

Abstract
The terminal components of complement C5b-C9 can cause significant injury to cardiac allografts. Using C6-deficient rats, we have found that the rejection of major histocompatibility (MHC) class I-incompatible PVG.R8 (RT1.AaBu) cardiac allografts by PVG.1U (RT1.AuBu) recipients is particularly dependent on C6. This model was selected to determine whether tissue injury results from C6 produced by macrophages, which are a conspicuous component of infiltrates in rejecting transplants. We demonstrated that high levels of C6 mRNA are expressed in isolated populations of macrophages. The relevance of macrophage-produced C6 to cardiac allograft injury was investigated by transplanting hearts from PVG.R8 (C6−) donors to PVG.1U (C6−) rats which had been reconstituted with bone marrow from PVG.1U (C6+) rats as the sole source of C6. Hearts grafted to hosts after C6 reconstitution by bone marrow transplantation underwent rejection characterized by deposition of IgG and complement on the vascular endothelium together with extensive intravascular aggregates of P-selectin-positive platelets. At the time of acute rejection, the cardiac allografts contained extensive perivascular and interstitial macrophage infiltrates. RT-PCR and in situ hybridization demonstrated high levels of C6 mRNA in the macrophage-laden transplants. C6 protein levels were also increased in the circulation during rejection. To determine the relative contribution to cardiac allograft rejection of the low levels of circulating C6 produced systemically by macrophages, C6 containing serum was passively transferred to PVG.1U (C6−) recipients of PVG.R8 (C6−) hearts. This reconstituted the C6 levels to about 3 to 6% of normal values, but failed to induce allograft rejection. In control PVG.1U (C6−) recipients that were reconstituted with bone marrow from PVG.1U (C6−) donors, C6 levels remained undetectable and PVG.R8 cardiac allografts were not rejected. These results indicate that C6 produced by macrophages can cause significant tissue damage.
Increasing evidence indicates that antibody and complement can contribute to the rejection processes of allografts. 1 We have demonstrated that a deficiency of the terminal complement component C6, 2,3 which prevents assembly of the membrane attack complex (MAC), can delay acute allograft rejection from 7 to 10 days to more than 3 weeks in rat strain combinations that differ at major and minor histocompatibility antigens. 4 The contribution of C6 to acute graft rejection can be even more profound in rat strains differing only at major histocompatibility (MHC) Class I antigens. 5
The liver has been identified as the primary site of synthesis of circulating complement components including C6. 6 In rats, orthotopic liver transplants from C6-sufficient donors restore circulating C6 to >90% of donor levels within 14 days. 3,7,8 Conversely, an extrahepatic source of C6 is evident when livers are transplanted from C6-deficient donors to normal recipients; following this procedure, C6 levels remain at 30 to 40% of pretransplantation levels for more than 100 days after surgery. 3,7,8 Bone marrow transplants from C6 sufficient donors to C6 deficient recipients demonstrated that hematopoietically derived cells are a source of least a portion of this extrahepatic C6. 3,7
Selected complement components can be synthesized by mononuclear phagocytes, fibroblasts, endothelial cells, gastrointestinal and genitourinary epithelial cells, and adipocytes in vitro. 6,9,10 Since the first reports on the biosynthesis of complement by mononuclear phagocytes, 11 it has been documented that mononuclear phagocytes can synthesize all of the classical pathway components and most of the alternative pathway proteins. 12,13 However, evidence that complement synthesized by mononuclear phagocytes contributes to tissue injury is limited. 9
In our previous experiments using complement-sufficient rats, extensive macrophage infiltrates were found within the acutely rejecting cardiac allografts in addition to severe vascular injury. 5 The aim of the present study is to determine whether C6 produced by macrophages causes significant tissue injury.
Materials and Methods
Animals
The derivation of PVG congenic rat strains with a C6 deficiency has been described previously. 5 The 9- to 12-week-old male PVG.R8 (RT1.AaBu) and PVG.1U (RT1.AuBu) used in these studies are mismatched at MHC Class I antigens. C6 levels in the sera were checked by a sandwich enzyme-linked immunosorbent assay (ELISA) and the MHC phenotype of the rats was confirmed by flow cytometry (see below). All animals were cared for in accordance with National Institutes of Health guidelines under the supervision of qualified veterinarians.
C6 Level Analysis
The ELISA for detection of C6 was performed using mouse anti-rat C6 monoclonal antibody 3G11 (a gift from Dr. W. Couser, University of Washington, Seattle, WA) to coat 96-well plates. After blocking the uncoated portions of the wells with PBA (phosphate buffered saline (PBS) containing 0.2% bovine serum albumin and 0.02% NaN3), serial dilutions of rat serum samples were incubated in the wells for 1 hour. Following 3 washes with PBA, bound rat C6 was detected with a goat anti-human C6 antibody (Calbiochem, La Jolla, CA), which cross-reacts with rat C6, followed by sequential incubation with biotin-conjugated donkey anti-goat IgG antibody (Jackson ImmunoReseach, West Grove, PA), horseradish peroxidase-conjugated streptavidin (Zymed, South San Francisco, CA) and the substrate o-phenylenediamine (Sigma Chemical Co., St. Louis, MO).
MHC Phenotyping
The MHC phenotype of all PVG.R8 and PVG.1U rats was confirmed preoperatively by labeling peripheral blood leukocytes (PBL) with MN4 (American Type Culture Collection, Manassas, VA), an IgG mouse monoclonal antibody to RT1.Aa antigen expressed by PVG.R8 rat lymphocytes and OX3 (ECACC, Port Down, Salisbury, UK), an IgG mouse monoclonal antibody to RT1.Bu antigen expressed by PVG.R8 and PVG.1U rat B lymphocytes. The bound monoclonal antibodies were detected with FITC-conjugated rat anti-mouse IgG (Jackson ImmunoResearch). After incubating for 30 minutes at 4°C, the cells were washed twice, then resuspended in PBA and measured by a FACScan flow cytometer (Becton Dickinson, Mountain View, CA).
Bone Marrow Transplantation
One day before grafting, bone marrow recipients underwent total body irradiation of 1000 rad at 84.7 rad/minute from a dual source 137Cs animal irradiator (Atomic Energy of Canada Ltd., Kanata, ON). Donor rats were killed by CO2 asphyxiation at 4 to 6 weeks of age. Marrow was flushed from the femora, tibiae, and humeri with RPMI 1640 (Flow Laboratories, McLean, VA). The marrow cell suspension was passed through a 21-gauge needle to remove clumps and debris, then washed once in RPMI 1640 and adjusted to 60 × 10 6 nucleated cells/ml. Each recipient was injected with 1 ml of bone marrow in the dorsal tail vein.
Heterotopic Heart Transplantation
Hearts from male PVG.R8 (C6−) rats were transplanted heterotopically into male PVG.1U (C6−) rats under methoxyfluorane (Pitman-Moore, Inc., Mundelein, IL) inhalational anesthesia. 4 Ice-cold cardioplegic solution (Abbott Laboratories, North Chicago, IL) was injected into the aortic root of the donor heart to metabolically arrest the heart before removal. The donor aorta and pulmonary artery were anastomosed to the recipient infrarenal aorta and inferior vena cava, respectively, in an end-to-side fashion with 8–0 prolene suture (Ethicon, Inc., Somerville, NJ), and then the abdomen was closed in layers. Cardiac graft function was evaluated daily by abdominal palpation until rejection, which was defined as total cessation of contractions. Rejection was confirmed by direct visualization and histological examination of the allograft.
Measurement of Alloantibodies
Alloantibodies were measured by flow cytometry on single cell suspensions of cervical lymph nodes from PVG.R8 rats as described previously. 4,5 Briefly, the cells were incubated with 50 μl of diluted sera (1:4, 1:16, 1:64, 1:256). The washed cells were reacted with 50 μl of PBA containing a mixture of FITC-conjugated goat anti-rat IgG and phycoerythrin conjugated goat anti-rat IgM (Jackson ImmunoResearch). The cells were analyzed using a FACScan flow cytometer (Becton-Dickinson).
Preparation of RNA and RNA Quality
Total cellular RNA was isolated from snap-frozen native and transplanted hearts that were homogenized in TRIZOL (Gibco, Grand Island, NY), a monophasic solution of phenol and guanidinium isothiocyanate. This method is an improvement of the single-step RNA isolation method developed by Chomczynski and Sacchi. 14 The quantity of RNA in samples was evaluated by the presence of β-actin housekeeping RNA. The isolation of RNA was performed according to the manufacturer’s guidelines.
RT-PCR
RNA samples (5 μg) were treated with DNase I, amplification grade (Gibco), and reverse-transcribed into cDNA in reaction mixtures containing 0.5 μl RNasin (40 U/ml; Promega, Madison, WI), 2.5 μl 10 mmol/L dNTP (Pharmacia Biotech, Piscataway, NJ), 1 μg oligo-dT12–18 (Promega), 400 U (Molony murine leukemia virus) reverse transcriptase (Bethesda Research Lab., Bethesda, MD), 10 μl 5× reverse transcriptase buffer, and diethyl pyrocarbonate (DEPC) water to a final volume of 50 μl. The reverse transcription reactions were carried out at 37°C for 90 minutes, heat-inactivated at 65°C for 10 minutes, and cooled for 3 minutes. PCRs were set up by using 5 μl of cDNA (the equivalent of 50 ng of RNA), 5 μl of 10× amplification buffer, 3 μl of 25 mmol/L MgCl2, 3 μl of 2.5 mmol/L dNTP, 1 μl of 10 μmol/L of each primer, 0.25 μl of 5 U/ml Taq DNA polymerase (Promega), and dH2O to a final volume of 50 μl. This mixture was overlaid with 100 μl of light mineral oil (Sigma). The following sense and antisense oligonucleotide primers were used (direction 5′ to 3′): β-actin, CTATCGGCAATGAGCGGTTC and CTTAGGAGTTGGGGGTGGCT; rat C6, GGGGCAAGTATGACCTTCTC and TGGGGACCGTTTTTCACAGT. According to the varying contents of specific cDNA and varying amplification efficiencies, the samples were subjected to different cycle numbers and annealing temperatures that were optimized empirically for each primer pair: 30 cycles, 63°C (β-actin); 35 cycles, 57°C (C6). The PCR amplification program was designed for the initial denaturation of cDNA at 94°C for 2 minutes, then cDNA was amplified for the specified number of cycles, each consisting of 1 minute at 94°C, 1 minute at the annealing temperature, and 1 minute for extension at 72°C. The final cycle extension was increased by 7 additional minutes at 72°C. PCRs were performed in a Hybaid OmniGene thermocycler (Hybaid Ltd., Woodbridge, NJ).
Competitive Template RT-PCR
Competitive templates (CT) for rat C6 and β-actin were designed to contain the same cDNA sequence as the gene of interest except for deletion of 90 to 100 bp within the competitor DNA. Using CT as internal standards in RT-PCR allows the amplification of both the wild-type (WT) cDNA and the CT in the same reaction with the gene-specific primers. The individual products are separated and analyzed on the basis of size. To determine the amount of cDNA present in each sample, the same amplification technique was used first to measure the expression of the housekeeping gene β-actin. Samples of WT cDNA equivalent to 50 ng total RNA from each individual RT reaction product were adjusted to contain equal concentrations of cDNA, based on the expression of β-actin in the sample. For each gene 5 μl of the normalized RT product was coamplified with a constant amount of the gene-specific CT. The relative amounts of WT cDNA in the various samples were determined by calculating their respective sample intensity ratios of WT cDNA/CT DNA. Finally, all samples were normalized against the respective β-actin WT cDNA/CT DNA ratio. This normalization controls for the quantity of cDNA loaded in all samples. PCR samples were separated on 2% agarose gel. The intensity of ethidium bromide luminescence was measured with CCD image sensor using EagleSight 3.0 Software (Stratagene, La Jolla, CA). This software provides basic analysis of the relative densities of gel images, which represent two-dimensional arrays of pixels. Gel images were further analyzed and quantified using NIH Image 1.54 program.
Cloning and Characterization of a Rat C6 cDNA
The fragment of rat C6 cDNA was obtained using a rat liver cDNA library as a template and degenerate primers designed for the most conservative sequences of human C6 protein. The following degenerate sense and antisense oligonucleotide primers were used to obtain 636-bp fragment (direction 5′ to 3′): GAYTTYGGNACNCAYTAYTTY and CCARCANCCCCAYTGNCCRTC, positions 345 and 540 of the homologous human amino acid sequence, respectively. The second pair of degenerate sense and antisense oligonucleotide primers were used in a nested PCR technique with a 636-bp fragment as a template (direction 5′ to 3′): TTYATHCARGGNGCNGARAA and TGYTTYTCRCARTTYTCNCCRTA. The 636-bp fragment of rat C6 was subcloned in the plasmid pCRII using TA cloning kit (Invitrogen, Carlsbad, CA). The resultant rat C6 cDNA was sequenced, and found to be homologous to human C6 at the DNA level and deduced protein level (BLAST).
Isolation of Macrophages, Lymphocytes, and Neutrophils for RT-PCR Studies of C6 mRNA
Granulocytes were isolated from heparinized whole blood. The leukocyte-rich buffy coat was collected after incubation of 10 ml of blood with 1.25 ml of dextran T500 solution (450 mg dextran and 450 mg glucose in 10 ml 0.9% NaCl; Pharmacia LKB, Uppsala, Sweden) for 45 minutes at room temperature. Contaminating erythrocytes were lysed using a hypotonic lysing buffer (Sigma) and the cells were washed twice before labeling. Peritoneal macrophages were harvested by peritoneal lavage, using 20 ml of sterile PBS. Lymphocytes were separated from single-cell suspension of lymph nodes. Cells isolated from the peritoneum and lymph nodes were washed twice and labeled.
The following monoclonal antibodies were used for immunofluorescence analysis and cell sorting: R73-FITC (fluorescein-conjugated mouse IgG1 anti-TCR αβ, Pharmingen, San Diego, CA), RLN-9D3 (mouse IgG2a anti-rat B cell, Caltag, Burlingame, CA), W3/25-PE (phycoerythrin-conjugated mouse IgG1 anti-rat CD4, Caltag), HIS48 (mouse IgM anti-rat granulocytes, Caltag). Macrophages from the peritoneum were labeled with W3/25-PE; T and B lymphocytes from lymph nodes were labeled with R73-FITC or RLN-9D3; and granulocytes from PBL were labeled with HIS48. These antibodies (100 μl) were added for 1 hour at 4°C and then washed twice in PBA. Cells reacted with unlabeled IgG primary antibodies were labeled with 100 μl of FITC-conjugated F(ab′)2 fragments of goat anti-mouse IgG-Fc (Jackson Immuno-Research Laboratories) at 1:100 dilution. Cells reacted with HIS48 were labeled with 100 μl of FITC-conjugated F(ab′)2 fragments of goat anti-mouse IgM μ chain specific antibodies (Jackson ImmunoResearch Laboratories). After 30 minutes’ incubation at 4°C, the cells were washed twice, and positively labeled cells were separated by fluorescent-activating cell sorting on a EPICS 752 (Coulter, Hialeah, FL). The purity of sorted populations of lymphocytes, macrophages, and granulocytes was greater than 98% by cytospin preparations. Total cellular RNA was isolated from purified populations of lymphoid cells using RNeasy Mini Kit (Qiagen, Santa Clara, CA). RNAs were reverse-transcribed into cDNA and analyzed for the expression of C6 as described above.
Histological Evaluation of Tissue Sections
At the time of sacrifice, adjacent full cross-sections of rejected cardiac grafts were frozen in OCT or fixed in 10% formalin in PBS. Formalin-fixed tissue was embedded in paraffin and sectioned at 7 μm. Rejection was assessed on sections that were stained with hematoxylin and eosin. Macrophage infiltration was evaluated by immunoperoxidase staining with monoclonal antibody ED1 that recognizes a cytoplasmic antigen in monocytes and most macrophages, 15 and P-selectin expression was assessed with an affinity-purified polyclonal rabbit antibody to P-selectin (PharMingen, San Diego, CA).
IgM, IgG, C3, and C3d were detected by immunofluorescent stains on frozen sections of the grafts. The primary antibodies to IgG and C3d were unlabeled, purified mouse monoclonal IgG antibodies. The anti-C3d was produced to human C3d (Quidel, San Diego, CA). It also binds to iC3b, but not to C3b or whole C3. Staining for C3d eliminates background staining of unactivated C3, which is present in high concentrations in plasma and interstitial fluids. This reagent cross-reacts with rat C3 split products. The secondary antibody was an FITC-labeled polyclonal rat antibody to mouse IgG (Jackson ImmunoResearch). C3 was detected directly with fluorescein-conjugated goat anti-rat C3 (ICN, Aurora, OH). IgM was stained directly with cyanine 3-conjugated goat anti-rat IgM (Jackson ImmunoResearch).
In Situ Hybridization of C6 mRNA
In situ hybridization with digoxigenin-labeled sense and antisense C6 riboprobes was performed on formalin-fixed, paraffin-embedded tissue sections. A 387-bp fragment of the C6 cDNA sequence was generated in RT-PCR using RNA from rat liver and the following sense and antisense primers, direction 5′ to 3′: GCCGCCAGGAGCTACAGAAC, and GGGCTTTCCTGAGGTTGTTC, respectively. The resultant cDNA fragment of C6 was subcloned into the expression vector BlueScript II (pCR-Script Amp, Stratagene, La Jolla, CA) by standard techniques. Sense and antisense riboprobes were generated by in vitro transcription with a commercially available kit (Boehringer Mannheim, Indianapolis, IN) using T7 and Sp6 RNA polymerases. After transcription, the probes were ethanol precipitated, dissolved in DEPC-treated dH2O, and stored at −80°C. Yields of the labeled probe were estimated by means of an enzyme-linked immunoassay using a commercially available kit (Boehringer Mannheim) by comparing the intensity of the colorimetric reaction of a dilution series of labeled control RNA (10 ng, 1 ng, 100 pg, 10 pg, 1 pg) and sample probe RNA dotted onto a positively charged nylon membrane. Tissue sections were dehydrated in a graded series of ethanol. After prehybridization for 1 hour, hybridization solution containing 5 ng/ml of either DIG-labeled antisense or sense probe was added to each section. After applying coverslips, the slides were heated at 60°C for 5 minutes on a heating block to denature the target sequences and then incubated in the moist chamber for approximately 18 hours at 37°C. Following hybridization, the coverslips were removed and the sections were washed to remove unbound probe. Finally to achieve high stringency hybridization, the sections were washed in 0.2× SSC/0.1% SDS for 20 minutes at 54°C. Hybridization was detected with sheep anti-digoxigenin antibody-alkaline phosphatase conjugate (polyclonal Fab fragments, Boehringer Mannheim) visualized by 5-bromo-4-chloro-3-indolyl phosphate and nitro blue tetrazolium salt (Boehringer Mannheim).
Results
Experimental Design
C6-deficient PVG.1U rats were reconstituted with bone marrow from congenic C6-sufficient rats. After recovery of bone marrow function, these rats received cardiac allografts from C6-deficient PVG.R8 donors. Using C6-deficient rats as heart donors eliminated endothelial cells, fibroblasts, myocytes, and passenger leukocytes as potential sources of C6 production. Thus, marrow-derived cells were the sole source of C6 in the experimental animals. The control C6-deficient PVG.1U recipients were reconstituted with bone marrow from syngeneic C6-deficient rats. These control animals also received cardiac allografts from C6-deficient PVG.R8 donors. As a result, they underwent the same procedures but had no source of C6.
Effects of C6 on PVG.R8 Cardiac Allografts in PVG.1U Recipients
Unmodified, C6-sufficient PVG.1U (RT1.AuBu) recipients rejected cardiac allografts from MHC class I incompatible PVG.R8 (RT1.AaBu) donors between 6 and 7 days (n = 8). In contrast, PVG.R8 cardiac allografts survived longer than 30 days in C6-deficient PVG.1U recipients (n = 5). This finding is consistent with reports that acute rejection of cardiac allografts in this rat strain combination is antibody-dependent, as demonstrated by passive serum transfer studies. 16,17
Verification that PVG.1U (C6+) and PVG.1U (C6−) Rats Are Histocompatible
To ascribe differences in graft survival to differences in C6 production rather than differences in histocompatibility, skin grafts were exchanged between representative PVG.1U (C6−) and PVG.1U (C6+) rats. First- and second-set skin grafts were fully accepted in both directions with no signs of rejection for over 180 days. Moreover, the PVG.1U (C6−) rats that received bone marrow transplants from PVG.1U (C6+) donors never showed any signs of graft failure or of graft versus host disease in 6 to 14 weeks of observation (n = 6). Skin grafts and bone marrow grafts are the two most stringent tests of histocompatibility and these results argue that no unexpected antigenic differences were introduced to the PVG.1U rats during derivation of the C6-deficient line. 5
C6 Restoration after Bone Marrow Transplantation
We have shown previously that bone marrow transplants from congenic C6-sufficient donors can restore a low level of circulating C6 in PVG rats. 3,7 Therefore, we monitored the engraftment of the bone marrow transplants by measuring C6 levels in the serum by ELISA. Serum samples were obtained 1 day before treatment and then weekly beginning 14 days after bone marrow transplantation.
As shown in Figure 1 ▶ , circulating C6 levels increased from undetectable levels to 2 to 5% of normal C6 levels between 4 and 8 weeks after bone marrow transplantation from PVG.1U (C6+) donors to PVG.1U (C6−) recipients (n = 6). In control PVG.1U (C6−) recipients (n = 8) that were reconstituted with bone marrow from PVG.1U (C6−) donors, C6 levels remained undetectable (Figure 2) ▶ .
Figure 1.

A: Survival of PVG.R8 (C6−) cardiac allografts in irradiated PVG.1U (C6−) recipients after reconstitution with PVG.1U(C6+) bone marrow. Circulating C6 levels as measured by ELISA increased from undetectable levels to 2 to 5% of normal C6 levels between 4 and 8 weeks after bone marrow transplantation. Survival of individual cardiac allografts from PVG.R8 (C6−) is demarcated by length of bars after individual symbols. Acute rejection was accompanied by a 2.4-fold increase in circulating C6 levels (thicker lines). B: Relative concentration of C6 mRNA recovered from individual cardiac allografts at the time of sacrifice. The amount of C6 mRNA recovered from a cardiac transplant to a PVG.1U (C6+) serves as a positive control (+, far right). Vertical bars indicate the range of 3 separate assays.
Figure 2.

Survival of PVG.R8 (C6−) cardiac allografts in irradiated PVG.1U (C6−) recipients after reconstitution with PVG.1U (C6−) bone marrow. Circulating C6 levels as measured by ELISA remained undetectable. All of the cardiac allografts from PVG.R8 (C6−) were beating vigorously at the time of sacrifice (marked by †).
Survival of Cardiac Allografts in Bone Marrow Reconstituted Recipients
Hearts were allografted from PVG.R8 (C6−) to PVG.1U (C6−) recipients at two intervals after reconstitution with PVG.1U (C6+) bone marrow (Figure 1) ▶ . Hearts grafted to hosts 4 weeks after reconstitution survived 13, 25, and >50 days (n = 3). Hearts transplanted 10 to 12 weeks after reconstitution were rejected acutely (7–8 days; n = 3). The control PVG.R8 (C6−) hearts that were transplanted to PVG.1U (C6−) recipients at 4 (n = 4) or 10 to 12 (n = 4) weeks after reconstitution with PVG.1U (C6−) bone marrow functioned vigorously until sacrifice (Figure 2) ▶ . Macroscopically, these control hearts were normal in color and size, whereas the experimental hearts were hemorrhagic and distended.
C6 and Antibody Responses to Cardiac Allografts in Bone Marrow Reconstituted Recipients
The antibody responses elicited by cardiac transplants did not differ between the PVG.1U rats that were reconstituted with bone marrow from C6-sufficient or C6-deficient donors. However, the IgM and IgG alloantibody responses that were elicited by hearts transplanted 4 weeks after bone marrow reconstitution were late (Figure 3) ▶ . These data indicate that the lymphocyte compartment did not recover function completely by 4 weeks after bone marrow reconstitution.
Figure 3.

IgM (upper panel) and IgG (lower panel) alloantibodies in the circulation after PVG.R8 cardiac allograft to irradiated PVG.1U (C6−) recipients after reconstitution with PVG.1U (C6+) or PVG.1U (C6−) bone marrow. Alloantibodies were measured by flow cytometry on PVG.R8 lymphoid cells. Serial dilutions of serum were analyzed. For clarity of presentation, only the 1:16 dilution is depicted.
Markedly different C6 responses were stimulated by the cardiac transplants. Hearts transplanted to PVG.1U (C6−) recipients reconstituted with PVG.1U (C6−) bone marrow stimulated no detectable C6 production. In contrast, increases in circulating C6 were stimulated by hearts transplanted to PVG.1U (C6−) recipients reconstituted with PVG.1U (C6+) bone marrow. Acute rejection was accompanied by a two- to fourfold increase in circulating C6 levels (Figure 1) ▶ . This increase in C6 production was down-regulated quickly. Within a week after rejection was completed, circulating C6 levels returned toward pretransplantation values.
Deposition of Antibody and Complement in Cardiac Allografts
Immunofluorescent stains on frozen sections of the cardiac allografts demonstrated deposition of IgM, IgG, C3, and C3d on the endothelium of arteries, capillaries, and veins in cardiac allografts to both control and experimental recipients. In areas of vascular injury there was subendothelial and perivascular deposition of IgG and C3d. These stains indicate that the alloantibodies elicited by the transplants had bound to the target antigens and activated the early components of the complement cascade in both the control and experimental recipients (Figure 4) ▶ .
Figure 4.

Immunofluorescent stains for C3d on the endothelium of arteries, capillaries and veins in cardiac allografts to control (C6−) and C6 reconstituted (C6+) recipients. In the reconstituted C6+ recipients, subendothelial and perivascular depositions of C3d are evident in areas of vascular injury (arrows). The inset demonstrates at higher magnification the disruption of the endothelial cell layer (arrow) and deposition of C3d in the media of an artery.
Macrophage Infiltrates in Cardiac Transplants
PVG.R8 cardiac allografts to both control and experimental PVG.1U (C6−) recipients contained extensive perivascular and interstitial mononuclear cell infiltrates 1–2 weeks after transplantation. Immunoperoxidase stains for the macrophage marker ED1 confirmed that macrophages were a major component of the infiltrates in both the control and experimental recipients (Figure 5, A and C) ▶ .
Figure 5.
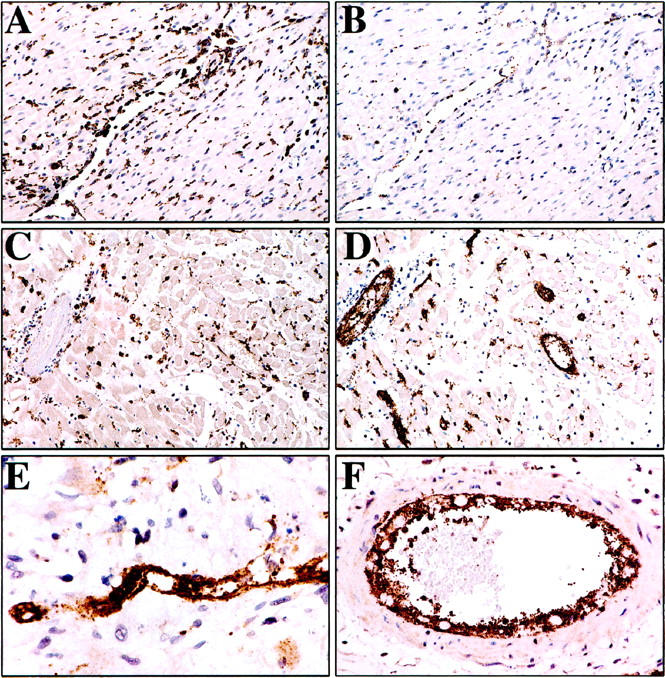
A and B: Control PVG.R8 cardiac allograft 7 days after transplantation to PVG.1U (C6−) recipient reconstituted with bone marrow from a PVG.1U (C6−) rat. Immunoperoxidase stains for ED1 demonstrate extensive perivascular macrophage infiltrates (A), but only moderate expression of P-selectin on endothelial cells and minimal platelet aggregation (B). Experimental PVG.R8 cardiac allograft 7 days after transplantation to PVG.1U (C6−) recipient reconstituted with bone marrow from a PVG.1U (C6+) rat (C-F). Immunoperoxidase stains for ED1 delineates extensive perivascular and interstitial macrophage infiltrates (C). Immunoperoxidase stains for P-selectin on a sequential section demonstrates expression on endothelial cells and several layers of aggregated platelets in vessels of all sizes at low power (D) and in a capillary (E) and large artery (F) at higher magnification.
P-Selectin Release in Cardiac Transplants
Vascular endothelial cell injury and platelet aggregation were a conspicuous feature of cardiac allograft rejection in PVG.1U (C6−) recipients reconstituted with bone marrow from PVG.1U (C6+). Immunoperoxidase stains demonstrated that these endothelial cells and platelets stained intensely for P-selectin (Figure 5, D–F) ▶ . P-selectin-positive platelet aggregates filled the lumens of capillaries (Figure 4E) ▶ , and several layers of P-selectin-positive platelets were attached to the endothelial cells of both arteries and veins (Figure 5F) ▶ .
This was in marked contrast to hearts transplanted to PVG.1U (C6−) recipients reconstituted with bone marrow from PVG.1U (C6−). Only moderate expression of P-selectin was detected on endothelial cells in these cardiac allografts to control recipients and this was associated with minimal platelet aggregation (Figure 5B) ▶ .
C6 mRNA in Cardiac Transplants
Competitive template RT-PCR analyses were performed on isolated leukocytes to determine potential sources of C6 mRNA from bone marrow-derived cells. Macrophages, neutrophils, and T and B lymphocytes were separated by fluorescent-activated cell sorting of resident peritoneal cells, peripheral blood cells, and lymph node cells, respectively (see Materials and Methods). Only macrophages had detectable amounts of mRNA for C6 by competitive template RT-PCR analysis. This technique demonstrated that unactivated macrophages produced amounts of mRNA similar to liver cells (Figure 6) ▶ . In situ hybridization on histological sections from the rejected cardiac allografts confirmed that macrophages located in the perivascular, interstitial, subendocardial, and epicardial infiltrates were the only cells with detectable cytoplasmic staining for C6 mRNA (Figure 7) ▶ .
Figure 6.

Competitive template RT-PCR analyses performed on liver and macrophages. Macrophages were separated by fluorescent activated cell sorting of resident peritoneal cells. Constant amounts of wild-type (WT) cDNA were mixed with twofold serial dilutions of competitor template (CT) cDNA in successive lanes (starting with 2.2 and ending with 0.07 pg/sample). Macrophages and liver cells produced similar amounts of mRNA for C6.
Figure 7.
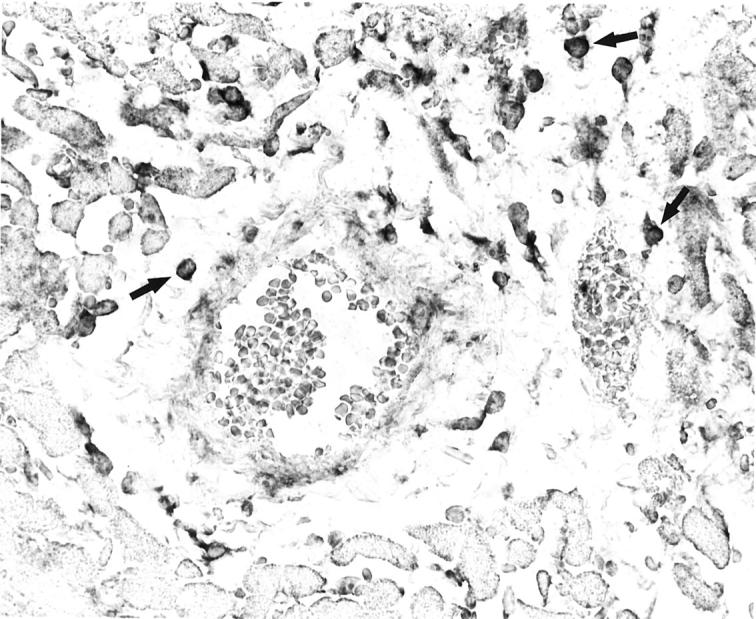
In situ hybridization on histological section from the rejected cardiac allografts demonstrates cytoplasmic staining for C6 mRNA in perivascular and interstitial macrophages (cells with darkly stained cytoplasm, see arrows for examples).
Competitive template RT-PCR analysis of sections from the apex of the cardiac transplants documented abundant quantities of mRNA for C6 in hearts harvested at the time of or within a week after acute rejection (Figure 1B) ▶ . In contrast, the one long-term surviving heart contained scant infiltrating macrophages and undetectable levels of C6 mRNA in the transplant at the time of harvest (Figure 1B) ▶ .
Passive Transfer of C6 to Cardiac Transplant Recipients
Although the RT-PCR and in situ hybridization studies demonstrate an association between the amounts of C6 mRNA expressed by the macrophages infiltrating rejected cardiac allografts and the degree of vascular injury, macrophages in other tissues are also capable of producing C6, as evidenced by the low levels of C6 demonstrated in the circulation before transplantation.
To determine the relative contribution to cardiac allograft rejection of these low levels of circulating C6 produced by macrophages, PVG.R8 (C6−) hearts were transplanted to PVG.1U (C6−) recipients (n = 4). Beginning on the day of transplantation, 1 ml of fresh serum from PVG.1U (C6+) donors was injected intravenously every day. This reconstituted the C6 levels to about 3 to 6% of normal values as measured by ELISA on days 3, 7, and 14 after transplantation. All 4 hearts transplanted to serum-reconstituted recipients continued beating until the recipients were sacrificed for histological evaluation 7 and 14 days after transplantation.
Significant perivascular and interstitial mononuclear cell infiltrates were present at both 7 and 14 days after transplantation, but only focal vascular endothelial injury and platelet aggregation was evident. Immunoperoxidase stains demonstrated localized areas of capillaries that contained P-selectin-positive platelet aggregates. These were more frequent at 14 days than 7 days. The expression of P-selectin in Weibel-Palade bodies of endothelial cells was prominent in many arteries, but translocation of P-selectin to the plasma membrane was minimal in arteries. These data indicate that the low levels of C6 demonstrated in the circulation before transplantation contribute to vascular injury but are not sufficient in themselves to cause complete graft rejection.
Discussion
Although production of complement by extrahepatic sources has been proposed to be important in the expansion of inflammatory responses, direct evidence of the magnitude of this mechanism and the cells responsible for complement production is sparse. 4,18,19 The assessment of macrophage production of complement components in normal animals is complicated by the fact that numerous extrahepatic sites of biosynthesis have been identified for individual complement components. In fact, endothelial cells, fibroblasts, and myocytes are among the cells that are capable of synthesizing several complement components. 6,9,10,12,13,20 Of direct relevance to our studies is the report that C6 can be synthesized by human endothelial cells as well as by monocytes and macrophages. 21 C6 mRNA has also been detected in tissue samples from myocardial infarcts in humans, but whether this was derived from the parenchymal tissue or reactive macrophages was not investigated. 22
Macrophages are a conspicuous component of many inflammatory processes including the infiltrates that characterize reperfusion injury, 18 acute rejection, 23,24 and chronic dysfunction 25,26 of transplants. The capacity of macrophages to produce an array of inflammatory mediators in vivo has been demonstrated by immunohistochemistry, PCR, and in situ hybridization. 18,23-26 Establishing the function of mediators produced by macrophages has relied largely on studying freshly isolated monocytes or mononuclear cell lines in culture. As a result, data on the actual contribution of individual mediators produced by macrophages to the pathogenesis of inflammation in vivo are limited.
In the present studies, rats genetically deficient in C6 were reconstituted with bone marrow from congenic C6 sufficient rats. These rats received cardiac allografts from C6 deficient donors. This choice of donor eliminated endothelial cells, fibroblasts, myocytes and so-called passenger leukocytes as a potential source of the C6 production. With marrow-derived cells as the sole source of C6, the cardiac allografts incurred irreversible acute rejection. In situ hybridization on histological sections from the rejected cardiac allografts demonstrated C6 mRNA in the cytoplasm of macrophages that were located in the perivascular, interstitial, subendocardial, and epicardial infiltrates. Competitive template RT-PCR analysis documented abundant quantities of mRNA for C6 in hearts harvested at or within 1 week after acute rejection. Moreover, the macrophage production of C6 was extremely responsive to the inflammatory cytokines stimulated by the rejection process. This was reflected by the two- to fourfold increase in circulating C6 levels stimulated by hearts transplanted 10 to 12 weeks after bone marrow reconstitution.
MAC would be predicted to have multiple effects in the vascular and extravascular compartments. Deposition of sublytic quantities of MAC on endothelial cells and platelets in vitro initiates a calcium-dependent series of activation responses. MAC stimulates the translocation of P-selectin from platelet α granules to the plasma membrane and the secretion of mediators from storage granules. 27,28 Similarly, MAC causes the translocation of P-selectin from Weibel-Palade bodies in endothelial cells to the plasma membrane and the release of von Willebrand factor. 29 In addition, MAC-stimulated endothelial cells have been found to synthesize and release interleukin-1 and tissue factor. 30 These responses to MAC would be predicted to cause an acute transformation of an anticoagulant to a procoagulant environment.
Our model demonstrates the in vivo consequences of the effects of MAC on endothelial cells and platelets. Platelet aggregation was sparse in hearts transplanted to C6-deficient rats, but was a prominent feature in hearts transplanted to PVG.1U (C6−) recipients reconstituted with bone marrow from PVG.1U (C6+) rats. C6 reconstitution was associated with increased P-selectin expression on the vascular endothelial cells of the allograft and on the platelets attached to the endothelial cells of arteries, capillaries, and veins.
Infiltration of macrophages into the perivascular and interstitial compartments of the inflamed tissue provides a mechanism to concentrate complement production in extravascular locations. By migrating through the vascular endothelial cell barrier where there is a high expression of complement regulatory molecules, the macrophages can ferry active complement components directly into the parenchyma. This may be of critical importance for complement components such as C6 that are present in limiting concentrations in the circulation. Limited concentrations of C6 can thwart lysis of nucleated target cells in vitro 31 because multiple MAC lesions are required to cause lysis of nucleated cells and MAC channels have a half-life of approximately 1 minute on nucleated cells. 32
Our studies demonstrated that the C6 produced by the macrophages caused extensive tissue injury as evidenced by vascular endothelial injury and platelet aggregation as well as interstitial edema and hemorrhage. This finding of in vivo functional activity is particularly important because monocyte-derived C3 has been reported to have only 10% of the hemolytic activity of plasma C3, which is primarily hepatocyte-derived. 13
In summary, we have shown previously that hepatic biosynthesis is responsible for the majority of C6 in the circulation. 3 This study demonstrates that functional C6 produced by macrophages can be a significant factor in tissue injury as exemplified by allograft rejection.
Acknowledgments
We gratefully acknowledge the expertise of James Flook, who separated our fluorescence-labeled cell samples on the EPICS 752, and Pamela Cady and Louvinia Jones for excellent immunofluorescent stains.
Footnotes
Address reprint requests to Zhiping Qian, M.D., Department of Pathology, Ross Research Room 664D, The Johns Hopkins University School of Medicine, 720 Rutland Avenue, Baltimore, MD 21205-2196. E-mail: ZPQIAN@jhmi.edu. Address correspondence to William M. Baldwin III,
Supported by National Institutes of Health grants AI42387 and HL56091.
References
- 1.Baldwin WM, III, Pruitt SK, Brauer RB, Daha MR, Sanfilippo F: Complement in organ transplantation: contribution to inflammation, injury and rejection. Transplantation 1995, 59:797-808 [PubMed] [Google Scholar]
- 2.Brauer RB, Baldwin WM, III, Daha MR, Pruitt SK, Sanfilippo F: The use of C6-deficient rats to evaluate the mechanism of hyperacute rejection of discordant cardiac xenografts. J Immunol 1993, 151:7240-7248 [PubMed] [Google Scholar]
- 3.Brauer RB, Baldwin WM, III, Wang D, et al: Hepatic and extrahepatic biosynthesis of complement factor C6 in the rat. J Immunol 1994, 153:3168-3176 [PubMed] [Google Scholar]
- 4.Brauer RB, Baldwin WM, III, Ibrahim S, Sanfilippo F: The contribution of terminal complement components to acute and hyperacute allograft rejection in the rat. Transplantation 1995, 59:288-293 [PubMed] [Google Scholar]
- 5.Qian Z, Jakobs FM, Pfaff-Amesse T, Sanfilippo F, Baldwin WM, III: Complement contributes to the rejection of complete and Class I MHC incompatible cardiac allografts. J Heart Lung Transplant 1998, 17:470-478 [PubMed] [Google Scholar]
- 6.Perlmutter DH, Colten HR: Complement: molecular genetics. Gallin JI Goldstein IM Snyderman R eds. Inflammation: Basic Principles and Clinical Correlates. 1992, :pp 81-102 Raven Press, New York [Google Scholar]
- 7.Brauer RB, Lam TT, Wang D, Horwitz L, Hess AD, Klein AS, Sanfilippo F, Baldwin WM, III: Extrahepatic synthesis of C6 in the rat is sufficient for complement-mediated hyperacute rejection of a guinea pig cardiac xenograft. Transplantation 1995, 59:1073-1076 [DOI] [PubMed] [Google Scholar]
- 8.Timmerman JJ, van Dixhoorn MGA, Schraa EO, van Gijlswijk-Jansen DJ, Muizert Y, van Es LA, Daha MR: Complement C6 and C2 biosynthesis in syngeneic PVG/c- and PVG/c+ rat stains. Scand J Immunol 1997, 46:366-372 [DOI] [PubMed] [Google Scholar]
- 9.Morgan BP, Gasque P: Extrahepatic complement biosynthesis: where, when and why? Clin Exp Immunol 1997, 107:1-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Volanakis JE: Transcriptional regulation of complement genes. Annu Rev Immunol 1995, 13:277-305 [DOI] [PubMed] [Google Scholar]
- 11.Stecher VJ, Morse JH, Thorbecke GJ: Sites of production of primate serum proteins associated with complement system. Proc Soc Exp Biol Med 1967, 124:433-438 [DOI] [PubMed] [Google Scholar]
- 12.Johnson E, Hetland G: Mononuclear phagocytes have the potential to synthesize the complete functional complement system. Scand J Immunol 1988, 27:489-493 [DOI] [PubMed] [Google Scholar]
- 13.McPhaden AR, Whaley K: Complement biosynthesis by mononuclear phagocytes. Immunol Res 1993, 12:213-232 [DOI] [PubMed] [Google Scholar]
- 14.Chomczynski P, Sacchi N: Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 1987, 162:156-159 [DOI] [PubMed] [Google Scholar]
- 15.Damoiseaux JG, Dopp EA, Calame W, Chao D, MacPherson GG, Dijkstra CD: Rat macrophage lysosomal membrane antigen recognized by monoclonal antibody ED1. Immunology 1994, 83:140-147 [PMC free article] [PubMed] [Google Scholar]
- 16.Gracie JA, Bolton EM, Porteous C, Bradley JA: T cell requirements for the rejection of renal allografts bearing an isolated class I MHC disparity. J Exp Med 1990, 172:1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morton AL, Bell EB, Bolton EM, Marshall HE, Roadknight C, McDonagh M, Bradley JA: CD4+ T cell-mediated rejection of major histocompatibility complex class I-disparate grafts: a role for alloantibody. Eur J Immunol 1993, 23:2078-2084 [DOI] [PubMed] [Google Scholar]
- 18.Takada M, Nadeau KC, Shaw GD, Marquette KA, Tilney NL: The cytokine-adhesion molecule cascade in ischemia/reperfusion injury of the rat kidney. Inhibition by a soluble P-selectin ligand. J Clin Invest 1997, 99:2682-2690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Timmerman JJ, van Dixhoorn MG, Schraa EO, van Gijlswijk-Jansen DJ, Muizert Y, van Es LA, Daha MR: Extrahepatic C6 is as effective as hepatic C6 in the generation of renal C5b-9 complexes. Kidney Int 1997, 51:1788-1796 [DOI] [PubMed] [Google Scholar]
- 20.Legoedec J, Gasque P, Jeanne JF, Fontaine M: Expression of the complement alternative pathway by human myoblasts in vitro: biosynthesis of C3, factor B, factor H and factor I. Eur J Immunol 1995, 25:3460-3466 [DOI] [PubMed] [Google Scholar]
- 21.Johnson E, Hetland G: Human umbilical vein endothelial cells synthesize functional C3, C5, C6, C8 and C9 in vitro. Scand J Immunol 1991, 33:667-671 [DOI] [PubMed] [Google Scholar]
- 22.Yasojima K, Schwab C, McGeer EG, McGeer PL: Human heart generates complement proteins that are upregulated and activated after myocardial infarction. Circ Res 1998, 83:860-869 [DOI] [PubMed] [Google Scholar]
- 23.Armstrong HE, Bolton EM, McMillan I, Spencer SC, Bradley AJ: Prolonged survival of actively enhanced rat renal allografts despite accelerated cellular infiltration and rapid induction of both class I and class II MHC antigens. J Exp Med 1987, 165:891-907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Russell ME, Adams DH, Wyner LR, Yamashita Y, Halnon NJ, Karnovsky MJ: Early and persistent induction of monocyte chemoattractant protein 1 in rat cardiac allografts. Proc Natl Acad Sci USA 1993, 90:6086-6090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adams DH, Russell ME, Hancock WW, Sayegh MH, Wyner LR, Karnovsky MJ: Chronic rejection in experimental cardiac transplantation: studies in the Lewis-F344 model. Immunol Rev 1993, 134:5-19 [DOI] [PubMed] [Google Scholar]
- 26.Hancock WW, Whitley WD, Tullius SG, Heemann UW, Wasowska B, Baldwin WM, III, Tilney NL: Cytokines, adhesion molecules, and the pathogenesis of chronic rejection of rat renal allografts. Transplantation 1993, 56:643-650 [DOI] [PubMed] [Google Scholar]
- 27.Sims PJ, Wiedmer T: The response of human platelets to activated components of the complement system. Immunol Today 1991, 12:338-342 [DOI] [PubMed] [Google Scholar]
- 28.Sims PJ, Wiedmer T: Induction of cellular procoagulant activity by the membrane attack complex of complement. Semin Cell Biol 1995, 6:275-282 [DOI] [PubMed] [Google Scholar]
- 29.Hattori R, Hamilton KK, McEver RP, Sims PJ: Complement proteins C5b-C9 induce secretion of high molecular weight multimers of endothelial von Willebrand factor and translocation of granule membrane protein GMP-140 to the cell surface. J Biol Chem 1989, 264:9053-9060 [PubMed] [Google Scholar]
- 30.Saadi S, Holzknecht RA, Patte CP, Stern DM, Platt JL: Complement-mediated regulation of tissue factor activity in endothelium. J Exp Med 1995, 182:1807-1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koski CL, Ramm LE, Hammer CH, Mayer MM, Shin ML: Cytolysis of nucleated cells by complement: cell death displays multi-hit characteristics. Proc Natl Acad Sci USA 1983, 80:3816-3820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramm LE, Whitlow MB, Koski CL, Shin ML, Mayer MM: Elimination of complement channels from the plasma membranes of U937, nucleated mammalian cell line: temperature dependence of the elimination. J Immunol 1983, 131:1411-1415 [PubMed] [Google Scholar]