclein was detected in the formic acid fraction, with no statistical difference in this fraction between MSA and controls. The amount of α-synuclein in S4 was significantly increased in MSA compared to controls. The partitioning of α-synuclein immunoreactivity was similar in SND and OPCA (Figure 4b) ▶ .
Figure 4.

a: Quantitative analysis of α-synuclein immunoreactivity in 7 cases of MSA and 4 controls. The only significant difference between MSA and controls was in the amount of α-synuclein immunoreactivity in the S4 fraction. b:There were no significant differences between SND and OPCA with respect to partitioning of α-synuclein immunoreactivity.
Analysis of the ratio of α-synuclein in the various fractions rather than absolute amount revealed significant differences between MSA and controls for S2, S3, and S4 fractions. A greater proportion of the α-synuclein immunoreactivity was in the SDS-soluble and less in the buffer- and Triton-soluble fractions (S2MSA 45 ± 15% versus S2ctl 79 ± 1%, P < 0.01; S3MSA 8 ± 5% versus S3ctl 17 ± 8%, P < 0.05; S4MSA 44 ± 14% versus S4ctl 4 ± 0.2%, P < 0.01).
Relation of Partitioning of α-Synuclein to the Number of GCI in the Basal Ganglia
Counts were made of GCI in the posterior putamen in 7 MSA cases on which quantitative immunoblots were obtained from adjacent slabs of frozen brain. The total α-synuclein immunoreactivity in the various fractions was plotted versus the number of GCI (Figure 5) ▶ . Although the relationship was not robust, there was a trend (R 2 = 0.15) for decreased α-synuclein immunoreactivity in Triton X-100-extractable fraction and GCI. Despite greater magnitude differences in S2 and S4 fractions, these changes were not associated with even weak relationships between α-synuclein immunoreactivity and number of GCI (S2 R 2 = 0.02; S4 R 2 = 0.06). This observation suggests that the major differences in the partitioning of α-synuclein in MSA compared to controls (with greater amounts of α-synuclein in SDS-soluble fractions in MSA) are not due merely to shift of α-synuclein to an insoluble form abundant in inclusion bodies. Rather, the findings suggest that there are likely complex factors that account for the abnormalities in α-synuclein partitioning in MSA that are not easily accounted for solely by histopathological lesions. There was a somewhat stronger trend (R 2 = 0.24) for the cases with more GCI to have decreased synaptophysin immunoreactivity in the basal ganglia. This is consistent with the idea that neurodegeneration, with neuronal and synaptic loss, is greater in cases with more GCI. Further support for the complexity of α-synuclein partitioning abnormalities in MSA was obtained in an analysis of regional anatomical distribution of α-synuclein.
Figure 5.

Correlation plots of synaptophysin (from S3) and α-synuclein immunoreactivity in various factions and total with respect to number of GCI in an adjacent section of basal ganglia. None of the regression lines produced a robust linear relation. For α-synuclein the best relation was for increasing GCI and increased formic acid-extractable α-synuclein, but these measurements are based on amounts 100- to 1000-fold lower and must be interpreted cautiously.
Regional Distribution of α-Synuclein in MSA and Control Brains
Immunoreactivity of α-synuclein in brain fractions (S2 + S3, S4, and S5) was measured in 10 regions of an MSA brain compared with corresponding fractions from a control brain (Figure 6) ▶ . Adjacent regions from this same brain were used for immunocytochemistry and counts of GCI (see below). Immunoblotting of S2 + S3 fractions revealed abundant α-synuclein in all brain regions. Among the regions examined, white matter from corpus callosum and the anterior thalamus had the lowest α-synuclein immunoreactivity. Variations in the amount of α-synuclein per unit weight of tissue between different brain regions in part may be due to differences in the density of synaptic terminals in the regions, because α-synuclein is predominantly a presynaptic protein. Thus, white matter and thalamus, with relatively lower synaptic density than other regions examined, had lower levels of α-synuclein.
Figure 6.

Regional distribution of α-synuclein immunoreactivity with respect to brain fractions in a single MSA (A, B, and C) and a normal individual (D, E, and F). The fractions are as follows: S2 + S3 (A and D); S4 (B and E) and S5 (C and F). The anatomical regions and number of GCI (± SE) in the MSA cases are as follows: Lane 1: amygdala (1.7 ± 0.9); Lane 2: hippocampus (1.7 ± 0.9); Lane 3: corpus callosum (31.3 ± 3.3); Lane 4: thalamus (22.7 ± 2.1); Lane 5: cingulate cortex (11.3 ± 1.7); Lane 6: striatum (180 ± 17); Lane 7: temporal cortex (1.7 ± 1.3); Lane 8: globus pallidus (134 ± 31); Lane 9: insular cortex (1.7 ± 0.9); Lane 10: frontal cortex (3.7 ± 0.5).
Immunoblotting of the SDS-soluble fraction (S4) revealed α-synuclein in all brain regions from the MSA case, but not in the corresponding fractions from the normal control brain. Another distinction between the MSA and control brains was the presence of α-synuclein immunoreactivity in high molecular weight species in most of the MSA brain regions examined. This observation held true even for brain regions with only a few GCI, such as the hippocampus and amygdala. In fact, the most striking observation of this regional neuroanatomical study was the seeming lack of association between the number of GCI detected with α-synuclein immunocytochemistry and the α-synuclein profile determined with immunochemical methods. Thus, areas with numerous GCI (Figure 6 ▶ , Lanes 3, 4, 5, 6, and 8) were indistinguishable from regions with only a few GCI (Figure 6 ▶ , Lanes 1, 2, 7, 9, and 10) in terms of the immunoblotting profile.
The SDS-soluble (S4) and formic acid-soluble fractions (S5) had detectable or abundant α-synuclein in 9 of the 10 regions in MSA, which was absent in corresponding controls. In addition to the 19-kd band, high molecular species of α-synuclein were detected in some regions (Figure 7 ▶ , Lanes 4, 6, 8, and 10), which tended to be regions with many GCI.
Figure 7.

The filter from Figure 6C ▶ was reprobed with a monoclonal antibody to ubiquitin, which also revealed heterogeneous high molecular weight species.
Comparison of Ubiquitin Immunoreactivity to α-Synuclein in SDS-Soluble Fractions
To determine whether the high molecular weight smearing could be related to ubiquitination, the blot of the S4 fraction was reprobed with a monoclonal antibody to ubiquitin (Figure 7) ▶ . Ubiquitin immunoreactivity was readily detected in the S4 fractions from the10 regions studied. Similar to α-synuclein, ubiquitin-immunoreactive species were not resolved into sharp bands, but rather appeared on the immunoblots as high molecular weight smears. The distribution of ubiquitin immunoreactivity on immunoblots overlapped but was not identical to the high molecular weight smears noted with antibodies to α-synuclein. These findings are compatible with the idea that some of the higher molecular weight α-synuclein species are ubiquitinated. This would be consistent with the immunocytochemical colocalization of α-synuclein and ubiquitin in neuronal and glial inclusions of MSA.
Discussion
GCI in MSA
Since the introduction of the unifying concept of MSA to encompass the apparently disparate clinicopathological entities of SND and OPCA, there have been few insights into the pathogenesis of MSA. 31 Postmortem studies have been largely descriptive and have not provided information about disease pathogenesis or supportive evidence for the concept of MSA. The discovery of GCI in 1989 opened a new avenue of investigation and added support to the notion that sporadic OPCA and SND were indeed a single entity. 7 The present studies support this unifying concept of MSA in that we could find no statistically significant differences in the density and distribution of GCI or in biochemical changes in α-synuclein when comparing SND and OPCA cases.
The specificity of GCI has been adequately defended and the weight of current evidence suggests that although glial inclusions can be found in other neurodegenerative diseases, GCI are specific to MSA. 7 The studies we have undertaken to characterize our antibodies to α-synuclein support this conclusion. The antibodies used in the present study were specific and sensitive for Lewy bodies and Lewy neurites and the neuronal and glial inclusions in MSA. None of the glial inclusions in other neurodegenerative diseases, including progressive supranuclear palsy, Pick’s disease, and corticobasal degeneration, had α-synuclein immunoreactivity, 32 which is similar to findings in previous studies. 29 The GCI of MSA were double stained for ubiquitin and less consistently with markers for oligodendroglia, in particular C4d. They were negative for GFAP and thus consistent with oligodendroglial inclusions. These results confirm and extend previous studies in that C4d immunoreactivity has not previously been reported in GCI, but has been detected in pathological oligodendroglia in other disorders. 33 It is unclear if the presence of complement protein immunoreactivity has any pathogenetic significance or if it merely represents a fortuitous marker for diseased oligodendrocytes in a variety of neurodegenerative disorders.
Biochemical Composition of GCI
The present results strongly support the hypothesis that the inclusion bodies in MSA are composed of α-synuclein. Direct analysis of purified GCI has not been attempted, and the relevance of the biochemical alterations observed in MSA with respect to the composition of GCI must, therefore, be interpreted with caution. Only one other study has evaluated biochemical changes in α-synuclein in MSA. 20 In that study, Tu et al identified changes in solubility of α-synuclein in MSA by differential centrifugation on sucrose density gradients, rather than solubility in chaotropic agents, as we have done. Sucrose gradient centrifugation has been used successfully to purify macromolecular aggregates such as neurofibrillary tangles or Lewy bodies. It is therefore reasonable to assume that α-synuclein in the denser sucrose gradient fractions may correspond to fractions enriched in GCI, although this was never addressed. The direct relationship observed between the amount of insoluble α-synuclein immunoreactivity and the number of GCI in white matter, but not gray matter, would support this conclusion.
The present study, however, suggests more fundamental changes in α-synuclein solubility in MSA that may not be directly related to inclusion body formation. In particular, the most significant change we observed was an increase of α-synuclein in the SDS-soluble fraction and the presence of heterogeneous high molecular α-synuclein species in the same fraction. These changes correlated poorly with GCI and were actually observed in brain regions that had very few GCI. Only the formic acid fraction showed an expected direct relationship with GCI. Given that other filamentous lesions also contain formic acid-extractable proteins, these findings may suggest that as a marker for α-synuclein abnormalities, the GCI may be only the tip of the iceberg.
Occult Pathology in Neurodegenerative Disorders
It should come as no surprise that antibody-based methods can disclose alterations in neurodegenerative diseases that are not apparent with routine histological methods. Examples are legion and include α-synuclein-immunoreactive neuronal inclusions in MSA 16 and Lewy neurites in the hippocampus in LBD, 34 neither of which were described before immunocytochemical methods were used to characterize these disorders. The study of white matter pathology in MSA by antibody methods recently reported by Matsuo and coworkers is another instructive example. 5 Matsuo and coworkers used antibodies to specific subregions of MBP in immunoblot and immunocytochemical studies of MSA. The MBP epitope was expressed in degenerating, but not normal, myelin. Using these antibodies, the authors showed that white matter pathology in MSA was more widespread than had been visible even with special histochemical stains for myelin. Of particular interest to the present study is the fact that myelin pathology was detected in brain regions that did not have GCI. Using different types of antibodies and different methods, we also found abnormalities in MSA brain regions with few or no GCI. The combined results of these studies suggest that the pathology of MSA is more subtle and widespread than has been apparent with routine methods and that GCI may be more of an epiphenomenon than a direct causative link to neurodegeneration.
Given that major biochemical alterations in α-synuclein in MSA correlate poorly with GCI, it will be important to explore the nature of alterations in α-synuclein that may account for these observations. In addition, future studies will be needed to focus on the normal function of α-synuclein in neuronal and glial cells, the subcellular distribution of the molecule, and alterations in disease states. Because recent evidence suggests that MSA is associated with subtle myelin pathology, it is also important to determine whether α-synuclein has a function in myelinating glia. Direct analyses of α-synuclein from diseased brains, with particular attention to possible covalent modifications in addition to ubiquitination, are also important for understanding disease pathogenesis.
Finally, the present studies suggest that it may be possible to develop a biochemical fingerprint for the synucleinopathies, even in the absence of inclusion bodies. Regardless of the explanation of the abnormal fractionation profile, one may operationally define a synucleinopathy as a disorder with increased ratio of SDS-soluble to buffer-soluble α-synuclein. In preliminary studies of LBD we have noted that α-synuclein is also increased in SDS-soluble fractions, but the magnitude of increase was less than for MSA. Given that synuclein-related pathology is much greater in MSA than in LBD, we hypothesize that observations are quantitative rather than qualitative. For example, it is not unusual to detect literally hundreds of GCI within a small area of affected tissue, but it is rare to find a similar lesion density in LBD. (Only in the amygdala can one find small foci with nearly comparable densities of Lewy bodies.) This potential biochemical signature of the synucleinopathies may find its greatest utility in animal models. Because changes in α-synuclein solubility may not bear direct relation to inclusion bodies, it may be possible to use this ratio as a means of analyzing transgenic mice that may fail to form inclusion bodies as a biochemical monitor of the disease phenotype.
Footnotes
Address reprint requests to Dennis W. Dickson, M.D., Neuropathology, Birdsall 317, Mayo Clinic Jacksonville, 4500 San Pablo Road, Jacksonville, FL 32224. E-mail: dickson.dennis@mayo.edu.
References
- 1.Graham JG, Oppenheimer DR: Orthostatic hypertension and nicotine insensitivity in a case of multiple system atrophy. J Neurol Neurosurg 1969, 32:28-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wenning GK, Tison F, Ben Shlomo Y, Daniel SE, Quinn NP: Multiple system atrophy: a review of 203 pathologically proven cases. Movement Disorders 1997, 12:133-147 [DOI] [PubMed] [Google Scholar]
- 3.Bandmann O, Sweeney MG, Daniel SE, Wenning GK, Quinn N, Marsden CD, Wood NW: Multiple-system atrophy is genetically distinct from identified inherited causes of spinocerebellar degeneration. Neurology 1997, 49:1598-1604 [DOI] [PubMed] [Google Scholar]
- 4.Bower JH, Maraganore DM, McDonnell SK, Rocca WA: Incidence of progressive supranuclear palsy and multiple system atrophy in Olmsted County, Minnesota, 1976 to 1990. Neurology 1997, 49:1284-1288 [DOI] [PubMed] [Google Scholar]
- 5.Matsuo A, Akiguchi I, Lee GC, McGeer EG, McGeer PL, Kimura J: Myelin degeneration in multiple system atrophy detected by unique antibodies. Am J Pathol 1998, 153:735-744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Papp MI, Kahn JE, Lantos PL: Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy-Drager syndrome). J Neurol Sci 1989, 94:79-100 [DOI] [PubMed] [Google Scholar]
- 7.Lantos PL: The definition of multiple system atrophy: a review of recent developments. J Neuropathol Exp Neurol 1998, 57:1099-1111 [DOI] [PubMed] [Google Scholar]
- 8.Arima K, Murayama S, Mukoyama M, Inose T: Immunocytochemical and ultrastructural studies of neuronal and oligodendroglial cytoplasmic inclusions in multiple system atrophy. 1. Neuronal cytoplasmic inclusions. Acta Neuropathol 1992, 83:453-460 [DOI] [PubMed] [Google Scholar]
- 9.Kato S, Nakamura H: Cytoplasmic argyrophilic inclusions in neurons of pontine nuclei in patients with olivopontocerebellar atrophy: immunohistochemical and ultrastructural studies. Acta Neuropathol 1990, 79:584-594 [DOI] [PubMed] [Google Scholar]
- 10.Horoupian DS, Dickson DW: Striatonigral degeneration, olivopontocerebellar atrophy and “atypical” Pick disease. Acta Neuropathol 1991, 81:287-295 [DOI] [PubMed] [Google Scholar]
- 11.Braak H, Braak E: Cortical and subcortical argyrophilic grains characterize a disease associated with adult onset dementia. Neuropathol Appl Neurobiol 1989, 15:13-26 [DOI] [PubMed] [Google Scholar]
- 12.Yamada T, McGeer PL: Oligodendroglial microtubular masses: an abnormality observed in some human neurodegenerative diseases. Neurosci Lett 1995, 120:163-166 [DOI] [PubMed] [Google Scholar]
- 13.Arima K, Nakamura M, Sunohara N, Ogawa M, Anno M, Izumiyama Y, Hirai S, Ikeda K: Ultrastructural characterization of the tau-immunoreactive tubules in the oligodendroglial perikarya and their inner loop processes in progressive supranuclear palsy. Acta Neuropathol 1997, 93:558-566 [DOI] [PubMed] [Google Scholar]
- 14.Chin SSM, Goldman JE: Glial inclusions in CNS degenerative diseases. J Neuropathol Exp Neurol 1996, 55:499-508 [DOI] [PubMed] [Google Scholar]
- 15.Cairns NJ, Atkinson PF, Hanger DP, Anderton BH, Daniel SE, Lantos PL: Tau protein in the glial cytoplasmic inclusions of multiple system atrophy can be distinguished from abnormal tau in Alzheimer’s disease. Neurosci Lett 1997, 230:49-52 [DOI] [PubMed] [Google Scholar]
- 16.Dickson DW, Liu W-K, Farrer M, Mehta N, Uitti R, Mark M, Zimmerman T, Golbe L, Sage J, Hardy J, Yen S-H: Glial cytoplasmic inclusions (GCI) of multiple system atrophy (MSA) contain α-synuclein (abstract). Movement Disorders 1998, 13(suppl. 2):128 [Google Scholar]
- 17.Arima K, Ueda K, Sunohara N, Arakawa K, Hirai S, Nakamura M, Tonozuka-Uehara H, Kawai M: NACP/α-synuclein immunoreactivity in fibrillary components of neuronal, and oligodendroglial cytoplasmic inclusions in the pontine nuclei in multiple system atrophy. Acta Neuropathol 1998, 96:439-444 [DOI] [PubMed] [Google Scholar]
- 18.Gai WP, Power JH, Blumbergs PC, Blessing WW: Multiple-system atrophy: a new α-synuclein disease? Lancet 1998, 352:547-548 [DOI] [PubMed] [Google Scholar]
- 19.Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M: Filamentous α-synuclein inclusions link multiple system atrophy with Parkinson’s disease, and dementia with Lewy bodies. Neurosci Lett 1998, 251:205-208 [DOI] [PubMed] [Google Scholar]
- 20.Tu P-h, Galvin JE, Baba M, Giasson B, Tomita T, Leight S, Nakajo S, Iwatsubo T, Trojanowski JQ, Lee V M-Y: Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple system atrophy brains contain insoluble α-synuclein. Ann Neurol 1998, 44:415-422 [DOI] [PubMed] [Google Scholar]
- 21.Wakabayashi K, Hayashi S, Kakita A, Yamada M, Toyoshima Y, Yoshimoto M, Takahashi H: Accumulation of α-synuclein/NACP is a cytopathological feature common to Lewy body disease and multiple system atrophy. Acta Neuropathol 1998, 96:445-452 [DOI] [PubMed] [Google Scholar]
- 22.Wakabayashi K, Yoshimoto M, Tsuji S, Takahashi H: Alpha-synuclein immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy. Neurosci Lett 1998, 249:180-182 [DOI] [PubMed] [Google Scholar]
- 23.Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA, Kittel A, Saitoh T: The precursor protein of non-Aβ component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 1995, 14:467-475 [DOI] [PubMed] [Google Scholar]
- 24.Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT, Jr: NACP, a protein implicated in Alzheimer’s disease, and learning, is natively unfolded. Biochemistry 1996, 35:13709-13715 [DOI] [PubMed] [Google Scholar]
- 25.Irizarry MC, Kim T-W, McNamara M, Tanzi RE, George JM, Clayton DF, Hyman BT: Characterization of the precursor protein of the non-Aβ component of senile plaques (NACP) in the human central nervous system. J Neuropathol Exp Neurol 1996, 55:889-895 [DOI] [PubMed] [Google Scholar]
- 26.Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VM, Trojanowski JQ, Iwatsubo T: Aggregation of α-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am J Pathol 1998, 152:879-884 [PMC free article] [PubMed] [Google Scholar]
- 27.Lowry OH, Rosenbrough NJ, Farr AL, Randall RJ: Protein measurement with the Folin phenol reagent. J Biol Chem 1951, 193:265-272 [PubMed] [Google Scholar]
- 28.Laemmli UK: Cleavage of structural protein during assembly of the head of bacteriophage T4. Nature 1970, 227:680-685 [DOI] [PubMed] [Google Scholar]
- 29.Takeda A, Mallory M, Sundsmo M, Honer W, Hansen L, Masliah E: Abnormal accumulation of NACP/α-synuclein in neurodegenerative disorders. Am J Pathol 1998, 152:367-372 [PMC free article] [PubMed] [Google Scholar]
- 30.Inoue M, Yagishita S, Ryo M, Hasegawa K, Amano N, Matsushita M: The distribution and dynamic density of oligodendroglial cytoplasmic inclusions (GCIs) in multiple system atrophy: a correlation between the density of GCIs and the degree of involvement of striatonigral and olivopontocerebellar systems. Acta Neuropathol 1997, 93:585-591 [DOI] [PubMed] [Google Scholar]
- 31.Castellani R: Multiple system atrophy: clues from inclusions. Am J Pathol 1998, 153:671-676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dickson DW, Farrer MJ, Mehta ND, Perez-Tur J, Tiseo P, Yen S-H, Hardy J: Antibodies to non-amyloid component of plaques (NACP) specifically label Lewy bodies and Lewy neurites, but not other inclusions in neurodegenerative diseases (abstract). J Neuropathol Exp Neurol 1998, 57:516 [Google Scholar]
- 33.Yamada T, McGeer PL: Oligodendroglial microtubular masses: an abnormality observed in some human neurodegenerative diseases. Neurosci Lett 1990, 120:163-166 [DOI] [PubMed] [Google Scholar]
- 34.Dickson DW, Ruan D, Crystal H, Mark MH, Davies P, Kress Y, Yen S-H: Hippocampal degeneration differentiates diffuse Lewy body disease (DLBD) from Alzheimer’s disease: light and electron microscopic immunocytochemistry of CA2–3 neurites specific to DLBD. Neurology 1991, 41:1402-1409 [DOI] [PubMed] [Google Scholar]