

Abstract
Cyclooxygenase-1 (Cox-1) and Cox-2 convert arachidonic acid to prostaglandin H2, the precursor of other prostaglandins and thromboxanes, eicosanoids important in vascular pathophysiology. However, knowledge of the expression of cyclooxygenases within atherosclerotic lesions is scant. This study tested the hypothesis that human atheroma and nonatherosclerotic arteries express the two Cox isoforms differentially. Cox-1 mRNA and protein localized on endothelial and medial smooth muscle cells of normal arteries (n = 5), whereas Cox-2 expression was not detectable. In contrast, atheromatous (n = 7) lesions contained both Cox-1 and Cox-2, colocalizing mainly with macrophages of the shoulder region and lipid core periphery, whereas smooth muscle cells showed lower levels, as demonstrated by immunohistochemical and in situ hybridization analysis. Furthermore, microvascular endothelium in plaques showed notable staining for both isoforms. In accord with immunohistochemical studies, Western blot analysis of protein extracts from normal arteries revealed constitutive Cox-1, but not Cox-2, expression. Extracts of atheromatous lesions, however, contained both Cox-1 and Cox-2 protein, detected as two immunoreactive proteins of approximately 70 and 50 kd. Macrophages expressed the short form of Cox-1/-2 constitutively after several days of in vitro culture, rather than the 70-kd protein. These results shed new light on the inflammatory pathways that operate in human atheroma. In particular, the expression of Cox-2 in atheromatous, but not in unaffected, arteries has therapeutic implications, given the advent of selective Cox-2 inhibitors.
During the last three decades many studies have addressed the roles of eicosanoids, such as prostacyclin (PGI2), thromboxane A2 (TXA2), and prostaglandin E2 (PGE2), in vascular pathophysiology. 1-4 The actions of PGI2 include inhibition of platelet aggregation and vasodilatation, reduction of cholesterol accumulation, and inhibition of vascular smooth muscle cell (SMC) proliferation. 5-7 TXA2 action is linked to the inositol-phosphate pathway and causes vasoconstriction and platelet aggregation. 8 PGE2 inhibits cholesterol esterification and can increase cholesterol synthesis by negative feedback. 9 Furthermore, PGE2 augments matrix metalloproteinase (MMP) expression in MΦ, 10 enzymes considered crucial for vulnerable plaque evolution.
Cyclooxygenases convert arachidonic acid to prostaglandin G/H2 and, hence, regulate eicosanoid synthesis. Cyclooxygenase-1 (Cox-1), originally purified from bovine vesicular glands 11 and cloned from sheep vesicular glands, 12-14 is constitutively expressed in many human tissues, eg, stomach, kidney, platelets, and the central nervous system. 15 Interestingly, the prostaglandin production in these tissues varies and correlates with the degree of inflammation, despite constitutive prostaglandin G/H synthase expression. Differential screening eventually revealed the presence of a second, inducible cyclooxygenase, called prostaglandin endoperoxide synthase-2 or Cox-2. 16,17 Induction of this isoform by several mediators, including proinflammatory cytokines, such as interleukin (IL)-1, tumor necrosis factor (TNF), interferon γ, endotoxin, growth factors, or shear stress implied a function for Cox-2 in both inflammation and regulation of cell growth, as reviewed elsewhere. 18,19 Recent reports further implicate cyclooxygenase products in the regulation of angiogenesis 20 and of apoptosis. 21,22 In vitro, Cox-2 is inducible in fibroblasts 23 and mucosal cells, 24 as well as in the atheroma-associated cell types endothelial cells (EC), SMC, and macrophages (MΦ). 25-28 In addition, IL-1 induces Cox-2 in human saphenous vein and internal mammary arteries segments in organ culture. 29 Even though human atherosclerotic lesions contain several of the above listed mediators of cyclooxygenase induction, 30 we possess no information concerning the expression of Cox-1 and/or Cox-2 in atheromatous tissue.
Both cyclooxygenases have a molecular weight of approximately 70 kd, share 70% identity in their amino acid sequence, and possess similar three-dimensional structures. 18,31,32 Even though the inducible enzyme Cox-2 closely resembles in structure and catalytic activity the constitutive Cox-1, the two isoforms have important differences in substrate and inhibitor selectivity, their intracellular location, and their biological function. 18 Both enzymes use arachidonic acid equally well. Cox-2, however, converts other fatty acid substrates, such as linolenic or linoleic acid, more efficiently. Furthermore, Cox-2 is less sensitive to aspirin inhibition than is Cox-1. 33,34 Aspirin inhibits Cox-1 in platelets, reducing thrombotic potential, probably via decreased production of prostaglandins, such as thromboxane A2. 35 The variation in the biological function of Cox isoforms may relate to their subcellular localization: Cox-1 primarily in the endoplasmic reticulum and Cox-2 in both the endoplasmic reticulum and the perinuclear space. 36
Despite the recognition that the Cox-1 and Cox-2 isoforms have distinct regulation, we have little knowledge of their relative importance in atherogenesis. This issue is particularly important given both the increased recognition of the inflammatory nature of atheroma, which might induce the proinflammatory isoform Cox-2, and the current availability of pharmacological agents that inhibit specifically this isoform. The present study, therefore, tested the hypothesis that human atherosclerotic lesions exhibit augmented Cox-2 expression.
Materials and Methods
Materials
Human recombinant IL-1β and TNFα were obtained from Endogen (Cambridge, MA), Escherichia coli endotoxin lipopolysaccharide (LPS) from Sigma (St. Louis, MO). Recombinant human CD 40 ligand (rCD40L) was prepared as described previously. 37 Goat polyclonal antibodies against human Cox-1 and Cox-2 as well as recombinant human Cox-1 and Cox-2 blocking peptides were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The respective mouse anti-human Cox-1 and Cox-2 monoclonal antibodies were provided by Cayman Chemicals (Ann Arbor, MI). Control mAb and rabbit Ig used for immunohistochemistry were obtained from PharMingen (La Jolla, CA).
Cell Isolation and Culture
Human vascular EC were isolated from saphenous veins by collagenase treatment (1 mg/ml; Worthington Biochemicals, Freehold, NJ) and cultured in dishes coated with fibronectin (1.5 μg/cm 2 New York Blood Center Reagents, New York, NY). Cells were maintained in medium 199 (BioWhittaker, Walkersville, MD), supplemented with 1% penicillin/streptomycin (BioWhittaker), 5% fetal bovine serum (FBS; Atlanta Biologicals, Norcross, GA), 50 μg/ml heparin (Sigma) and ECGF (endothelial cell growth factor; Pel-Freez Biological, Rogers, AK). Human vascular SMC were isolated from human saphenous veins by explant outgrowth 38 and cultured in DMEM (BioWhittaker) supplemented with 1% L-glutamine (BioWhittaker), 1% penicillin/streptomycin, and 10% FBS. Both cell types were subcultured after trypsinization in 0.5% trypsin (Worthington Biochemicals)/0.2% EDTA (EM Science, Gibbstown, NJ) in 75-cm 2 culture flasks (Becton Dickinson, Franklin Lakes, NJ), and used throughout passages two to four. Culture media and FBS contained <40 pg endotoxin/ml as determined by the chromogenic Limulus amoebocyte assay (QLC-1000; BioWhittaker). EC and SMC were characterized by immunostaining with anti-von Willebrand factor and anti-SMC α-actin antibody (Dako, Carpinteria, CA), respectively. Both cell types were cultured 24 hours before the experiment in media lacking FBS: vascular EC were cultured in M199 supplemented with 0.1% human serum albumin and vascular SMC in insulin/transferrin (IT) medium, as described previously. 39
Mononuclear phagocytes were isolated by density gradient centrifugation, using Lymphocyte Separation Medium (Organon-Teknika, Durham, NC), and subsequent counterflow elutriation from freshly prepared human peripheral blood mononuclear cells (PBMC) obtained from leukopacs of healthy donors (kindly provided by Dr. B. Rollins, Dana Farber Cancer Institute, Boston, MA). Mononuclear phagocytes were either used directly for the experiments (monocytes) or cultured for 1, 3, or 9 days (MΦ) in RPMI 1640 containing 2% human serum (Sigma). The purity of monocytes and MΦ was ≥96%, as determined by fluorescence-activated cell sorter analysis (anti-human CD68 mAb FITC, PharMingen). For certain studies, MΦ were stimulated in RPMI 1640 lacking serum.
Immunohistochemistry
Surgical specimens of human carotid atheroma and aorta were obtained by protocols approved by the Human Investigation Review Committee at the Brigham and Women’s Hospital. Serial cryostat sections (5 μm) were cut, air dried onto microscope slides (Fisher Scientific, Pittsburgh, PA), and fixed in acetone at −20°C for 5 minutes. Sections were pre-incubated with phosphate-buffered saline (PBS) containing 0.3% hydrogen peroxidase activity. The sections were then incubated for 90 minutes with primary or control (mouse myeloma protein MOPC-21, Sigma) antibody, diluted in PBS supplemented with 5% appropriate serum. After washing three times in PBS, sections were incubated with the respective biotinylated secondary antibody (45 minutes; Vector, Burlingame, CA) followed by avidin-biotin-peroxidase complex (Vectastain ABC kit, Vector), and antibody binding was visualized with 3-amino-9-ethyl carbazole (Vector) according to the recommendations provided by the supplier. For colocalization of Cox-1 or Cox-2 with the respective cell type, double-immunofluorescence staining was performed. The anti-human Cox-1 and Cox-2 Ab (1:200) was applied for 90 minutes, followed by biotinylated anti-mouse/goat secondary antibody for 45 minutes and Texas red-conjugated streptavidin (Amersham, Arlington Heights, IL). After application of the avidin/biotin blocking kit (Vector), anti-muscle actin mAb for SMC (Enzo Diagnostics, New York, NY), anti-CD31 mAb for EC (1:400, Dako), or anti-CD68 mAb for MΦ (1:600, Dako) were added and sections incubated overnight at 4°C. Subsequently, biotinylated horse-anti-mouse secondary antibodies were applied for 30 minutes, followed by streptavidin-FITC (Amersham).
In Situ Hybridization
In situ hybridization was performed according to the instructions of the manufacturer (Hyb-Probe, Shandon/Lipshaw, Pittsburgh, PA). Briefly, frozen tissue sections obtained as described above were fixed in cold acetone, air-dried, and incubated with a mixture of FITC-labeled Cox-1 (5′-GTGACCTTGTACCGATCGGAAAGAACATCG-3′; 5′-TACGAAGTCGTTCGTCGGGAGGTGAGGTCG3′; 5′-CAACCGAGGTTTGACGAGGGTA GTAAGGAA-3′) or random oligomers in hybridization-buffer (30% formamide, 0.6 mol/L NaCl2, 10% dextran sulfate, 50 mmol/L Tris (pH7.5), 0.1% sodium-pyro-phosphate, 0.2% Ficoll, 5 mmol/L EDTA) for 10 minutes at 65°C and subsequently for 2 hours at 37°C in a moist chamber. Finally, slides were washed 3 times and forwarded to the immunological reaction employing alkaline phosphatase-conjugated rabbit Fab′ anti-FITC (30 minutes) and Nitroblue tetrazolium/5-bromo-4-chloro-3-indoyl phosphate chromogen solution (1 hour).
Biochemical Analysis of Human Atherosclerotic Lesions
Frozen tissue from five nonatherosclerotic arteries and seven atheromatous carotid plaques were homogenized (IKA-Labortechnik, Dortmund, Germany, Ultra-turrax T 25) and lysed (0.3 mg tissue/ml lysis buffer) as described previously. 40 The lysates were clarified (16,000 × g, 15 minutes) and the protein concentration for each tissue extract as well as for the cell culture samples was determined using a bicinchoninic acid protein assay according to the instructions of the manufacturer (Pierce, Rockford, IL).
Western Blot Analysis
Tissue extracts (50 μg total protein/lane), cell extracts (20 μg total protein/lane), and culture supernatants (10×) were separated by standard sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) under reducing conditions and blotted to polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA) using a semidry blotting apparatus (0.8 mA/cm2, 30 minutes; Bio-Rad). Blots were blocked and first and second monoclonal antibodies were diluted in 5% defatted dry milk/PBS/0.1% Tween 20. After 1 hour of incubation with the respective primary antibody, blots were washed three times (PBS/0.1% Tween) and the secondary, peroxidase-conjugated goat-anti-mouse antibody (Jackson Immunoresearch, West Grove, PA) was added for another 1 hour. Finally, the blots were washed (20 minutes, PBS/0.1% Tween 20) and immunoreactive proteins were visualized using the Western blot chemiluminescence system (New England Nuclear, Boston, MA).
Results
Human Atherosclerotic Lesions Express both Cox-1 and Cox-2 Protein in Situ
Normal arterial tissue (n = 5) and atherosclerotic lesions (n = 7) contained immunostainable Cox-1 (Figure 1) ▶ . In contrast, nonatherosclerotic arterial tissue had little or no Cox-2. Interestingly, staining for Cox-2 was abundant in atheromatous lesions compared to normal arterial tissue. Within the lesion, Cox-1 and Cox-2 accumulated in the shoulder region of the lesions as well as the periphery of the lipid core, areas also staining for MΦ (anti-CD68, data not shown). Immunofluorescence double-labeling associated the expression of Cox-1 in normal tissue with vascular EC and SMC (data not shown). Within the atherosclerotic lesion, both cyclooxygenase isoforms colocalized with EC and SMC, but showed brightest signals in MΦ (Figure 2, 3) ▶ ▶ . The endothelium of plaque microvessels also showed prominent Cox-1 and Cox-2 staining (Figure 4) ▶ . Preincubation of the Cox-antibodies with the respective peptide inhibited staining, indicating the specificity of the signals obtained (data not shown). Immunohistochemical analysis performed with the polyclonal or the monoclonal anti-Cox-1/-2 antibody showed similar results. Tissues showed no staining with an irrelevant IgG1 antibody (Figure 1 ▶ , bottom left panel).
Figure 1.

Expression of Cox-1 and Cox-2 in normal arterial and atherosclerotic plaque tissue. Frozen sections of nonatherosclerotic human arteries and atheromatous plaques were stained for Cox-1 and Cox-2 employing horseradish-peroxidase-mediated immunohistochemistry (red reaction product). No immunoreactivity was observed in tissue stained with the respective control IgG1 antibody (control). The lumen of the artery is at the top of each photomicrograph. Analysis of five normal aortic tissue and seven atheroma obtained from different donors showed similar results.
Figure 2.
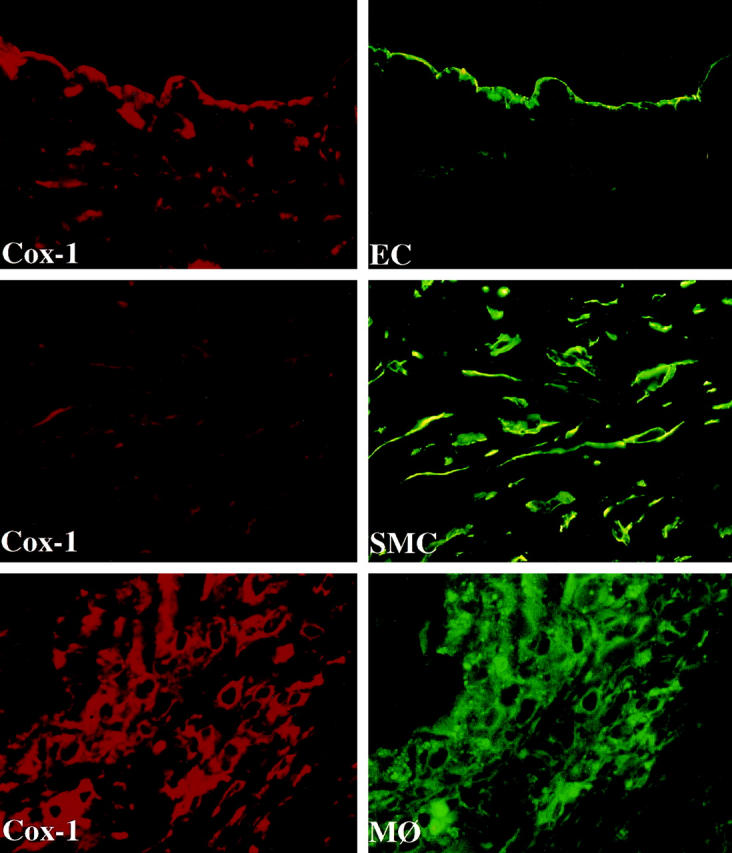
Colocalization of Cox-1 with endothelial cells (EC), smooth muscle cells (SMC), and macrophages (MΦ) in human atheroma. High power views (×400) of frozen sections of human carotid lesions showed specific staining for Cox-1 (red staining) on human vascular EC, SMC, and MΦ within the atheroma. Cell types were characterized by immunofluorescence-double labeling (green staining) as described in Materials and Methods. The lumen of the artery is at the top of each photomicrograph. Analysis of atheroma obtained from five different donors showed similar results.
Figure 3.

Colocalization of Cox-2 with EC, SMC, and MΦ in human atheroma. High power views (×400) of frozen sections of human carotid lesions showed specific staining for Cox-2 (red staining) on human vascular EC, SMC, and MΦ within the atheroma. Cell types were characterized by immunofluorescence-double staining (green staining) as described in Materials and Methods. The lumen of the artery is at the top of each photomicrograph. Analysis of atheroma obtained from five different donors showed similar results.
Figure 4.

Colocalization of Cox-1 and Cox-2 with microvascular endothelium. High power views (×400) of frozen sections of human carotid lesions showed specific staining for Cox-1 or Cox-2 (red staining), respectively, on microvascular endothelium. Cell types were characterized by immunofluorescence double-staining (green staining) as described in Materials and Methods. Analysis of atheroma obtained from five different donors showed similar results.
Human Atherosclerotic Lesions Express Cox-1 mRNA in Situ
Further characterization of the variation in cell-type specific expression of Cox-1 in nonatherosclerotic and atheromatous arterial tissue used in situ hybridization. In accord with the immunohistochemical data, Cox-1 transcripts localized in EC and SMC of the normal vessel wall (Figure 5) ▶ . Within human atherosclerotic lesions, however, Cox-1 mRNA (although expressed in EC and SMC as well) localized most prominently in the shoulder region of the lesion and the periphery of the lipid core, areas characterized by immunohistochemistry on adjacent sections as smooth muscle cell-poor and MΦ-rich (data not shown). Furthermore, SMC within the tunica media underlying the lesion as well as in sections with normal vessel morphology, stained strongly for Cox-1 transcripts. In situ hybridization with negative control probes yielded no signal.
Figure 5.
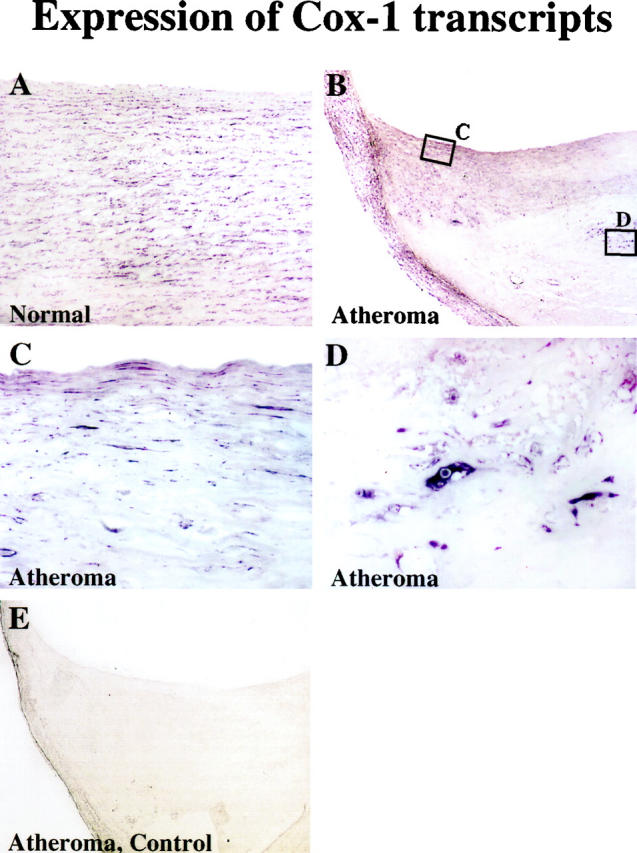
Human Cox-1 mRNA colocalizes with EC, SMC, and MΦ of the atherosclerotic lesion. In situ hybridization analysis for Cox-1 expression in nonatherosclerotic (A) and atheromatous plaque (B-D) tissue. Higher magnification (×400) revealed colocalization of Cox-1 transcripts with MΦ- as well as EC- and SMC-like cell types within the lesion. Negative control probes yielded no detectable signal (E). Analysis of three nonatherosclerotic arteries and three atheroma from different donors showed similar results.
Western Blot Analysis of Cox-1 and Cox-2 Expression in Human Atherosclerotic Plaques
Western blot analysis performed on extracts of the surgical specimens using antibodies identical to those used for the immunohistochemistry studies revealed immunoreactive Cox-1 in both normal arterial and atherosclerotic tissue (Figure 6 ▶ , right panel). The analysis demonstrated two major immunoreactive proteins, migrating at 70 kd and 50 kd. The 50-kd band was more pronounced in atherosclerotic tissue extracts. The higher molecular weight band comigrated with the prominent immunoreactive protein obtained with lysates of unstimulated as well as IL-1β/TNFα stimulated EC and SMC. The lower molecular weight band, however, was particularly prominent in lysates of MΦ, compared to EC and SMC. Supernatants of either unstimulated or stimulated EC, SMC or MΦ did not contain Cox-1. In accord with the immunohistochemical studies, Western blot analysis revealed no or only little immunoreactive Cox-2 in extracts of nonatherosclerotic tissue, but showed markedly increased immunoreactive Cox-2 protein in atheromatous lesions. As in the case of Cox-1, we observed two immunoreactive Cox-2 proteins with approximate molecular weights of 70 kd and 50 kd. Also similar to the studies on Cox-1 expression, the higher molecular weight Cox-2 band obtained with tissue extracts comigrated with the prominent immunoreactive protein detected in lysates of IL-1β/TNFα-stimulated EC and SMC, whereas the lower molecular weight form was the prominent band in extracts of activated MΦ derived from peripheral blood monocytes after nine days of in vitro culture. Lysates of unstimulated MΦ, but not of EC or SMC cultures, expressed immunoreactive Cox-2 protein. Furthermore, IL-1/TNFα stimulated EC, but not SMC and MΦ cultures released immunoreactive Cox-2. Besides IL-1β/TNFα, classic mediators of Cox-2 expression, we also used rCD40L as a stimulus, resulting in the increased levels of Cox-2, but not Cox-1, in all three cell types (Figure 6) ▶ . These findings agree with a recent study demonstrating that CD40 engagement up-regulates Cox-2 in human lung fibroblasts. 23
Figure 6.

Human atherosclerotic lesions express immunoreactive forms of Cox-1 and Cox-2. Protein extracts (Lys; 20 μg/lane) and supernatants (SN; 50 μl, ×10) of EC, SMC, and MΦ, cultured 24 hours in the absence (None) or presence of IL-1β/TNFα (10 ng/50 ng/ml), rCD40L (10 μg/ml), or endotoxin (100 ng LPS/ml), as well as tissue extracts (50 μg/lane) of nonatherosclerotic tissue (Normal) and atheromatous plaques (Atheroma), were separated by standard SDS-PAGE under reducing conditions and analyzed by Western blotting for Cox-1 and Cox-2 expression. For specificity control, atheroma tissue extract (sample 4) was analyzed by Western blotting performed with the Cox-1 or Cox-2 antibody preincubated for 30 minutes at 37°C with the respective blocking peptide (10 μg/ml). Arrowheads at left indicate the positions of the molecular weight markers. Analysis of four normal tissues as well as four atheromatous lesions from eight different donors showed similar results.
To explore further the pattern of Cox-1 and Cox-2 expression in MΦ, we analyzed whether MΦ differentiation might regulate expression of the observed immunoreactive bands. Freshly isolated peripheral blood monocytes as well as MΦ derived from monocytes after 1 day of in vitro culture, incubated for 24 hours with medium or LPS, expressed Cox-1 constitutively with a molecular weight of 70 kd (Figure 7) ▶ . After 3 and, more markedly, after 9 days of in vitro culture, a constitutively expressed second immunoreactive Cox-1 and Cox-2 protein was detected with an molecular weight of approximately 50 kd. Western blot analysis on these lysates for Cox-2 revealed that the inducibility of the 70-kd form diminishes with increased time in culture of the MΦ. In contrast, the lower molecular weight form, only moderately expressed in freshly isolated or 1-day cultured monocyte-derived MΦ, rose with time of culture. Stimulation of MΦ with rCD40L yielded findings similar to those described above for LPS-stimulated cultures (data not shown).
Figure 7.

Differential expression of Cox-1 and Cox-2 during MΦ differentiation. Extracts of freshly isolated (0 days) as well as 1-, 3-, or 9-day cultured peripheral blood monocytes, cultured 24 hours in the absence (None) or presence (LPS) of endotoxin (100 ng/ml), were separated by standard SDS-PAGE under reducing conditions and analyzed by Western blotting for Cox-1 and Cox-2 expression. Arrowheads at left indicate the positions of the molecular weight markers. Analysis of MΦ extracts from three different donors showed similar results.
Discussion
Cyclooxygenases catalyze the conversion of arachidonic acid to eicosanoids, which mediate a variety of biological actions involved in vascular pathophysiology. 1-10,20-22 Despite the findings that cyclooxygenases can modulate functions crucial in atherogenesis, little is known of the relative contribution of the two Cox isoforms. This study demonstrates in situ the expression of Cox-1 and Cox-2 within human atherosclerotic lesions, but only of constitutive Cox-1 in undiseased arteries. The constitutive expression of Cox-1 is further supported by comparable amounts of immunoreactive protein in protein extracts of either tissue. (Whether the lower-molecular-weight form is an isoform or a processing product is unknown and will be a subject of future investigations.) Our findings further correspond with the constitutive expression of Cox-1 in most tissues and cells, 15 and with the lack of Cox-2 expression in freshly prepared saphenous veins and internal mammary arteries as well as umbilical veins and arteries. 27,29 Finally, Rimarachin et al demonstrated that the expression of Cox-2 after in vivo vascular injury extends over many days, whereas the expression of Cox-1 remains unchanged. 41 However, the present finding of heterogeneous Cox-1 expression in SMC within the atheromatous vessel is unanticipated. MΦ of the shoulder region and the lipid core periphery, not SMC, contain most of the Cox-1 protein within the lesion. In contrast, medial SMC underlying the plaque as well as those in adjacent sections with normal morphology exhibit prominent Cox-1 staining. This finding may have functional importance, because different cell types can regulate the production of different eicosanoids. Endothelium predominantly releases prostaglandin I2, 42 a potent inhibitor of platelet activation and cholesterol accumulation, 5,6,43 whereas MΦ, not present in normal arterial tissue, produce an array of prostanoids, including prostaglandin E2 and thromboxane A2, 28 considered the more atherogenic eicosanoids. The in vivo finding that prostacyclin agonists suppress MΦ atherogenic activity and thus inhibit the development of early atherosclerosis 44 heightens the relevance of MΦ-derived eicosanoids. Interestingly, previous studies demonstrated that cyclooxygenase products, such as PGI2, but not PGE2, augment cholesteryl ester hydrolase activity, whereas PGE2, but not PGI2, inhibits Acyl-CoA cholesterol acyl-transferase activity in human vascular SMC, 45,46 highlighting a possible lipid accumulation-reducing (hence anti-atherogenic) function of elevated Cox-2 expression. Future studies are needed to determine whether and how enhanced cyclooxygenase expression functionally affects atherogenesis and the net effect in vivo of the interplay between the anti- and pro-atherogenic products of cyclooxygenases, particularly with regard to the vascular wall-associated, Cox-2-mediated synthesis of prostacyclin, a potent vasodilator and endogenous inhibitor of platelet aggregation. 47
The constitutive expression of Cox-1 in diseased as well as undiseased arterial tissue, however, implicates a more physiological rather than inflammatory role of this enzyme with homeostatic functions, as recently reviewed elsewhere. 48
Interestingly, both Cox-1 and Cox-2 predominantly localize with lesional MΦ, a finding that agrees with observations in abdominal aortic aneurysms, where MΦ also represent the majority of Cox-expressing cells. 49 Increased Cox-2 expression within the lesion, a site of chronic inflammation, further agrees with reports describing Cox-2 expression in atheroma-associated cells, including EC, SMC, and MΦ, 25-28 on stimulation with proinflammatory cytokines such as IL-1 and TNFα, mediators found within human atherosclerotic lesions. 30 We recently demonstrated the presence of another inflammatory pathway in the atherosclerotic plaque, the CD40-CD40L receptor-ligand pair, 50 which modulates atheroma-associated functions in vitro 39,50-52 and in vivo. 53 We therefore tested whether CD40 ligation affects the expression of either cyclooxygenase in vascular cells. Our finding that recombinant CD40L potently stimulates the expression of cyclooxygenase-2 in EC, SMC, and MΦ agrees with the recently published study of Zhang et al, 23 demonstrating that CD40 engagement up-regulates this isoform in human lung fibroblasts. The colocalization of the enzyme with CD40-positive lesional EC, SMC, and MΦ (GK Sukhova, U Schönbeck, unpublished observations) supports the potential importance of CD40/CD40L in the regulation of Cox-2 within human atheroma.
Some eicosanoids may play a protective role in cardiovascular and inflammatory diseases, as they reduce adhesion molecule expression, platelet activation, and SMC proliferation. 5-7,43,54 In contrast, studies with Cox inhibitors indicated a proinflammatory effect of Cox-2. 55,56 An intriguing and novel potential proatherogenic mechanism of cyclooxygenase products is supported by the very recent demonstration by Tsujii et al 20 that Cox-1 activity in EC modulates angiogenesis. Neovessel formation, furthermore, required the presence of Cox-2, mediating the synthesis of angiogenic factors. The formation of neovessels may contribute to the evolution of the plaque. 57 Indeed, the microvascular endothelium prominently expressed both Cox isoforms, raising the possibility that parallel presence of Cox-1 and Cox-2 within the lesion contributes to the formation of new blood vessels, thus allowing the plaque to expand.
Prostanoids also have potent actions on vascular SMC, regulating contractility, cholesterol metabolism, and proliferation. 5-7 Increased expression of cyclooxygenases might thus contribute to the accumulation of lipids in lesional SMC (and MΦ), favoring formation of SMC- and MΦ-derived foam cells within atheroma. On the other hand, antiproliferative and antimigratory 58 actions of Cox products on human vascular SMC, in combination with our finding that the expression of lesional cyclooxygenases depends mainly on the content of MΦ, suggests potential contributions of the enzymes to the evolution of a lesion toward an SMC-depleted and MΦ-enriched, and thus more vulnerable, plaque. Interestingly, prostaglandin E2, a predominant eicosanoid of MΦ, induces, 10 whereas PGI2, the predominant arachidonic acid product in vascular cells, inhibits 58 the expression of MMPs, enzymes considered crucial in the degradation of plaque stability. Our previous description 40 of various MMPs in regions reported here as Cox-positive and found to be MΦ-enriched suggest that such regulation of MMP expression by Cox products may operate in vivo. Furthermore, we found by Western blot analysis that morphologically stable (SMC-enriched and MΦ-depleted) plaques expressed substantially less Cox-1, Cox-2, and MMPs compared to lesions with more unstable features (U Schönbeck, GK Sukhova, unpublished observation). Finally, prostaglandins inhibit the production of extracellular matrix macromolecules, such as fibronectin and type I and III collagen, further favoring plaque fragility. 59
It remains to be determined how cyclooxygenase products mediate their actions. Two classes of prostaglandin receptors can transduce signals on binding of the ligand: the G-coupled cytoplasmic receptors 60 and the nuclear peroxisome proliferator-activated receptor (PPAR) class. 61 The two Cox isoforms may exert different functions 18 because of their location in separate subcellular compartments. 36 As Cox-2, but not Cox-1, 36 localizes in the perinuclear region, its product may have more ready access to nuclear receptors. One Cox product, prostaglandin J2, is a potent ligand for the PPAR-γ, 61,62 a nuclear receptor that forms parts of a transcriptional complex after ligand binding. 63 We and others have previously demonstrated that atheroma-associated cells express PPARs, that ligation by prostaglandin J2 regulates atheroma-associated gene expression within these cells, and that PPARs are expressed within human atherosclerotic lesions. 58,61,62,64
In summary, this study demonstrates the expression of both Cox-1 and Cox-2 by EC, SMC, and particularly by MΦ within human atherosclerotic lesions. Although the in vivo function of the two isoforms remains to be determined, atherogenic rather than anti-atherogenic effects may prevail. The present findings indicate new potential inflammatory pathways in the evolution of atherosclerotic lesions, which have therapeutic implications in view of the recent availability of selective Cox-1 and particularly Cox-2 inhibitors. Conclusions from our findings on the potential role of Cox-2 inhibitors can only be speculative in nature. However, these findings suggest, in combination with previous reports that selective inhibition of Cox-2 results in profound suppression of PGE2 65 andsystemic prostacyclin biosynthesis, 66 mediators mostly considered anti-atherogenic, that future clinical trials may have to consider the possibility of proatherogenic effects during treatment with Cox-2-specific inhibitors.
Acknowledgments
We thank Maria Muszynski, Eugenia Shvartz, Irina Chulsky, and Elissa Simon-Morrissey (Brigham and Women’s Hospital) for their skillful technical assistance.
Footnotes
Address reprint requests to Peter Libby, M.D., Vascular Medicine and Atherosclerosis Unit, Cardiovascular Division, Department of Medicine, Brigham and Women’s Hospital and Harvard Medical School, 221 Longwood Avenue, LMRC 307, Boston, MA 02115. E-mail: plibby@rics.bwh.harvard.edu.
Supported in part by grants of the National Heart, Lung and Blood Institute to Dr. Peter Libby (HL-56985) and performed during the tenure of the Paul Dudley White fellowship of the American Heart Association by Dr. Uwe Schönbeck.
References
- 1.Sinzinger H, Feigl W, Silberbauer K: Prostacyclin generation in atherosclerotic arteries. Lancet 1979, 2:469-470 [DOI] [PubMed] [Google Scholar]
- 2.FitzGerald GA, Smith B, Pedersen AK, Brash AR: Increased prostacyclin biosynthesis in patients with severe atherosclerosis and platelet activation. N Engl J Med 1984, 310:1056-1068 [DOI] [PubMed] [Google Scholar]
- 3.Rush DS, Kerstein MD, Bellan JA, Knoop SM, Mayeux PR, Hyman AL, Kadowitz PJ, McNamara DB: Prostacyclin, thromboxane A2, and prostaglandin E2 formation in atherosclerotic human carotid arteries. Arteriosclerosis 1988, 8:73-78 [DOI] [PubMed] [Google Scholar]
- 4.Mehta JL, Lawson D, Mehta P, Saldeen T: Increased prostacyclin and thromboxane A2 biosynthesis in atherosclerosis. Proc Natl Acad Sci USA 1988, 88:4511-4515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moncada S, Gryglewski R, Bunting S, Vane JR: An enzyme isolated from arteries reforms prostaglandin endoperoxides to an unstable substance that inhibits platelet aggregation. Nature 1976, 263:663-665 [DOI] [PubMed] [Google Scholar]
- 6.Hajjar DP, Pomerantz KB: Signal transduction in atherosclerosis: integration of cytokines and the eicosanoid. FASEB J 1992, 6:2933-2941 [DOI] [PubMed] [Google Scholar]
- 7.Huttner JJ, Gwebu ET, Panganamala RV, Milo GE, Cornwell DG, Sharma HM, Geer JC: Fatty acids and their prostaglandin derivatives: inhibitors of proliferation in aortic smooth muscle cells. Science 1977, 197:289-291 [DOI] [PubMed] [Google Scholar]
- 8.Lefer AM, Smith EF, Araki H, Smith JB, Aharony D, Claremon DA, Magolda RL, Nicolaou KC: Dissociation of vasoconstrictor and platelet aggregatory activities of thromboxane by carbocyclic thromboxane A2, a stable analog of thromboxane A2. Proc Natl Acad Sci USA 1980, 77:1706-1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berberian PA, Ziboh VA, Hsia SL: Inhibition of cholesterol esterification in rabbit aorta by prostaglandin E2. Atherosclerosis 1977, 27:213-220 [DOI] [PubMed] [Google Scholar]
- 10.Corcoran ML, Stetler-Stevenson WG, Brown PD, Wahl LM: Interleukin 4 inhibition of prostaglandin E2 synthesis blocks interstitial collagenase, and 92-kd Type IV collagenase/gelatinase production by human monocytes. J Biol Chem 1992, 267:515-519 [PubMed] [Google Scholar]
- 11.Miyamoto T, Ogino N, Yamamoto S, Hayaishi O: Purification of prostaglandin endoperoxide synthetase from bovine vesicular gland microsomes. J Biol Chem 1976, 251:2629-2636 [PubMed] [Google Scholar]
- 12.DeWitt DL, Smith WL: Primary structure of prostaglandin G/H synthase from sheep vesicular gland determined from the complementary DNA sequence. Proc Natl Acad Sci USA 1988, 85:1412-1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Merlie JP, Fagan D, Mudd J, Needleman P: Isolation and characterization of the complementary DNA for sheep seminal vesicle prostaglandin endoperoxide synthase (cyclooxygenase). J Biol Chem 1988, 263:3550-3553 [PubMed] [Google Scholar]
- 14.Yokoyama C, Takai T, Tanabe T: Primary structure of sheep prostaglandin endoperoxide synthases deduced from cDNA sequence. FEBS Lett 1988, 231:347-351 [DOI] [PubMed] [Google Scholar]
- 15.O’Neill GP, Ford-Hutchinson AW: Expression of mRNA for cyclooxygenase-1 and -2 in human tissues. FEBS Lett 1993, 330:156-160 [DOI] [PubMed] [Google Scholar]
- 16.Simmons DL, Levy DB, Yannoni Y, Erikson RL: Identification of a phorbol-ester-repressible v-src-inducible gene. Proc Natl Acad Sci USA 1989, 86:1178-1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hla T, Nielson K: Human cyclo-oxygenase-2 cDNA. Proc Natl Acad Sci USA 1992, 89:7384-7388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Williams CS, DuBois RN: Prostaglandin endoperoxidase synthase: why two isoforms? Am J Physiol 1996, 270:393-400 [DOI] [PubMed] [Google Scholar]
- 19.Vane JR, Bakhle YS, Botting RM: Cyclooxygenases 1 and 2. Annu Rev Pharmacol Toxicol 1998, 38:97-120 [DOI] [PubMed] [Google Scholar]
- 20.Tsujii M, Kawano S, Tsuji S, Sawaoka H, Hori M, DuBois RN: Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell 1998, 93:705-716 [DOI] [PubMed] [Google Scholar]
- 21.Tsujii M, DuBois RN: Alterations in adhesion and apoptosis in epithelial cells overexpressing prostaglandin endoperoxide synthase-2. Cell 1995, 83:493-501 [DOI] [PubMed] [Google Scholar]
- 22.Lu X, Xie W, Reed D, Bradshaw WS, Simon DS: Nonsteroidal antiinflammatory drugs cause apoptosis and induce cyclooxygenases in chicken embryo fibroblasts. Proc Natl Acad Sci USA 1995, 92:7961-7965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y, Cao HJ, Graf B, Meekins H, Smith TJ, Phipps RP: CD40 engagement up-regulates cyclooxygenase-2 expression, and prostaglandin E2 production in human lung fibroblasts. J Immunol 1998, 160:1053-1057 [PubMed] [Google Scholar]
- 24.Romano M, Ricci V, Memoli A, Tuccillo C, Di Popolo A, Sommi P, Acquaviva AM, Blanco CDV, Bruni CB, Zarrilli R: Helicobactor pylori up-regulates cyclooxygenase-2 mRNA expression and prostaglandin E2 synthesis in MKN 28 gastric mucosal cells in vitro. J Biol Chem 1998, 273:28560-28562 [DOI] [PubMed] [Google Scholar]
- 25.Arias-Negrete S, Kelley K, Chadee K: Proinflammatory cytokines regulate cyclooxygenase-2 expression in human macrophages. Biochem Biophys Res Commun 1995, 208:582-589 [DOI] [PubMed] [Google Scholar]
- 26.Ristimäki A, Garfinkel S, Wessendorf J, Maciag T, Hla T: Induction of cyclooxygenase-2 by interleukin-1α. J Biol Chem 1994, 269:11769-11775 [PubMed] [Google Scholar]
- 27.Öst M, Uhl E, Carlsson M, Gidlöf A, Söderkist P, Sirsjö A: Expression of mRNA for phospholipase A2, cyclooxygenases, and lipoxygenases in cultured human umbilical vascular endothelial and smooth muscle cells and in biopsies from umbillical arteries and veins. J Vasc Res 1998, 35:150-155 [DOI] [PubMed] [Google Scholar]
- 28.Fu JY, Masferre JL, Seibert K, Raz A, Needleman P: The induction and suppression of prostaglandin H2 synthase in human monocytes. J Biol Chem 1990, 265:16737-16740 [PubMed] [Google Scholar]
- 29.Bishop-Bailey D, Pepper JP, Haddad E-B, Newton R, Larkin SW, Mitchell JA: Induction of cyclooxygenase-2 in human saphenous vein and internal mammary artery. Arterioscler Thromb Vasc Biol 1997, 17:1644-1648 [DOI] [PubMed] [Google Scholar]
- 30.Libby P, Galis ZS: Cytokines regulate genes involved in atherogenesis. Ann NY Acad Sci 1995, 748:158-168 [DOI] [PubMed] [Google Scholar]
- 31.Picot D, Loll PJ, Garavito RM: The X-ray crystal-structure of the membrane protein prostaglandin H2-synthase-1. Nature 1994, 367:243-249 [DOI] [PubMed] [Google Scholar]
- 32.Kurumbail RG, Stevens AM, Gierse JK, McDonald JJ, Stegeman RA, Pak JY, Gildehaus D, Miyashiro JM, Penning TD, Seibert K, Isakson PC, Stallings WC: Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature 1996, 384:644-648 [DOI] [PubMed] [Google Scholar]
- 33.Mancini JA, O’Neill GP, Bayly C, Vickers PJ: Mutation of serine-516 human prostaglandin G/H synthase-2 to methionine or aspirin acetylation of this residue stimulates 15-R-HETE synthesis. FEBS Lett 1994, 342:33-37 [DOI] [PubMed] [Google Scholar]
- 34.Lecomte M, Laneuville O, Ji C, DeWitt DL, Smith WL: Acetylation of human prostaglandin endoperoxide synthase-2 by aspirin. J Biol Chem 1994, 269:13207-13215 [PubMed] [Google Scholar]
- 35.Vane JR: Inhibition of prostaglandin synthesis as a mechanism of action for the aspirin-like drugs. Nature 1971, 231:232-235 [DOI] [PubMed] [Google Scholar]
- 36.Morita J, Schindler MS, Regier MK, Otto JC, Hori T, DeWitt SL, Smith WL: Different intracellular locations for prostaglandin endoperoxide synthase-1 and -2. J Biol Chem 1995, 270:10902-10908 [DOI] [PubMed] [Google Scholar]
- 37.Mazzei GJ, Edgerton MD, Losberger C, Lecoanet-Henchoz S, Graber P, Durandy A, Gauchat J-F, Bernard A, Allet B, Bonnefoy J-Y: Recombinant soluble trimeric CD40 ligand is biologically active. J Biol Chem 1995, 270:7025-7028 [DOI] [PubMed] [Google Scholar]
- 38.Ross R, Kariya B: Morphogenesis of vascular smooth muscle in atherosclerosis and cell structure. Handbook of Physiology, The Cardiovascular System. Edited by DF Bohr, AP Somlyo, HY Sparks. Bethesda, MD, American Physiological Society, 1980, pp 66–91
- 39.Schönbeck U, Mach F, Bonnefoy JY, Loppnow H, Flad HD, Libby P: Ligation of CD40 activates interleukin 1β-converting enzyme (caspase-1) activity in vascular smooth muscle and endothelial cells and promotes elaboration of active interleukin 1β. J Biol Chem 1997, 272:19569-19574 [DOI] [PubMed] [Google Scholar]
- 40.Galis ZS, Sukhova GK, Lark MW, Libby P: Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest 1994, 94:2493-2503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rimarachin JA, Jacobson JA, Szabo P, Maclouf J, Creminon C, Weksler BB: Regulation of cyclooxygenase-2 expression in aortic smooth muscle cells. Arterioscler Thromb 1994, 14:1021-1031 [DOI] [PubMed] [Google Scholar]
- 42.Mitchell JA, Akarasereenont P, Thiemermann C, Flower RJ, Vane JR: Selectivity of nonsteroidal anti-inflammatory drugs as inhibitors of constitutive and inducible cyclooxygenase. Proc Natl Acad Sci USA 1993, 90:11693-11697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sneddon JM, Vane JR: Endothelium-derived relaxing factor reduces platelet adhesion to bovine endothelial cells. Proc Natl Acad Sci USA 1988, 85:2800-2804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kowala MC, Mazzucco CE, Hartl KS, Seiler SM, Warr GA, Abid S, Grove RI: Prostacyclin agonists reduce early atherosclerosis in hyperlipidimic hamsters: octimibate and BMY 42393 suppress monocyte chemotaxis, macrophage cholesteryl ester accumulation, scavenger receptor, activity, and tumor necrosis factor production. Arterioscl Thromb 1993, 13:435-444 [DOI] [PubMed] [Google Scholar]
- 45.Hajjar DP, Weksler BB: Metabolic activity of cholesteryl esters in aortic smooth muscle cells is altered by prostaglandins I2 and E2. J Lipid Res 1983, 24:1176-1185 [PubMed] [Google Scholar]
- 46.Hajjar DP, Marcus AJ, Etingin OR: Platelet-neutrophil-smooth muscle cell interactions: lipoxygenase-derived mono- and dihydroxy acids activate cholesteryl ester hydrolysis by the cyclic AMP dependent protein kinase cascade. Biochemistry 1989, 28:8885-8891 [DOI] [PubMed] [Google Scholar]
- 47.Moncada S, Vane JR: The role of prostacyclin in vascular tissue. Fed Proc 1979, 38:66-71 [PubMed] [Google Scholar]
- 48.Lipsky PE: Role of cyclooxygenase-1 and -2 in health and disease. Am J Orthop 1999, 28:8-12 [PubMed] [Google Scholar]
- 49.Holmes DR, Wester W, Thompson RW, Reilly JM: Prostaglandin E2 synthesis, and cyclooxygenase expression in abdominal aortic aneurysms. J Vasc Surg 1997, 25:810-815 [DOI] [PubMed] [Google Scholar]
- 50.Mach F, Schönbeck U, Sukhova GK, Bourcier T, Bonnefoy JY, Pober JS, Libby P: Functional CD40 ligand is expressed on human vascular endothelial cells, smooth muscle cells, and macrophages: implications for CD40-CD40 ligand signaling in atherosclerosis. Proc Natl Acad Sci USA 1997, 1936, 94:1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mach F, Schönbeck U, Bonnefoy JY, Pober JS, Libby P: Activation of monocyte/macrophage functions related to acute atheroma complication by ligation of CD40: induction of collagenase, stromelysin, and tissue factor. Circulation 1997, 96:396-399 [DOI] [PubMed] [Google Scholar]
- 52.Schönbeck U, Mach F, Sukhova GK, Murphy C, Bonnefoy JY, Fabunmi RP, Libby P: Regulation of matrix metalloproteinase expression in human vascular smooth muscle cells by T lymphocytes: a role for CD40 signaling in plaque rupture? Circ Res 1997, 81:448-454 [DOI] [PubMed] [Google Scholar]
- 53.Mach F, Schönbeck U, Sukhova GK, Atkinson E, Libby P: Reduction of atherosclerosis in mice by inhibition of CD40 signalling. Nature 1998, 394:200-203 [DOI] [PubMed] [Google Scholar]
- 54.Bishop-Bailey D, Burke-Gaffney A, Hellewell PG, Pepper JP, Mitchell JA: Cyclo-oxygenase-2 regulates inducible ICAM-1, and VCAM-1 expression in human vascular smooth muscle cells. Biochem Biophys Res Commun 1998, 249:44-47 [DOI] [PubMed] [Google Scholar]
- 55.Husain S, Andrews NP, Mulcahy D, Panza JA, Quyyumi AA: Aspirin improves endothelial dysfunction in atherosclerosis. Circulation 1998, 97:716-720 [DOI] [PubMed] [Google Scholar]
- 56.Masferrer JL, Zweifel B, Manning PT, Hauser SD, Leahy KM, Smith WG, Isakson PG, Seibert K: Selective inhibition of inducible cyclooxygenase-2 in vivo is antiinflammatory and non-ulcerogenic. Proc Natl Acad Sci USA 1994, 91:3228-3232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moulton KS, Heller E, Konerding MA, Flynn E, Palinski W, Folkman J: Angiogenesis inhibitors endostatin or TNP-470 reduce intimal neovascularization and plaque growth in apolipoprotein E-deficient mice. Circulation 1999, 99:1726-1732 [DOI] [PubMed] [Google Scholar]
- 58.Marx N, Schönbeck U, Lazar MA, Libby P, Plutzky J: Peroxisome proliferator-activated receptor γ activators inhibit gene expression and migration in human vascular smooth muscle cells. Circ Res 1998, 83:1097-1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Varga J, Diaz-Perez A, Rosenbloom J, Jimenez SA: PGE2 causes a coordinate decrease in the steady state levels of fibronectin, and types I, and III procollagen mRNAs in normal human dermal fibroblasts. Biochem Biophys Res Commun 1987, 147:1282-1288 [DOI] [PubMed] [Google Scholar]
- 60.Breyer MD, Jacobson HR, Breyer RM: Functional and molecular aspects of renal prostaglandin receptors. J Am Soc Nephrol 1996, 7:7-18 [DOI] [PubMed] [Google Scholar]
- 61.Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM: 15-Deoxy-delta-12,14-prostaglandin-J2 is a ligand for the adipocyte determination factor PPARγ. Cell 1995, 83:803-812 [DOI] [PubMed] [Google Scholar]
- 62.Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM: A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor γ and promotes adipocyte differentiation. Cell 1995, 83:813-819 [DOI] [PubMed] [Google Scholar]
- 63.Mangelsdorf DJ, Evans RM: The RXR heterodimers, and orphan receptors. Cell 1995, 83:841-850 [DOI] [PubMed] [Google Scholar]
- 64.Marx N, Sukhova G, Murphy C, Libby P, Plutzky J: Macrophages in human atheroma contain PPARγ: differentiation-dependent PPARγ expression and reduction of MMP-9 activity through PPARγ activation in mononuclear phagocytes. Am J Pathol 1998, 153:17-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wallace JL, Chapman K, McKnight W: Limited anti-inflammatory efficacy of cyclo-oxygenase-2 inhibition in carrageenan-airpouch inflammation. Br J Pharmacol 1999, 126:1200-1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McAdam BF, Catella-Lawson F, Mardini IA, Kapoor S, Lawson JA, FitzGerald GA: Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2. Proc Natl Acad Sci USA 1999, 96:272-277 [DOI] [PMC free article] [PubMed] [Google Scholar]