

Abstract
Germline mutations in PTEN, encoding a dual-specificity phosphatase on 10q23.3, cause Cowden syndrome (CS), which is characterized by a high risk of breast and thyroid cancers. Loss of heterozygosity of 10q22–24 markers and somatic PTEN mutations have been found to a greater or lesser extent in a variety of sporadic component and noncomponent cancers of CS. Among several series of sporadic breast carcinomas, the frequency of loss of flanking markers around PTEN is approximately 30 to 40%, and the somatic intragenic PTEN mutation frequency is <5%. In this study, we analyzed PTEN expression in 33 sporadic primary breast carcinoma samples using immunohistochemistry and correlated this to structural studies at the molecular level. Normal mammary tissue had a distinctive pattern of expression: myoepithelial cells uniformly showed strong PTEN expression. The PTEN protein level in mammary epithelial cells was variable. Ductal hyperplasia with and without atypia exhibited higher PTEN protein levels than normal mammary epithelial cells. Among the 33 carcinoma samples, 5 (15%) were immunohistochemically PTEN-negative; 6 (18%) had reduced staining, and the rest were PTEN-positive. In the PTEN-positive tumors as well as in normal epithelium, the protein was localized in the cytoplasm and in the nucleus (or nuclear membrane). Among the immunostain negative group, all had hemizygous PTEN deletion but no structural alteration of the remaining allele. Thus, in these cases, an epigenetic phenomenon such as hypermethylation, -ecreased protein synthesis or increased protein degradation may be involved. In the cases with reduced staining, 5 of 6 had hemizygous PTEN deletion and 1 did not have any structural abnormality. Finally, clinicopathological features were analyzed against PTEN protein expression. Three of the 5 PTEN immunostain-negative carcinomas were also both estrogen and progesterone receptor-negative, whereas only 5 of 22 of the PTEN-positive group were double receptor-negative. The significance of this last observation requires further study.
The tumor suppressor gene PTEN, encoding a dual-specificity phosphatase, has been cloned and mapped to chromosome 10q23.3. 1-3 Germline PTEN mutations are found in the autosomal dominant Cowden syndrome (CS), which is characterized by multiple hamartomas involving many organ systems as well as an increased risk of developing breast and thyroid cancers. 4,5 Loss of heterozygosity (LOH) of markers at 10q23–25 is a frequent event (30–50%) in endometrial cancer, 6-9 glioblastoma, 10 and breast cancer. 11-13 Somatic intragenic mutations of PTEN are a frequent event in endometrial carcinomas, 6-9 malignant gliomas, 14-17 and melanomas. 18 However, unlike endometrial carcinoma and glioblastoma, only a very small fraction (<5%) of the 40% of primary breast cancers showing allelic loss in this region also have mutations in the remaining allele, 11-13,19 despite the fact that females with CS have a ≤50% lifetime risk of developing breast cancer. 5,20,21 In contrast to these analyses based on primary breast carcinomas, initial studies using breast cancer cell lines seemed to show that a large proportion have biallelic loss of PTEN. 1,3 Investigators, therefore, questioned whether loss of one PTEN allele (haploinsufficiency) is sufficient for tumorigenesis or whether inactivation of the second allele might occur through epigenetic rather than mutational events.
We report a study of PTEN expression using immunohistochemical methods in a series of 33 primary human breast tumors. This is a powerful method because it provides an internal control comparing the staining of tumor tissue to that of the adjacent normal breast tissue. We also began to explore the association of PTEN expression with genomic PTEN status and clinicopathological features.
Materials and Methods
Breast Carcinoma Samples
Paraffin blocks of 33 unselected sporadic primary ductal breast carcinomas were drawn from the files of the Kingston General Hospital (Kingston, ON, Canada). LOH analysis with seven microsatellite markers known to map to the 10q23 interval and flanking PTEN as well as PTEN mutation analysis have been performed previously. 13 Of the 33 women diagnosed with primary mammary adenocarcinomas, 4 were diagnosed before the age of 50. The tumors ranged in size from 1 to 6 cm. There were 2 well differentiated, 13 moderately differentiated, and 18 poorly differentiated tumors. Seven of the 13 women had regional lymph node involvement at presentation.
Immunohistochemistry
The monoclonal antibody 6H2.1 raised against the last 100 C-terminal amino acids of PTEN (Ziebold and Lees, unpublished) was used in all immunohistochemical analyses.
The tissue samples were fixed by immersion in 10% buffered formalin and embedded in paraffin according to standard procedures. 22 Four-millimeter sections were cut and mounted on Superfrost Plus slides (Fisher Scientific, Pittsburgh, PA). Immunostaining was performed essentially as described. 22-24 In summary, the sections were deparaffinized and hydrated by passing through xylene and a graded series of ethanol. Antigen retrieval was performed for 20 minutes at 98°C in 0.01 mol/L sodium citrate buffer, pH 6.4, in a microwave oven. Incubating the sections in 0.3% hydrogen peroxide for 30 minutes blocked endogenous peroxidase activity. After blocking for 30 minutes in 0.75% normal horse serum the sections were incubated with 6H2.1 (dilution 1:100) for 1 hour at room temperature. The sections were washed in Tris-buffered saline and then incubated with biotinylated horse anti-mouse IgG followed by avidin peroxidase using the Vectastain ABC elite kit (Vector Laboratories, Burlingame, CA). The chromogenic reaction was carried out with 3–3′ diaminobenzidine using nickel cobalt amplification, 25 which gives a black product. After counterstaining with Nuclear Fast Red (Rowley Biochemical, Danvers, MA) and mounting, the slides were evaluated under a light microscope. According to the amount of staining, the tumors were divided in three groups: the group assigned ++ showed increased or equal staining intensity compared to the corresponding normal tissue; the group assigned + had decreased staining intensity; and the group assigned − had no trace of staining.
A series of commercially available cell lines with known PTEN status was used as positive and negative controls to prove antibody specificity by immunohistochemistry: Balb C/3T3, Nalm6, DU145, MDA-MB-468, A172, and PC3 (see Results). In addition, the U2OS osteosarcoma cell line was transfected with full length PTEN cDNA expression construct as a further positive control.
Incubating the sections in the absence of antibody as well as preincubation during 2 hours at 37°C of the antibody with recombinant PTEN protein led to negative results (data not shown).
Western Blot Analysis
As biochemical proof of antibody specificity for PTEN, total protein lysates were obtained 26 (Dahia, 1999 #955) from a series of commercially available cell lines (American Type Culture Collection, Manassas, VA), for which PTEN status is known: MCF-7, T47D, MDA-MB-435S, ZR-75-1, BT-549, and MDA-MB-468 (see Results). In addition, as an additional positive control, the wild-type full length human PTEN cDNA sequence was cloned into the mammalian expression vector pUHD10-3, which contains a tetracycline-suppressible promoter (Gossen, 1992 #1065), and stably transfected into the MCF7/T-off (Clontech, Palo Alto, CA) breast cancer cell line. After 24 hours of tetracycline withdrawal for purposes of PTEN induction, protein lysates were collected for Western blot analysis as well. Western blot analysis was performed as previously described, 26 except that 6H2.1 was used at a 1:250 dilution. Control antibody was against α-tubulin and used at 1:1000 dilution.
LOH Analysis
All breast carcinoma samples have been previously evaluated for LOH with markers closely flanking, but not within, PTEN. 13 In the event that our immunohistochemical results seemed to be discordant with the molecular analyses, further LOH analysis was performed using markers within PTEN itself as previously described. 26,27 Potential hemizygosity at the PTEN locus was assessed by screening for a T/G polymorphism within PTEN intron 8 detected by differential digestion with the restriction endonuclease HincII as previously described 27 and the intragenic markers D10S2491, AFM086wg9, and D10S2492.
Results
Specificity of Monoclonal Antibody 6H2.1
Because this study relied on a monoclonal antibody, 6H2.1, specific recognition of PTEN by this antibody is crucial. Western blot analysis using a series of breast cancer lines with known PTEN status and the 6H2.1 anti-PTEN monoclonal antibody demonstrated the specificity of this antibody (Figure 1) ▶ . Western analysis of three PTEN+/+ lines, MCF-7, T-47D, and MDA-MB-435S, revealed a single band at the molecular weight predicted for PTEN. After induction of MCF-7/PTEN, increased expression of PTEN was evidenced by an increased band intensity (Figure 1) ▶ . In contrast, ZR-75-1, with a hemizygous deletion of PTEN and a missense mutation in the remaining allele, yielded a weak band of the expected size. BT-549 and MDA-MB-468, which are null for PTEN, had no signal. No nonspecific bands were noted. Control blot with anti-α-tubulin antibody revealed signals for all lines.
Figure 1.

Western blot of 7 breast cancer cell lines using the anti-PTEN monoclonal antibody 6H2.1 (left panel) and using the anti-α-tubulin antibody as a control (right panel). MCF-7, T-47D, and MDA-MB-435S have endogenous PTEN. BT-549 and MDA-MB-468 are PTEN-null. ZR-75-1 has monoallelic PTEN deletion and a missense mutation on the remaining allele. MCF-7/PTEN is the MCF-7 line transfected with a wild-type PTEN construct and a tetracycline-inducible promoter after withdrawal of tetracycline and, hence, induced expression of PTEN.
To test the suitability of the antibody for immunohistochemistry, we used PTEN-transfected U2OS cells as well as a series of cell lines expressing PTEN (Balb C/3T3, Nalm6, DU145) as positive controls. MDA-MB-468, a breast cancer cell line with a hemizygous deletion of PTEN and a truncating mutation of the remaining allele, A172, a glioblastoma cell line with loss of one PTEN allele and a truncating mutation in exon 2 of the remaining allele and PC3, a prostate cell line with homozygous deletion of PTEN, were used as negative controls (data not shown).
PTEN Immunohistochemistry in Primary Breast Carcinomas
Samples from 33 sporadic primary breast carcinomas, which had been examined previously for LOH of markers flanking PTEN as well as somatic PTEN mutations, 13 were subjected to immunohistochemical analysis using a monoclonal antibody, 6H2.1, raised against the terminal 100 amino acids of human PTEN. Of the 33 total cancers, 29 had accompanying normal tissue; in each of the 29 samples, the normal glandular epithelium showed immunoreactivity to 6H2.1. Interestingly, there was a distinctive staining pattern in the normal tissue. The myoepithelial cells of the normal ducts showed the strongest signal with a nuclear predominance (Figure 2B) ▶ . In contrast, the amount of staining in the epithelial cell layer was variable. Areas of epithelial ductal hyperplasia with and without atypia stained more strongly than the normal epithelia (Figure 2A) ▶ . Endothelial cells, especially within neovascular capillaries, and nerves showed strong PTEN expression and were useful as internal positive controls.
Figure 2.
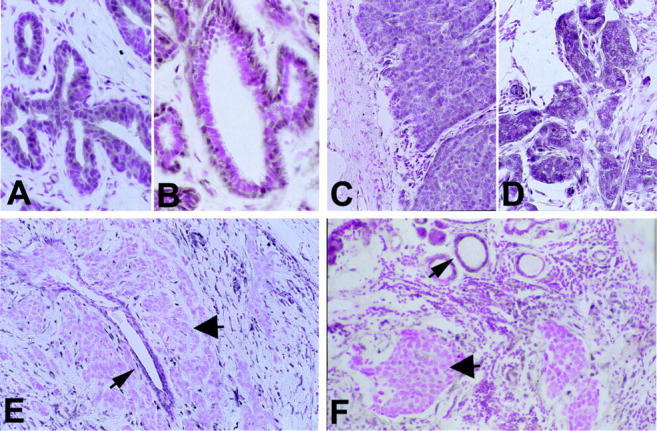
A: Ductal hyperplasia (case 58) with increased staining in the epithelial layer (original magnification, ×60). B: Normal breast glands (case 46) with predominantly nuclear staining in the myoepithelial layer (original magnification, ×60). C (case 48) and D (case 43): Ductal carcinoma with strong PTEN staining (++, original magnification, ×30). E: Ductal PTEN-negative carcinoma (arrowhead, case 58) and surrounding normal duct (arrow). Original magnification, ×30. F: Ductal PTEN-negative carcinoma (arrowhead, case 46) with non-neoplastic normal duct (arrow). Original magnification, ×30.
Of 33 breast carcinoma samples, 5 (15%) lost all PTEN immunoreactivity and showed negative immunostaining, graded − (Table 1 ▶ and Figure 2, E and F ▶ ). In each of these 5 cases, adjacent non-neoplastic glands (Figure 2F) ▶ as well as enclosed non-neoplastic ducts (Figure 2E) ▶ stained positively. Interestingly, the cells within the desmoplastic reaction surrounding each of these 5 carcinomas had high PTEN expression. Six of the 33 (18%) breast cancer specimens stained weakly, graded +, in comparison to the normal tissue (Table 1 ▶ and Figure 3 ▶ ). One of these tumors (Sample 40, Table 1 ▶ ) showed positive immunostaining in the intraductal component, whereas the adjacent invasive component lost almost all PTEN protein expression (Figure 3A) ▶ . The remaining 22 (66%) tumors stained positively, graded ++ (increased staining compared to normal glands). All these tumors showed homogeneous PTEN immunoreactivity throughout the examined section. PTEN immunoreactivity in these 22 tumors as well as their corresponding normal and hyperplastic breast tissue involved the cytoplasmic and nuclear (most likely nuclear membrane) compartment of the cells.
Table 1.
Correlation between PTEN Immunostaining and PTEN and/or 10q22-23 LOH
| PTEN Immuno ++ | PTEN Immuno + | PTEN Immuno − | |
|---|---|---|---|
| LOH 5′ Markers | 4* | 2 | 4 |
| LOH 3′ Markers | 1* | 2 | 5 |
| ROH Flank Markers | 18 | 2* | 0 |
| Total Tumors | 22 | 6 | 5 |
Correlation between PTEN immunostaining and LOH of 5′ and/or 3′ flanking markers.
Concordance 82%.
LOH, loss of heterozygosity; ROH, retention of heterozygosity.
*Apparent discordance 18%.
Figure 3.
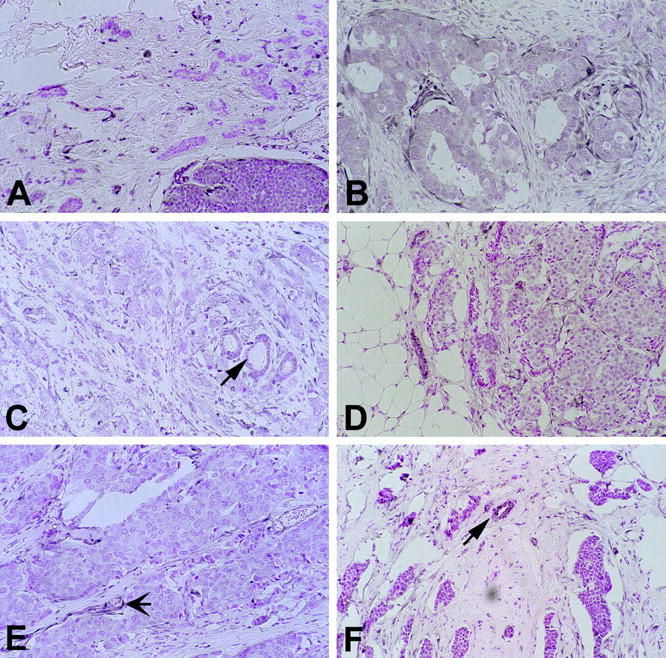
Cases with weak staining (arrows in C and F, non-neoplastic duct; arrow in E, blood vessel). A: Ductal carcinoma (case 40) showing no staining (graded −) in the invasive component (top) adjacent to immunostain-positive intraductal component (bottom). B: Case 66. C: Case 59. D: Case 57. E: Case 45. Original magnification, ×30.
Comparison of Immunohistochemical and Structural Mutation Data
Immunohistochemical evidence of PTEN expression was absent or weak in a total of 11 (33%) of 33 breast carcinomas. These breast carcinomas had been previously examined for LOH of markers flanking PTEN and also for intragenic PTEN mutations; 13 40% demonstrated LOH but there were no intragenic PTEN mutations or biallelic deletion. Whether there is a one-to-one concordance between molecular and immunohistochemical observations is further explored in this report.
LOH analysis for markers in the 10q22–24 interval was previously performed using seven microsatellite markers (centromeric to telomeric): D10S579, D10S215, D10S1765, D10S541, D10S1735, D10S1739, and D10S564. 13 PTEN lies between D10S1765 and D10S541, a genetic distance of 1 cM but a physical distance of only several hundred kilobasepairs. For purposes of this study, to compare the immunohistochemical data to the LOH data, PTEN was considered to be physically deleted only when one or more immediately flanking (informative) markers centromeric and telomeric of PTEN showed LOH. Using this strict and conservative interpretation for monoallelic PTEN deletion, 6 of the tumors were shown to have a loss of one allele of the PTEN gene, another 7 were shown to have a loss flanking one side of (which may or may not include) PTEN. For these latter 7 tumors, potential hemizygosity at the PTEN locus was further assessed by screening for a T/G polymorphism within PTEN intron 8 (IVS8+32T/G), detected by differential digestion with the restriction endonuclease HincII, and the intragenic polymorphic markers AFM086wg9, D10S2491, and D10S2492. AFM086wg9 lies in intron 2 of PTEN. The likely intragenic marker order is centromere − D10S2491 − AFM086wg9 − D10S2492/IVS8+32T/G − telomere (Marsh and Eng, unpublished).
Of the 5 breast carcinomas that exhibited no immunohistochemical evidence of PTEN expression (graded −), 4 showed extensive LOH of markers flanking PTEN and hence, PTEN itself (Table 1 ▶ , Column 3 and Table 3 ▶ ). The fifth carcinoma had LOH on the telomeric side (D10S541) of PTEN. Further molecular analysis revealed retention of heterozygosity at AFM086wg9 but LOH at the IVS8+32T/G polymorphism, suggesting hemizygous deletion of the 3′ end of PTEN. Therefore, all 5 breast carcinomas that had negative PTEN immunostaining also had hemizygous PTEN deletion (Table 3) ▶ . None of these 5 had biallelic deletion of PTEN nor did they have a second intragenic PTEN hit, ie, mutation of the remaining allele.
Table 3.
Summary of PTEN Expression by Immunohistochemistry Compared to Molecular Analysis
| PTEN Expression | LOH* | ROH |
|---|---|---|
| PTEN− | 5 | 0 |
| PTEN+ | 5 | 1 |
| PTEN++ | 1 | 21 |
*LOH of both flanking markers or a minimum of LOH of one intragenic marker.
Of the 6 carcinomas that had weak PTEN immunostaining, graded +, 4 had been previously shown to have LOH of markers flanking one side or the other of PTEN and 2 showed no LOH of flanking markers (Tables 2 and 3) ▶ ▶ . Further LOH analysis within PTEN revealed that the 4 carcinomas with LOH of markers flanking one side of the gene also had LOH of at least one of the intragenic markers (Table 2) ▶ . Thus, these 4 tumors with decreased immunostaining seemed to have hemizygous deletion of PTEN or at least part of it. In the remaining two carcinomas without LOH of markers immediately flanking the gene, further analyses within the gene were uninformative or showed retention of heterozygosity (Tumors 45 and 57, Tables 2 and 3 ▶ ▶ ). In all likelihood, PTEN might not be altered at the structural level in that particular tumor.
Table 2.
Analysis of Correlation between PTEN Immunostaining and PTEN Intragenic LOH in Cases with Decreased Immunostaining and Apparently Discordant Tumors
| Tumor | Immuno-staining score | PTEN | |||||
|---|---|---|---|---|---|---|---|
| 10q22-23 Markers | |||||||
| S1765 | S2491 | AFM086 | S2492 | IVS8 | S541 | ||
| 41 | ++ | LOH | NI | LOH | ROH | ROH | NI |
| 52 | ++ | LOH | NI | ROH | ROH | N/A | ROH |
| 53 | ++ | LOH | ROH | ROH | ROH | ROH | LOH |
| 50 | ++ | LOH | ROH | ROH | NI | ROH | NI |
| 40 | + | LOH | N/A | NI | N/A | LOH | ROH |
| 59 | + | LOH | N/A | LOH | N/A | LOH | ROH |
| 66 | + | ROH | NI | LOH | N/A | N/A | LOH |
| 57 | + | ROH | LOH | LOH | N/A | N/A | ROH |
| 55 | + | ROH | NI | NI | N/A | LOH | LOH |
| 45 | + | ROH | NI | ROH | NI | ROH | ROH |
Tumor numbers correspond to those of Feilotter et al. 13
LOH, loss of heterozygosity; ROH, retention of heterozygosity; NI, not informative (germline homozygosity at marker); N/A, not applicable or not done.
Among the remaining 22 carcinomas that showed immunohistochemical evidence of strong PTEN expression (increased staining compared to normal mammary glands), 18 (82%) showed no LOH and biallelic presence of PTEN was demonstrated (Table 1) ▶ . There were 4 tumors that seemed to be immunostained (grade ++), yet showed LOH flanking PTEN (Tables 2 and 3) ▶ ▶ . However, it should be noted that 3 of these 4 tumors had LOH of D10S1765 immediately centromeric of PTEN but with either retention of heterozygosity or noninformativeness at D10S541 immediately 3′ of the gene. Further LOH analysis within PTEN corroborates the previous observations (Table 2) ▶ : in tumor 6, 3′ markers within the gene showed retention of heterozygosity and a 5′marker (AFM086wg9) showed LOH; in tumor 5, where D10S1765 showed LOH, markers within the gene (AFM086wg9 and D10S2492) and 3′ of the gene (D10S541) all showed retention of heterozygosity. Similarly, tumor 9, which had LOH at D10S1765, had 3 of 4 intragenic markers with retention of heterozygosity. Tumor 53 was unusual in that both D10S1765 and D10S541 had LOH, although molecular analysis demonstrated all 4 intragenic markers with retained heterozygosity.
Correlation of PTEN Immunohistochemistry and Clinicopathological Parameters
PTEN immunostaining status was compared with such clinicopathological parameters as age at diagnosis, size of primary tumor, tumor grade, lymph node status, and estrogen receptor and progesterone receptor status. Because of the relatively small numbers, especially in the context of subset analyses, no conclusions could be drawn with confidence from our observed correlations. The most interesting association seemed to be that between PTEN expression and hormone receptor status (Table 4) ▶ . Three of the 5 carcinomas (67%) that had no PTEN protein were estrogen and progesterone receptor-negative compared to 5 of 22 (23%; P < 0.05 Fisher’s exact test) in the PTEN-immunopositive samples. All 6 carcinomas that had weak PTEN staining were estrogen and progesterone receptor-positive. Other trends are also noteworthy. Although there were only 2 grade I tumors, both had high PTEN expression. All 5 tumors that were 1.5 cm or smaller had high levels of PTEN protein.
Table 4.
Estrogen/Progesterone Receptor Status of Breast Carcinomas by PTEN Immunostaining Status
| PTEN IHC status | ER/PR − | ER/PR + |
|---|---|---|
| Negative (−) | 3 | 2 |
| Decreased (+) | 0 | 7 |
| Positive (++) | 5 | 16 |
ER, estrogen receptor; PR, progesterone receptor.
An equivocal positive receptor status (n = 5) was scored as a positive.
Discussion
In this first report of immunohistochemical analyses of PTEN expression in sporadic primary breast carcinomas, we found that 33% of these tumors had either no or decreased expression of PTEN, which generally appeared to correlate with structural monoallelic deletion of the gene. Although it is understandable that tumors with monoallelic loss of PTEN have decreased PTEN expression at the protein level, one must explain the 5 samples with no immunoreactivity and structural PTEN hemizygosity. None of these samples was found to have intragenic PTEN mutations in the remaining allele, either. It is more than plausible, therefore, that an epigenetic phenomenon, such as hypermethylation of the promoter region 28 and decreased protein synthesis or increased protein turnover, 26 might be inactivating the remaining allele. Similarly, for the tumor (case 45) with decreased staining but no structural PTEN deletion, similar hypotheses may be raised. Other explanations include point mutations in the putative promoter of the remaining allele or normal tissue contamination of the breast samples, thus giving pseudo-hemizygosity in the face of real homozygous deletion. The latter can be discarded because very careful microdissection of the carcinoma components was performed by a pathologist with extensive experience in microdissection. Further, since the pattern of all positive and negative tumors was homogeneous, regional PTEN deletions in tumor subclones are very unlikely. Conversely, the observation of rare immunopositive tumors (n = 4) which appear to have LOH of flanking markers can be plausibly explained as well: at least in 3 informative tumors, no deletion of the gene proper or no deletion of most of the 3′ end of the gene has occurred. Hence, the monoclonal antibody, which is raised against the C terminus of PTEN, would still immunostain these samples positively. In this situation, therefore, incomplete 5′ deletion of PTEN might still be associated with translation of a truncated immunocompetent PTEN protein. In summary, while structural deletion or mutation of PTEN can lead to decreased PTEN protein levels, other mechanisms which lead to complete loss of PTEN expression seem to be prominent as well, at least in the breast carcinoma model.
Whether loss of PTEN expression is an early or late event in breast carcinogenesis is still controversial, although preliminary reports suggest that it is a late event. 11 The observation that loss of PTEN expression is correlated with a negative estrogen and progesterone status and that both grade I tumors had strong PTEN expression also strengthen this hypothesis. There is no doubt that these latter clinicopathological observations need to be investigated further. Nonetheless, these data in toto argue that despite the observation that germline PTEN mutations cause Cowden syndrome, 4 somatic PTEN mutation or functional loss of PTEN expression is associated with tumor progression and not tumor initiation, at least in the breast cancer model. It is also clear from our and other data that breast carcinogenesis does not rely uniformly on the involvement of the PTEN pathway, although how PTEN plays a role in various aspects of normal development and in the pathogenesis of breast carcinoma is not straightforward.
Acknowledgments
We thank Jeff FitzGerald for technical assistance, Dr. Oliver Gimm for expert administrative assistance and many members of CE’s laboratory for critical review of the manuscript.
Footnotes
Address reprint requests to Charis Eng, Human Cancer Genetics Program, Ohio State University Comprehensive Cancer Center, 690C Medical Research Facility, 420 W. 12th Avenue, Columbus, Ohio 43210. E-mail: eng-1@medctr.osu.edu.
Supported in part by grants from the American Cancer Society (RPG-97-064-01-VM and RPG98-211-01-CCE), the Department of Defense Breast Cancer Research Program (DAMD17-98-1-8058), the Concert for the Cure (to C. E.), the National Cancer Institute (P30CA16058, Comprehensive Cancer Center), and the Canadian Breast Cancer Research Initiative (to L. M. M.). P. L. M. D. is a Postdoctoral Fellow of the Susan G. Komen Breast Cancer Research Foundation (to C. E.) and A. P. is a Fellow of the Lydia Hochstrasser-Stiftung, Zürich, Switzerland (to P. K.).
References
- 1.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang S, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittman M, Tycko B, Hibshoosh H, Wigler MH, Parsons R: PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275:1943-1947 [DOI] [PubMed] [Google Scholar]
- 2.Li D-M, Sun H: TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor B. Cancer Res 1997, 57:2124-2129 [PubMed] [Google Scholar]
- 3.Steck PA, Pershouse MA, Jasser SA, Yung WKA, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, Frye C, Hu R, Swedlund B, Teng DHF, Tavtigian SV: Identification of a candidate tumor suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet 1997, 15:356-362 [DOI] [PubMed] [Google Scholar]
- 4.Liaw D, Marsh DJ, Li J, Dahia PLM, Wang SI, Zheng Z, Bose S, Call KM, Tsou HC, Peacocke M, Eng C, Parsons R: Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet 1997, 16:64-67 [DOI] [PubMed] [Google Scholar]
- 5.Eng C, Peacocke M: PTEN and inherited hamartoma-cancer syndromes. Nat Genet 1998, 19:223. [DOI] [PubMed] [Google Scholar]
- 6.Levine RL, Cargile CB, Blazes MS, van Rees B, Kurman RJ, Ellenson LH: PTEN mutations and microsatellite instability in complex atypical hyperplasia, a precursor lesion to uterine endometrioid carcinoma. Cancer Res 1998, 58:3254-3258 [PubMed] [Google Scholar]
- 7.Maxwell GL, Risinger JL, Gumbs C, Shaw H, Bentley RC, Barrett JC, Berchuck A, Futreal PA: Mutation of the PTEN tumor suppressor gene in endometrial hyperplasias. Cancer Res 1998, 58:2500-2503 [PubMed] [Google Scholar]
- 8.Tashiro H, Blazes MS, Wu R, Cho KR, Bose S, Wang SI, Li J, Parsons R, Ellenson LH: Mutations in PTEN are frequent in endometrial carcinoma but rare in other common gynecological malignancies. Cancer Res 1997, 57:3935-3940 [PubMed] [Google Scholar]
- 9.Kong D, Suzuki A, Zou T-T, Sakurada A, Kemp LW, Wakatsuki S, Yokohama T, Yamakawa H, Furukawa T, Sato M, Ohuchi N, Sato S, Yin J, Want S, Abraham JM, Souza RF, Smolinksi KN, Meltzer SJ, Horii A: PTEN1 is frequently mutated in primary endometrial carcinomas. Nat Genet 1997, 17:143-144 [DOI] [PubMed] [Google Scholar]
- 10.Maier D, Zhang ZW, Taylor E, Hamou MF, Gratzl O, van Meir EG, Scott RJ, Merlo A: Somatic deletion mapping on chromosome 10 and sequence analysis of PTEN/MMAC1 point to the 10q25–26 region as the primary target in low-grade and high-grade gliomas. Oncogene 1998, 16:3331-3335 [DOI] [PubMed] [Google Scholar]
- 11.Bose S, Wang SI, Terry MB, Hibshoosh H, Parsons R: Allelic loss of chromosome 10q23 is associated with tumor progression in breast carcinomas. Oncogene 1998, 17:123-127 [DOI] [PubMed] [Google Scholar]
- 12.Singh B, Ittman MM, Krolewski JJ: Sporadic breast cancers exhibit loss of heterozygosity on chromosome segment 10q23 close to the Cowden disease locus. Genes Chromosomes Cancer 1998, 21:166-171 [PubMed] [Google Scholar]
- 13.Feilotter HE, Coulon V, McVeigh JL, Boag AH, Dorion-Bonnet F, Duboué B, Latham WCW, Eng C, Mulligan LM, Longy M: Analysis of the 10q23 chromosomal region and the PTEN gene in human sporadic breast carcinoma. Br J Cancer 1999, 79:718-723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dürr E-M, Rollbrocker B, Hayashi Y, Peters N, Meyer-Puttlitz B, Louis DN, Schramm J, Wiestler OD, Parsons R, Eng C, von Deimling A: PTEN mutations in gliomas and glioneuronal tumors. Oncogene 1998, 16:2259-2264 [DOI] [PubMed] [Google Scholar]
- 15.Wang SI, Puc J, Li J, Bruce JN, Cairns P, Sidransky D, Parsons R: Somatic mutations of PTEN in glioblastoma multiforme. Cancer Res 1997, 57:4183-4186 [PubMed] [Google Scholar]
- 16.Rasheed BKA, Stenzel TT, McLendon RE, Parsons R, Friedman AH, Friedman HS, Bigner DD, Bigner SH: PTEN gene mutations are seen in high-grade but not in low-grade gliomas. Cancer Res 1997, 37:4187-4190 [PubMed] [Google Scholar]
- 17.Boström J, LudwigCobbers JMJ, Wolter M, Tabatabai G, Weber RG, Lichter P, Collins VP, Reifenberger G: Mutation of the PTEN (MMAC1) tumor suppressor gene in a subset of glioblastomas but not in meningiomas with loss of chromosome arm 10q. Cancer Res 1998, 58:29-33 [PubMed] [Google Scholar]
- 18.Tsao HS, Zhang X, Benoit E, Haluska FG: Identification of PTEN/MMAC1 alterations in uncultured melanomas and melanoma cell lines. Oncogene 1998, 16:3397-3402 [DOI] [PubMed] [Google Scholar]
- 19.Rhei E, Kang L, Bogomoliniy F, Federici MG, Borgen PI, Boyd J: Mutation analysis of the putative tumor suppressor gene PTEN/MMAC1 in primary breast carcinomas. Cancer Res 1997, 57:3657-3659 [PubMed] [Google Scholar]
- 20.Starink TM, van der Veen JPW, Arwert F, de Waal LP, de Lange GG, Gille JJP, Eriksson AW: The Cowden syndrome: a clinical and genetic study in 21 patients. Clin Genet 1986, 29:222-233 [DOI] [PubMed] [Google Scholar]
- 21.Eng C: Genetics of Cowden syndrome: through the looking glass of oncology. Int J Oncol 1998, 12:701-710 [DOI] [PubMed] [Google Scholar]
- 22.Eng C, Thomas GA, Neuberg DS, Mulligan LM, Healey CS, Houghton C, Frilling A, Raue F, Williams ED, Ponder BAJ: Mutation of the RET proto-oncogene is correlated with RET immunostaining in subpopulations of cells in sporadic medullary thyroid carcinoma. J Clin Endocrinol Metab 1998, 83:4310-4313 [DOI] [PubMed] [Google Scholar]
- 23.Komminoth P, Roth J, Schroder S, Saremaiani P, Heitz PU: Overlapping expression of immunohistochemical markers and synaptophysin mRNA in pheochromocytomas and adenocortical carcinomas. Lab Invest 1995, 72:424-431 [PubMed] [Google Scholar]
- 24.Heitz PU, Kasper M, Polak JM, Kloppel G: Pancreatic endocrine tumors. Hum Pathol 1982, 13:263-271 [DOI] [PubMed] [Google Scholar]
- 25.Werner M, von Wasiekewski R, Komminoth P: Antigen retrieval, signal amplification and intensification in immunohistochemistry. Histochem Cell Biol 1996, 105:253-260 [DOI] [PubMed] [Google Scholar]
- 26.Dahia PLM, Aguiar RCT, Alberta J, Kum J, Caron S, Sills H, Marsh DJM, Freedman A, Ritz J, Stiles C, Eng C: PTEN is inversely correlated with the cell survival factor PKB/Akt and is inactivated by diverse mechanisms in haematologic malignancies. Hum Mol Genet 1999, in press [DOI] [PubMed]
- 27.Dahia PLM, Marsh DJ, Zheng Z, Zedenius J, Komminoth P, Frisk T, Wallin G, Parsons R, Longy M, Larsson C, Eng C: Somatic deletions and mutations in the Cowden disease gene, PTEN, in sporadic thyroid tumors. Cancer Res 1997, 57:4710-4713 [PubMed] [Google Scholar]
- 28.Whang YE, Wu X, Suzuki H, Reiter RE, Tran C, Vessella RL, Said JW, Isaacs WB, Sawyers CL: Inactivation of the tumor suppressor PTEN/MMAC1 in advanced human prostate cancer through loss of expression. Proc Natl Acad Sci USA 1998, 95:5246-5250 [DOI] [PMC free article] [PubMed] [Google Scholar]