

Abstract
To investigate genetic abnormalities associated with the development of thymic epithelial tumors, we performed microsatellite analysis of 26 thymomas belonging to three different World Health Organization types (A, B3, and C) using 48 repeats. The most frequent aberration seen was loss of heterozygosity (LOH) in the region 6q23.3-25.3 detected in 11 tumors (45.8% of informative cases). Further consistent LOHs were detected in regions 3p22-24.2, 3p14.2 (FHIT gene locus), 5q21 (APC), 6p21, 6q21-22.1, 7p21-22, 8q11.21-23, 13q14 (RB), and 17p13.1 (p53). Microsatellite instability was extremely rare, occurring in one type B3 thymoma only, although, at 12.5% of the analyzed loci. Comparing the allelotypes of the analyzed thymomas, we were able to identify two pathogenetic pathways these tumors develop along, characterized by the 6q23.3-25.3 and 5q21 LOHs, respectively. The APC aberration on 5q21 showed significant associations with LOH in the 3p22-24.2, 13q14, and 17p13.1 regions. Interestingly, type A thymomas presented with consistent LOH in the region 6q23.3-25.5 only, they did not reveal any aberrations in the APC, RB, and p53 gene loci or regions 3p22-24.2 and 8q11.21-23. The absence of these aberrations might be the reason for the well-known benign behavior of type A thymomas as compared to types B3 and C tumors.
The classification of tumors of the thymus has been one of the most controversial fields in tumor pathology of the last decade. 1-3 These neoplasms show considerable functional, histogenetic, and morphological complexity. Recently, several major conceptional advances in the understanding of thymomas have been made and a new World Health Organization-authorized histological classification of thymic epithelial tumors emerged (Table 1) ▶ . 4,5 Thymomas retaining morphological features of the normal thymus are now divided into two major types, A and B, according to the appearance of the neoplastic cells (spindle/oval versus round/polygonal shaped cells, respectively). Tumors combining these features are called type AB. Type B thymomas are further subdivided on the basis of an increasing epithelial cell/lymphocyte ratio and emergence of atypia in the neoplastic epithelial cells into three subtypes, B1, B2, and B3, respectively. Nonorganotypic thymic carcinomas exhibiting clear-cut atypia and resembling tumors arising outside the thymus form a separate group, type C thymomas. The new classification is a further development of a previous histological grouping of thymomas 6,7 used also as an important independent survival prognostic factor. 8-13 Because the new WHO types are almost equivalent to the previously described histogenetic thymoma subtypes, 14 it can be expected that this classification is suitable for prognosis determination as well.
Table 1.
Comparison of Different Classifications of Thymic Epithelial Tumors
| Clinicopathological classification | WHO type | Histogenetic classification |
|---|---|---|
| Benign thymoma* | A | Medullary thymoma |
| AB | Mixed thymoma | |
| Malignant thymomas, category I | B1 | Predominantly cortical thymoma |
| B2 | Cortical thymoma | |
| B3 | Well-differentiated thymic carcinoma | |
| Malignant thymomas, category II | C | Epidermoid keratinizing (squamous cell) carcinoma |
| Epidermoid nonkeratinizing carcinoma | ||
| Lymphoepithelioma-like carcinoma | ||
| Sarcomatoid carcinoma (carcinosarcoma) | ||
| Clear cell carcinoma | ||
| Basaloid carcinoma | ||
| Mucoepidermoid carcinoma | ||
| Undifferentiated carcinoma |
The WHO classification is compared with the clinicopathological classification of Levine and Rosai 48 and the histogenetic classification of Müller-Hermelink and co-workers. 6,7,49 The terminology of the category II thymic carcinoma subtypes follows the WHO proposal.
*The benign category is modified according to the postulate that type A and AB thymomas are clinically benign irrespective of invasiveness. Originally, all encapsulated thymomas were considered to be benign irrespective of their histology.
Only little is known about chromosomal aberrations in thymoma. Cytogenetic studies describing the karyotypes of these tumors are mostly case studies showing no recurrent aberrations. The only exception is translocation t(15;19), found in altogether three undifferentiated thymic carcinomas (type C thymoma). 15-17 Various gross genetic aberrations and their association with several different thymoma types were recently depicted in a comparative genomic hybridization (CGH) study. 18 The most prominent abnormalities described in that work were amplifications of genetic material on chromosome 1q and deletions on chromosomes 6 and 13q in type B3 thymomas; amplifications of regions on chromosomes 1q, 17q, and 18 and deletions on chromosomes 3p, 6, 16q, and 17p in type C thymomas. Striking was the paucity of chromosomal abnormalities in type A thymoma. Even less is known about specific molecular mechanisms underlying thymomagenesis and the role individual genes are playing in that process. Some studies have shown that inactivation of the CDKN2A (p16) and RB genes may play a role in the progression of thymoma and thymic carcinoma 19 and that BCL-2 overexpression correlates with aggressiveness of thymic epithelial neoplasms. 20 The high incidence of HRAS mutations in metastatic thymomas indicates that this aberration could be used as a possible marker of aggressive behavior. 21 Also nerve growth factor (NGF) receptors might be involved in the abnormal proliferation of neoplastic thymic epithelial cells as indicated by down-regulation of type 1 neurotrophic tyrosine kinase receptor TrkA and up-regulation of nerve growth factor receptor p75NGFR in thymic carcinomas. 22
However, all these molecular studies have usually narrowly focused on the role of a particular individual gene in the pathogenesis of the disease and did not investigate the sequence and relationship between the genetic abnormalities detected. The aforementioned CGH study identified several nonrandom losses or gains of genetic material suggesting the presence of tumor suppressor genes or oncogenes, respectively, whose function is altered during the process of malignant transformation. It identified the chromosomal regions involved, but not the individual genes. To do that, a method that can detect also amplifications or deletions in the range of tens of base pairs like microsatellite analysis has to be applied. This method has also the advantage of simultaneously detecting microsatellite instability caused by dysfunction of mismatch-repair genes recently described in epithelial cancer. 23 Therefore, we performed a limited genomic search with 48 highly informative microsatellite markers on 26 thymoma cases, including types A, B3, and C tumors, aiming to identify gene loci involved in the pathogenesis of thymomas. In this work, we characterize the genetic aberrations common in the analyzed thymomas and compare the new WHO classification with the grouping of thymomas based on our allelotype data.
Materials and Methods
Patients and Samples
Twenty-six thymic epithelial tumors lacking heavy lymphocyte contamination including 8 thymomas of WHO type A, 14 of WHO type B3, and 4 cases of type C from the thymoma collection at the Institute of Pathology in Würzburg, on whom fresh-frozen tissue (2 WHO type A, 7 type B3, and 2 type C) or formalin-fixed, paraffin-embedded tissue (6 WHO type A, 7 type B3, and 2 type C) was available, were selected for the study. The diagnosis was established according to the criteria of the new WHO Thymic Epithelial Tumor Classification 4 by morphological and immunophenotypic analyses of paraffin-embedded and fresh-frozen tissue sections using standard staining methods as described recently. 14,24 Clinical data on the analyzed thymoma patients are listed in Table 2 ▶ .
Table 2.
Clinical Data on 26 Thymic Epithelial Tumor Patients Analyzed in the Study
| Number | Age | Sex | TET type | Stage* |
|---|---|---|---|---|
| 28279 | 74 | M | A | II |
| 788 | 65 | M | A | I |
| 454 | 74 | F | A | I |
| 4560 | 57 | F | A | I |
| 7968 | 84 | F | A | I |
| 7336 | 68 | M | A | III |
| 5444 | 71 | M | A | I |
| 2901 | 66 | M | A | I |
| 6175 | 48 | M | B3 | IVb |
| 27920 | 45 | M | B3 | III |
| 10585 | 73 | F | B3 | II |
| 15904 | 72 | F | B3 | III |
| 13561 | 29 | M | B3 | II |
| 11859 | 42 | F | B3 | II |
| 20798 | 48 | M | B3 | II |
| 3759 | 46 | F | B3 | III |
| 8373 | 43 | F | B3 | III |
| 2846 | 53 | F | B3 | III |
| 6694 | 57 | M | B3 | II |
| 843 | 63 | M | B3 | IV |
| 4550 | 63 | M | B3 | III |
| 6902 | 69 | F | B3 | III |
| 16402 | 63 | M | C | II |
| 366 | 57 | F | C | Not available |
| 4349 | 50 | F | C | IVb |
| 6320 | 67 | M | C | IVb |
Microdissection and DNA Extraction
In each case, 20 serial 10-μm-thick tissue sections were cut. The first and last cuts were stained with hematoxylin and eosin to assure high tumor content and as guidance for the following dissection. The fresh-frozen tissue sections were visualized under microscope and an area showing thymoma was scraped using a blade. In a similar way, control genomic DNA was derived from separate tissue blocks not involved by the tumor. Paraffin-embedded, formalin-fixed tissue sections were additionally stained by Nuclear-Fast Red to precisely delineate tumor-containing areas and the collected tissue deparaffinized with xylene before digestion. DNA extraction was performed using proteinase K and phenol-chloroform according to routine molecular biology protocols. 25
Microsatellite Analysis
Primer sequences for the amplification of microsatellite repeats listed in Table 3 ▶ were retrieved from Genome Database (GDB, http://gdbwww.gdb.org). Polymerase chain reaction (PCR) primers were synthesized at MWG Biotech (Munich, Germany) and one oligonucleotide of each primer pair labeled with fluorescent dye phosphoramidites FAM, TAMRA, or HEX. Paired normal and tumor DNA samples from each patient were amplified with AmpliTaq Gold enzyme (Applied Biosystems, Foster City, CA) in multiplex PCR reactions using 50 ng of genomic DNA as template under conditions specified by GDB. Thirty cycles were performed in a MWG Primus Gold thermal cycler (MWG Biotech, Munich, Germany) in a total volume of 20 μl. Aliquots of the PCRs were then mixed with size standard and formamide, denatured, and subjected to electrophoresis on a 377 DNA Sequencer (Applied Biosystems, Foster City, CA). The automatically collected data were analyzed using the Genescan and Genotyper software as described in the manufacturer’s manual. Only patients heterozygous for a given locus were regarded to be informative, homozygosity and microsatellite instability rendered the particular locus uninformative for loss of heterozygosity (LOH) or amplification. In heterozygous cases, ratios of both alleles in normal and tumor tissues were calculated. If these ratios showed a difference of >20%, the locus was further evaluated for possible allelic imbalance. For determination of LOH or amplification, first the unchanged allele was identified (by comparison with other microsatellites showing no change in the same multiplex PCR), then the ratios of the allele showing decreased or increased signal to the unchanged allele were calculated, first for control DNA, then for the tumor. Increase of the ratio by 40% in the tumor as compared to the control was called amplification, decrease by 40% LOH. All aberrations were confirmed two times. Regional allelic imbalance (RAI) was defined as percentage of regions showing allelic imbalance (31 regions listed in Table 3 ▶ were investigated altogether). Patients on whom the results for at least two regions were not evaluable (not all formalin-fixed, paraffin-embedded tissue derived DNA could be amplified) were excluded from the comparison (case no. 6902 and no. 2901).
Table 3.
Forty-Eight Microsatellites Used in the Analysis of Thymomas, Their Chromosomal Locations, and Genes of Interest
| Microsatellites | Location | Gene |
|---|---|---|
| D3S1283 | 3p22-24.2 | Unknown |
| D3S4103, D3S1300, D3S659 | 3p14.2 | FHIT |
| D3S1276 | 3p12 | Unknown |
| D3S1212, D3S1229, D3S1580, D3S1314 | 3q26-27 | BCL-6 |
| D4S1566 | 4q32-33 | Unknown |
| D5S82, D5S346 | 5q21 | APC |
| D6S1666, D6S1560 | 6p21 | MHC |
| D6S1592, D6S447 | 6q21-22.1 | Unknown |
| D6S310, D6S441 | 6q23.3-25.3 | Unknown |
| D7S673 | 7p21-22 | Unknown |
| EGFR-CA | 7p12.1-12.3 | EGFR |
| D8S532 | 8q11.21-11.29 | Unknown |
| D8S384, C-MYC-CA | 8q22-24.13 | C-MYC |
| D9S2136, D9S1748 | 9p21 | p15/p16 |
| D10S2491 | 10q23 | PTEN |
| Wt 400/401 | 11p13 | WT1 |
| D11S987 | 11q13.3 | CyclinD1 |
| D11S2179, D11S1356 | 11q23-q24 | ATM, MLL |
| D12S88 | 12q14-21.1 | Unknown |
| D12S78 | 12q22-23 | Unknown |
| D13S153, D13S319 | 13q14 | RB |
| D14S267 | 14q32 | AKT1 |
| D15S231, D15S114 | 15q14-22 | Unknown |
| D16S663 | 16p13.3 | Unknown |
| D16S409 | 16p11.2-12 | Unknown |
| D16S411 | 16q12.1 | p130 |
| D16S402 | 16q23-24 | Unknown |
| TP53CA, p53p | 17p13.1 | p53 |
| chrnb1 | 17p11-12 | CHRNB1 |
| D18S35, D18S1127, D18S1129 | 18q21 | DCC, BCL-2 |
| D19S49 | 19q12 | Unknown |
CGH
CGH methods and results on 19 tumors from the investigated group were published previously. 18
Immunohistochemical Staining for hMSH2 and hMLH1
Immunohistochemical staining for hMSH2 (hybridoma clone FE11; Calbiochem, San Diego, CA) and hMLH1 (hybridoma clone G168-15; Pharmingen, San Diego, CA) was performed on pressure cooker-pretreated, formalin-fixed, paraffin-embedded tissue sections and visualized using standard immunoperoxidase technique. Normal tissue of the same patient served as a control. A case was considered positive for each antigen, when >80% of the tumor cells showed strong nuclear staining as compared to normal cells on the same slide.
Results
Microsatellite Analysis Shows Frequent Allelic Imbalance in All Analyzed Thymoma Types
Twenty-six thymoma cases listed in Table 2 ▶ including 8 WHO type A, 14 type B3, and 4 type C tumors were screened for loss or amplification of genomic DNA using 48 microsatellite repeats (Table 3) ▶ . The repeats were chosen to cover loci of known and putative oncogenes or tumor suppressor genes and regions showing frequent aberrations in cytogenetic and CGH studies on lung, head, and neck squamous cell carcinomas, 26-28 and hot spots of aberrations detected in a previous CGH study performed on part of the same material. 18 Out of 882 informative genotypes (not all formalin-fixed, paraffin-embedded tissue-derived DNA could be amplified), 105 (11.9%) showed LOH, 6 (0.7%) amplification of genomic DNA. Several regions on various chromosomes revealed allelic imbalance in at least three cases; the frequency of such aberrations is depicted in Figure 1 ▶ . To distinguish LOH from amplification, multiplex PCRs were performed using unaffected repeats as an internal control. The results were further confirmed by CGH in the majority of patients (Figure 2) ▶ . 18
Figure 1.

Chromosomal regions showing frequent allelic imbalance. The frequency of allelic imbalance found in chromosomal regions showing more than two aberrations was plotted in the diagram. Filled bars, LOH; open bars, amplifications of genomic DNA.
Figure 2.

LOH in the retinoblastoma gene locus as detected using the D13S153 microsatellite and confirmatory CGH for chromosome 13 in case no. 4550. A: DNA samples from normal and tumor tissues were amplified for the D13S153 (black) microsatellite in a PCR. The 224-bp allele of the D13S153 microsatellite showed signal strength decrease by 49% (arrow). B: Confirmatory CGH profile of chromosome 13 from the same tumor is shown on the right. The cut-off values for losses (0.80) and gains (1.25) are depicted as red and green lines, respectively.
Chromosome 6 Harbors Hot Spots of Aberrations
The most frequent LOH was found in the 6q23.3-25.3 chromosomal region (Table 3) ▶ as detected by markers D6S310 and D6S441 in 11 (45.8% of informative cases) tumors (3 type A, 5 type B3, and 3 type C thymomas, Figure 1 ▶ ). The second hot spot of deletions was located in the 6p21 region (assayed for by markers D6S1666 and D6S1560) containing the major histocompatibility (MHC) classes I and II gene loci, with seven (33.3% of informative analyses) tumors (two type A, four type B3, and one type C) showing an involvement. Other frequent LOHs were located in regions 3p22-24.2, 3p14.2 (FHIT gene locus), 5q21 (APC gene locus), 6q21-22.1, 7p21-22, 13q14 (RB gene locus), and 17p13.1 (p53 tumor suppressor gene locus). However, two thymomas (both type B3, patients no. 6175 and no. 10585) showed LOH with all repeats on chromosome 6 suggesting monosomy 6 (confirmed by fluorescence in situ hybridization later, unpublished results) to be the cause of LOH in these two cases. Similarly, two other type B3 tumors (no. 6175 and no. 15904) showed LOH with all nine microsatellite markers on chromosome 3, suggesting the presence of monosomy 3 in these patients.
Thymic Epithelial Tumors Show Two Distinct Patterns of Genetic Aberrations
Interesting associations emerged comparing aberrations common to the analyzed thymoma types. We could identify two groups of tumors characterized by specific mutually exclusive aberrations (Figure 3) ▶ . The first one consisted of 11 (45.8% of informative analyses) thymomas displaying the 6q23.3-25.3 LOH. Another group was made up by four (15% of informative analyses) tumors showing the APC tumor suppressor gene locus (5q21) deletion. The APC aberrations showed statistically significant associations with LOH in the 3p22-24.2 (chi-square test, Yates’ correction; P = 0.045), 13q14 (chi-square test, Yates’ correction; P = 0.0055), and 17p13.1 (chi-square test, Yates’ correction; P = 0.0003) regions. The rest of the analyzed thymomas displayed either other heterogeneous aberrations (seven cases) or no aberrations at all (four cases). Thymomas of all three WHO types were among the tumors showing the 6q23.3-25.3 deletion. In contrast, none of the type A patients was detected to have an LOH in the APC, RB, and p53 gene loci or the 3p22-24.2 and 8q11.21-11.23 regions. The 6q23.3-25.3 deletion occurred already in stage I disease and was present in all higher stages thereafter. The APC LOH was exclusively associated with later stages (III or IV) of disease.
Figure 3.
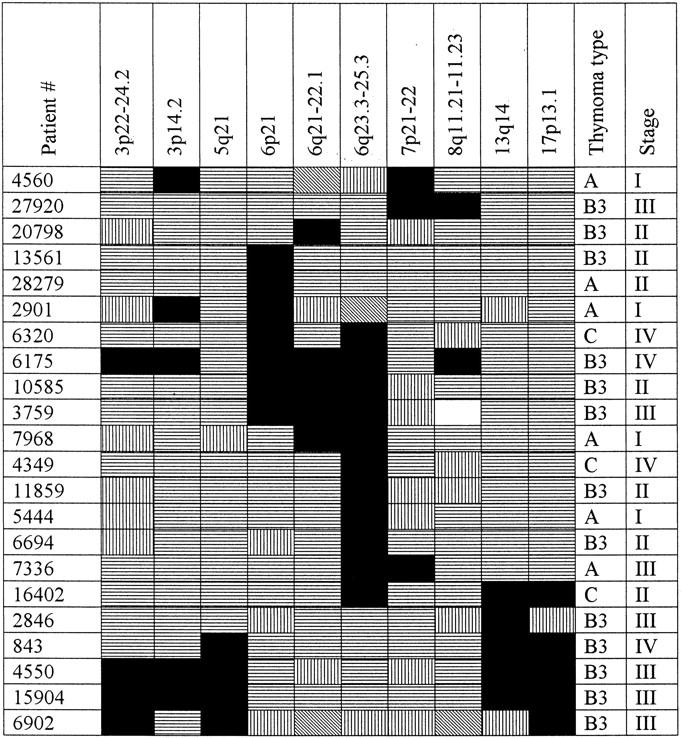
Pattern of genetic aberrations in thymic epithelial tumors. Microsatellite analysis results for 10 chromosomal regions harboring at least three allelic imbalances are displayed. Patients showing at least one aberration are listed in the first left column. Status of each locus is indicated: black, LOH; horizontal stripes, retention of heterozygosity; falling stripes, no amplificate; vertical stripes, homozygosity; and white, genomic amplification.
Late Stages of Disease Show Increased RAI Frequency and Broader Spectrum of Aberrations
We used the frequency of RAI (see Materials and Methods) displayed by these tumors to assess the degree of genomic instability associated with different thymoma stages, regardless of the WHO thymoma type (Figure 4) ▶ . Stage I thymomas showed RAI with mean of 4.1% and SD of 4.2%, only seven regions exhibiting allelic imbalances were detected. In Stage II, regions affected by aberrations numbered 8, the RAI increased to 8 ± 4.7%. In stage III, the number of regions showing aberrations rose to 19 including the APC tumor suppressor gene locus, the RAI showed an increase to 16 ± 14.8%. Only four patients presented with a stage IV thymoma and that seemed to influence the results of the statistical analysis; merely 15 various regions showed an allelic imbalance, the RAI frequency was 15.2 ± 10%. The RAI frequency difference between stages I and IV proved to be statistically significant (Mann-Whitney U test, P = 0.027).
Figure 4.

Increasing RAI frequency with higher stage of disease. Stage I thymomas showed the lowest RAI frequency values with mean of 4.1% and SD of 4.2%. Increasing RAI values were observed in later stages: stage II, 8 ± 4.7%; stage III, 16 ± 14.8%; and stage IV, 15.2 ± 10%. However, only the RAI frequency difference between stages I and IV was statistically significant (Mann-Whitney U test, P = 0.027).
Only One Thymic Epithelial Tumor Features MSI
Six (0.5%) of 1178 genotypes proved to be microsatellite instability (MSI)-positive in these tumors. Interestingly, all of the MSI-positive repeats detected were clustered in only one tumor, a type B3 thymoma (no. 11859). In this particular case, the frequency of MSI was low-level, with six (12.5%) repeats (D3S1300, D6S1666, D6S447, D8S532, D12S78, P53p) showing MSI. Immunohistochemical staining for protein products of the mismatch repair genes hMSH2 and hMLH1 was performed to investigate the cause of MSI in this case further. However, most of the tumor cells presented strong nuclear staining meaning the hMSH2 and hMLH1 proteins were present (results not shown). MSI in this tumor thus did not originate from aberrations in the hMSH2 or hMLH1 mismatch repair genes. Microscopic review of the histology of the case confirmed a typical pattern of a WHO type B3 thymoma without any unusual findings.
Discussion
Thymic epithelial tumors are the most frequent kind of neoplasms arising in the thymus. Recent establishment of a new WHO Thymic Epithelial Tumor Classification 4 ended a long period of confusion regarding the morphological diagnosis of these tumors. However, the possibility that there are characteristic genetic aberrations underlying individual thymoma types has not been exploited yet. Aberrations common in thymomas have not been characterized except for several nonspecific abnormalities described in a recent CGH work 18 and rare findings of t(15;19) reported in conventional cytogenetic studies. 15-17 Because of the heavy admixture of normal lymphocytes in the tumor, the thymomas are not easy to study. Using microdissection and focusing on thymoma entities in which a heavy normal cell admixture is not a serious problem, we analyzed 26 thymomas (8 WHO type A, 14 type B3, and 4 type C tumors) with 48 microsatellite repeats.
LOH in the 6q23.3-25.3 region was the most frequent aberration (11 cases, 45.8% of informative cases) detected in this work. Losses of genomic DNA in this region occurred in all three thymoma types and in all disease stages analyzed. They were the sole aberration in two patients, but mostly were accompanied by other allelic imbalances. Deletions and rearrangements involving chromosome 6q have been reported in a number of human malignancies, including breast carcinoma, 29 malignant melanoma, 30 renal cell carcinoma, 31 salivary gland adenocarcinoma, 32 ovarian carcinoma, 33 acute lymphoblastic leukemia, 34 and nodal non-Hodgkin’s lymphoma. 35 We described several hot spots of deletions on the long arm of chromosome 6 in extranodal gastric high-grade large B-cell lymphoma previously. 36 One of the hot spots was detected in the region 6q23.3-25.3 between the markers D6S310 to D6S441 used in this thymoma study as well. This region contains, in addition to tens of unidentified expressed sequence tag (ESTs), several already cloned genes, eg, IGF2R, MAP3K5, and ZAC. Interestingly, the ZAC tumor suppressor gene has been shown to inhibit tumor cell proliferation in vitro and in vivo in nude mice 37 and its expression was found to be lost or reduced in breast cancer cell lines and primary breast tumors. 38 Whatever gene is the target of deletions here, it seems to define a particularly important early event in thymoma pathogenesis.
A smaller but distinct group of four cases presented with LOH in the APC tumor suppressor gene locus in the chromosomal region 5q21. These tumors were WHO type B3 thymomas in late stages of disease. All four displayed additional 17p13.1 LOH (chi-square test, Yates’ correction; P = 0.0003) in the p53 tumor suppressor gene locus. Three revealed concomitant LOH in the 5q21 and 3p22-24.2 regions (chi-square test, Yates’ correction; P = 0.045), another three concomitant LOH in the 5q21 and 13q14 (retinoblastoma tumor suppressor gene) regions (chi-square test, Yates’ correction; P = 0.0055). APC aberrations, which generally lead to a truncated APC protein or take form of allele loss, can be detected in ∼75% of sporadic colorectal cancers, already in the earliest adenomas from which these cancers develop. 39,40 They are considered to be initiating events in colorectal cancerogenesis. Additional genetic aberrations are required for the progression from adenoma to carcinoma. One of the hallmarks of such a progression is the loss of function of the p53 tumor suppressor gene allowing cells to accumulate additional mutations throughout the genome. A significant minority of thymic epithelial tumors thus share a similar set of aberrations with colorectal cancer. To which extent this similarity reflects the similarity of pathogenetic pathways of both colorectal cancer and thymoma remains to be investigated. However, we did not find any APC aberrations in early thymomas of stage I or II, just the 6q aberrations. It is thus possible that these neoplasms exhibit a similar series of aberrations, but the succession of the aberration events, which aberrations are initiating and which then follow in later stages of disease, may be different.
The second most frequently detected aberration (seven tumors, 33.3% of informative cases) was LOH in the 6p21 region. This very well-mapped and already sequenced region contains among others the genes for tumor necrosis factor-α, CDKN1A, and MHC. Loss of MHC genes has been reported in numerous tumors recently. 41,42 The MHC class I and II genes encode highly polymorphic cell surface glycoproteins that bind antigenic peptides for presentation to CD4 and CD8 bearing cytotoxic T lymphocytes, respectively. 43 Because of the role of the MHC molecules in presenting immunogenic peptides to cytotoxic T cells, defects in the MHC genes expression allow the tumor cells to evade the immune response that might be expected to be generated after multiple mutations of genes, whose abnormal products are potential targets for the immune system. Allelic loss of the MHC genes may thus confer a clonal selective advantage, providing tumor cells with a mechanism to escape recognition by the immune defense system. 44 This selection pressure would explain why the MHC gene loss was present in all investigated thymomas with such a high frequency.
Generally, during progression from low- to high-grade disease or from early to later stages, tumors acquire additional genetic aberrations enabling them, at the end, to grow indefinitely in the absence of growth factors. 45 Such genetic aberrations are signs of genomic instability featured by almost all types of cancer. 46 We compared genomic instability in various stages of thymomagenesis (regardless of thymoma type) measuring the frequency of RAI, the percentage of regions showing any allelic imbalance. The RAI values showed a trend to increase with higher stage of disease (Figure 4) ▶ . Stage I tumors displayed mean RAI frequency of 4.1% with SD of 4.2% versus stage IV thymomas with RAI values of 15.2 ± 10% (Mann-Whitney U test, P = 0.027). The increasing genomic instability with higher stage of disease was also manifested by broader spectrum of detected aberrations, stage I cases showing just seven various aberrations, in contrast, stage III already 19. Comparing the RAI values of different thymoma types, type A tumors (considered to be the most benign of the thymomas) showed the lowest RAI level with mean of 4.5% and SD of 3.6%; on the contrary, type B3 thymomas revealed RAI values of 14.5 ± 12%. The RAI frequency difference between the former and latter proved to be statistically significant (Mann-Whitney U test, P = 0.021). However, the former were from 75% stage I tumors, the latter mixed stages II and III (33% stage II thymomas, 47% stage III; Table 2 ▶ ). It thus still has to be investigated (using appropriately matched tumors) if this RAI difference is because of the fundamentally different biological behavior associated with a particular thymoma type or to the fact that type A thymomas present mostly as stage I tumors and rarely progress beyond that stage.
In contrast to the abundance of LOH, considered to be a feature of karyotypic instability, only six genotypes showed microsatellite instability. Peculiarly, all MSI events occurred in just one thymoma. However, this tumor also showed only low-level MSI with 12.5% MSI-positive microsatellites. It did not display >30 to 40% of MSI-positive repeats typical for hereditary nonpolyposis colorectal cancer, in which MSI plays a significant role in the pathogenesis. Immunostains for the mismatch repair gene products hMSH2 and hMLH1 confirmed the presence of both proteins in the tumor. MSI in this case is thus not because of the deficiency of these mismatch repair proteins. The widespread genomic alterations in the form of LOHs and amplifications of genetic material and the low frequency of MSI detected in this study are rather substantial evidence for the mutator pathway having only a minor role in the pathogenesis of thymoma. A small fraction of many tumor types, in addition to those of the colon, display some level of MSI. 47 In most of these tumors, the instability is considerably less pronounced than that observed in hereditary nonpolyposis colorectal cancer and it is questionable if it is because of mismatch repair gene defects.
To identify specific genetic aberrations playing a major role in thymomagenesis, we compared the allelotypes of tumors generated in this study according to the thymoma stage and type (Figure 3) ▶ . The 6g23.3-25.3 LOH was present in all stages of disease; its prevalence ranged from 25% (stage I) to 75% (stage IV) of patients. It is the most frequent and early aberration in thymomas detected in this study. Additional genetic abnormalities appeared already in stage I, however, none of them occurred in more than one case. The APC abnormalities were evident in stage III for the first time. Notably, type B3 thymomas of stage III suffering LOH in the 5q21 region presented with somewhat higher RAI frequency than comparable tumors of the same type and stage displaying the 6g23.3-25.3 LOH. None of the 5q21 LOH-positive thymomas showed a concomitant 6q23.3-25.3 aberration; these two abnormalities seem to be mutually exclusive. These findings thus define two pathogenetic pathways in the development of thymomas: one characterized by the presence of the 6g23.3-25.3 LOH, the other by LOH in the APC tumor suppressor gene locus in the region 5q21 associated with 3p22-24.2, 13q14 (RB), and 17p13.1 (p53) LOHs. Dividing the analyzed tumors according to their WHO type, specific allelotype patterns could be assigned to individual thymoma groups. The 6q23.3-25.3 LOH was present in all three investigated thymoma types. In contrast, the LOH in the APC locus was found in type B3 only. Other abnormalities present exclusively in the types B3 and C were frequent LOH in the 13q14 and 17p13 regions, loci of the RB and p53 genes, respectively, in the 3p22-24.2 region, and less frequent LOH in the region 8q11.21-23. These five aberrations thus define the difference between type A thymoma on one side and types B3 and C on the other side. The difference in the pattern of genetic aberrations suffered by individual types of thymoma could be a reason for the different prognosis of type A as compared to type B3 tumors. Peculiarly, approximately two-thirds of type A thymomas present as stage I disease and only 5% of type A tumors extend into adjacent organs (stage III). The overall survival of type A thymoma patients approaches 100% at 5 and 10 years. 11,12 In contrast, type B3 thymoma is a malignant tumor presenting mostly at stage III (59%) or even IV (25%) with survival rates of 80% and 40 to 54% at 5 and 10 years, respectively. 8,13 There is thus a considerable difference in the survival of thymoma patients, which could be a result of different patterns of aberrations displayed by these tumors.
In summary, we show that karyotypic instability plays a decisive role in the development of type A, B3, and C thymomas. We characterize some of the common genetic aberrations occurring during thymomagenesis and describe two pathogenetic pathways these tumors develop along. The first one typified by the 6q23.3-25.3 LOH and common to types A, B3, and C type thymomas, the second one characterized by the APC LOH and displayed by WHO type B3 thymomas only. We describe a set of additional clonal aberrations featured by type B3 and C thymomas (but not type A thymomas): LOH in the RB, p53 loci, 3p22-24 and 8q11.21-23 regions. We suggest that the differences between the kind of genetic aberrations seen in type A thymoma on one side and types B3 and C thymoma on the other side might be mirrored in the differences in biological behavior displayed by these tumors. Type A thymomas being known as tumors with benign prognosis, types B3 and C thymic epithelial tumors displaying more aggressive behavior with accordingly shorter survival rates.
Footnotes
Address reprint requests to Petr Starostik, Institute of Pathology, Würzburg University, Luitpoldkrankenhaus, Josef-Schneider-Strasse 2, D-97080 Würzburg, Germany. E-mail: petr.starostik@mail.uni-wuerzburg.de.
Supported by a Visiting Scientist Award (no. 97833011 to R. Z.) from the Chinese Scholarship Council.
References
- 1.Harris NL, Muller-Hermelink HK: Thymoma classification. A siren’s song of simplicity. Am J Clin Pathol 1999, 112:299-303 [DOI] [PubMed] [Google Scholar]
- 2.Kornstein MJ: Thymoma classification: my opinion. Am J Clin Pathol 1999, 112:304-307 [DOI] [PubMed] [Google Scholar]
- 3.Suster S, Moran CA: Thymoma classification. The ride of the valkyries? Am J Clin Pathol 1999, 112:308-310 [DOI] [PubMed] [Google Scholar]
- 4.Rosai J, Sobin LH: Histological typing of tumors of the thymus. Rosai J Sobin LH eds. WHO International Histological Classification of Tumors. 1999, :pp 1-65 SpringerVerlag, Berlin [Google Scholar]
- 5.Müller-Hermelink HK, Marx A: Pathological aspects of malignant and benign thymic disorders. Ann Med 1999, 31(Suppl 2):S5-S14 [PubMed] [Google Scholar]
- 6.Marino M, Muller-Hermelink HK: Thymoma and thymic carcinoma. Relation of thymoma epithelial cells to the cortical and medullary differentiation of thymus. Virchows Arch A Pathol Anat Histopathology 1985, 407:119-149 [DOI] [PubMed] [Google Scholar]
- 7.Kirchner T, Schalke B, Buchwald J, Ritter M, Marx A, Muller-Hermelink HK: Well-differentiated thymic carcinoma. An organotypical low-grade carcinoma with relationship to cortical thymoma. Am J Surg Pathol 1992, 16:1153-1169 [PubMed] [Google Scholar]
- 8.Ho FC, Fu KH, Lam SY, Chiu SW, Chan AC, Muller-Hermelink HK: Evaluation of a histogenetic classification for thymic epithelial tumours. Histopathology 1994, 25:21-29 [DOI] [PubMed] [Google Scholar]
- 9.Kuo TT, Lo SK: Thymoma: a study of the pathologic classification of 71 cases with evaluation of the Muller-Hermelink system. Hum Pathol 1993, 24:766-771 [DOI] [PubMed] [Google Scholar]
- 10.Pan CC, Wu HP, Yang CF, Chen WY, Chiang H: The clinicopathological correlation of epithelial subtyping in thymoma: a study of 112 consecutive cases. Hum Pathol 1994, 25:893-899 [DOI] [PubMed] [Google Scholar]
- 11.Pescarmona E, Rendina EA, Venuta F, Ricci C, Ruco LP, Baroni CD: The prognostic implication of thymoma histologic subtyping. A study of 80 consecutive cases. Am J Clin Pathol 1990, 93:190-195 [DOI] [PubMed] [Google Scholar]
- 12.Quintanilla-Martinez L, Wilkins EW, Jr, Ferry JA, Harris NL: Thymoma—morphologic subclassification correlates with invasiveness and immunohistologic features: a study of 122 cases. Hum Pathol 1993, 24:958-969 [DOI] [PubMed] [Google Scholar]
- 13.Quintanilla-Martinez L, Wilkins EW, Jr, Choi N, Efird J, Hug E, Harris NL: Thymoma. Histologic subclassification is an independent prognostic factor. Cancer 1994, 74:606-617 [DOI] [PubMed] [Google Scholar]
- 14.Marx A, Muller-Hermelink HK: From basic immunobiology to the upcoming WHO-classification of tumors of the thymus. The Second Conference on Biological and Clinical Aspects of Thymic Epithelial Tumors and related recent developments. Pathol Res Pract 1999, 195:515-533 [DOI] [PubMed] [Google Scholar]
- 15.Kees UR, Mulcahy MT, Willoughby ML: Intrathoracic carcinoma in an 11-year-old girl showing a translocation t(15;19). Am J Pediatr Hematol Oncol 1991, 13:459-464 [DOI] [PubMed] [Google Scholar]
- 16.Kubonishi I, Takehara N, Iwata J, Sonobe H, Ohtsuki Y, Abe T, Miyoshi I: Novel t(15;19)(q15;p13) chromosome abnormality in a thymic carcinoma. Cancer Res 1991, 51:3327-3328 [PubMed] [Google Scholar]
- 17.Lee AC, Kwong YI, Fu KH, Chan GC, Ma L, Lau YL: Disseminated mediastinal carcinoma with chromosomal translocation (15;19). A distinctive clinicopathologic syndrome. Cancer 1993, 72:2273-2276 [DOI] [PubMed] [Google Scholar]
- 18.Zettl A, Strobel P, Wagner K, Katzenberger T, Ott G, Rosenwald A, Peters K, Krein A, Semik M, Muller-Hermelink HK, Marx A: Recurrent genetic aberrations in thymoma and thymic carcinoma. Am J Pathol 2000, 157:257-266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hirabayashi H, Fujii Y, Sakaguchi M, Tanaka H, Yoon HE, Komoto Y, Inoue M, Miyoshi S, Matsuda H: p16INK4, pRB, p53 and cyclin D1 expression and hypermethylation of CDKN2 gene in thymoma and thymic carcinoma. Int J Cancer 1997, 73:639-644 [DOI] [PubMed] [Google Scholar]
- 20.Chen FF, Yan JJ, Chang KC, Lai WW, Chen RM, Jin YT: Immunohistochemical localization of Mcl-1 and bcl-2 proteins in thymic epithelial tumours. Histopathology 1996, 29:541-547 [DOI] [PubMed] [Google Scholar]
- 21.Mukai K, Sato Y, Hirohashi S, Shimosato Y: Expression of ras p21 protein by thymoma. Virchows Arch B Cell Pathol Incl Mol Pathol 1990, 59:11-16 [DOI] [PubMed] [Google Scholar]
- 22.Parrens M, Labouyrie E, Groppi A, Dubus P, Carles D, Velly JF, de Mascarel A, Merlio JP: Expression of NGF receptors in normal and pathological human thymus. J Neuroimmunol 1998, 85:11-21 [DOI] [PubMed] [Google Scholar]
- 23.Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, Srivastava S: A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 1998, 58:5248-5257 [PubMed] [Google Scholar]
- 24.Dorfman DM, Shahsafaei A, Chan JK: Thymic carcinomas, but not thymomas and carcinomas of other sites, show CD5 immunoreactivity. Am J Surg Pathol 1997, 21:936-940 [DOI] [PubMed] [Google Scholar]
- 25.Sambrook J, Frish E, Maniatis T: Molecular Cloning. 1989. Cold Spring Harbor Press, Cold Spring Harbor
- 26.Mertens F, Johansson B, Hoglund M, Mitelman F: Chromosomal imbalance maps of malignant solid tumors: a cytogenetic survey of 3185 neoplasms. Cancer Res 1997, 57:2765-2780 [PubMed] [Google Scholar]
- 27.Petersen I, Bujard M, Petersen S, Wolf G, Goeze A, Schwendel A, Langreck H, Gellert K, Reichel M, Just K, du Manoir S, Cremer T, Dietel M, Ried T: Patterns of chromosomal imbalances in adenocarcinoma and squamous cell carcinoma of the lung. Cancer Res 1997, 57:2331-2335 [PubMed] [Google Scholar]
- 28.Speicher MR, Howe C, Crotty P, du Manoir S, Costa J, Ward DC: Comparative genomic hybridization detects novel deletions and amplifications in head and neck squamous cell carcinomas. Cancer Res 1995, 55:1010-1013 [PubMed] [Google Scholar]
- 29.Theile M, Seitz S, Arnold W, Jandrig B, Frege R, Schlag PM, Haensch W, Guski H, Winzer KJ, Barrett JC, Scherneck S: A defined chromosome 6q fragment (at D6S310) harbors a putative tumor suppressor gene for breast cancer. Oncogene 1996, 13:677-685 [PubMed] [Google Scholar]
- 30.Trent JM, Rosenfeld SB, Meyskens FL: Chromosome 6q involvement in human malignant melanoma. Cancer Genet Cytogenet 1983, 9:177-180 [DOI] [PubMed] [Google Scholar]
- 31.Morita R, Saito S, Ishikawa J, Ogawa O, Yoshida O, Yamakawa K, Nakamura Y: Common regions of deletion on chromosomes 5q, 6q, and 10q in renal cell carcinoma. Cancer Res 1991, 51:5817-5820 [PubMed] [Google Scholar]
- 32.Stenman G, Sandros J, Mark J, Edstrom S: Partial 6q deletion in a human salivary gland adenocarcinoma. Cancer Genet Cytogenet 1989, 39:153-156 [DOI] [PubMed] [Google Scholar]
- 33.Orphanos V, McGown G, Hey Y, Boyle JM, Santibanez-Koref M: Proximal 6q, a region showing allele loss in primary breast cancer. Br J Cancer 1995, 71:290-293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hayashi Y, Raimondi SC, Look AT, Behm FG, Kitchingman GR, Pui CH, Rivera GK, Williams DL: Abnormalities of the long arm of chromosome 6 in childhood acute lymphoblastic leukemia. Blood 1990, 76:1626-1630 [PubMed] [Google Scholar]
- 35.Offit K, Wong G, Filippa DA, Tao Y, Chaganti RS: Cytogenetic analysis of 434 consecutively ascertained specimens of non-Hodgkin’s lymphoma: clinical correlations. Blood 1991, 77:1508-1515 [PubMed] [Google Scholar]
- 36.Starostik P, Greiner A, Schultz A, Zettl A, Peters K, Rosenwald A, Kolve M, Muller-Hermelink HK: Genetic aberrations common in gastric high-grade large B-cell lymphoma. Blood 2000, 95:1180-1187 [PubMed] [Google Scholar]
- 37.Spengler D, Villalba M, Hoffmann A, Pantaloni C, Houssami S, Bockaert J, Journot L: Regulation of apoptosis and cell cycle arrest by Zac1, a novel zinc finger protein expressed in the pituitary gland and the brain. EMBO J 1997, 16:2814-2825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bilanges B, Varrault A, Basyuk E, Rodriguez C, Mazumdar A, Pantaloni C, Bockaert J, Theillet C, Spengler D, Journot L: Loss of expression of the candidate tumor suppressor gene ZAC in breast cancer cell lines and primary tumors. Oncogene 1999, 18:3979-3988 [DOI] [PubMed] [Google Scholar]
- 39.Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, Vogelstein B, Kinzler KW: APC mutations occur early during colorectal tumorigenesis. Nature 1992, 359:235-237 [DOI] [PubMed] [Google Scholar]
- 40.Lane DP: Cancer. p53, guardian of the genome. Nature 1992, 358:15-16 [DOI] [PubMed] [Google Scholar]
- 41.Jimenez P, Canton J, Collado A, Cabrera T, Serrano A, Real LM, Garcia A, Ruiz-Cabello F, Garrido F: Chromosome loss is the most frequent mechanism contributing to HLA haplotype loss in human tumors. Int J Cancer 1999, 83:91-97 [DOI] [PubMed] [Google Scholar]
- 42.Amiot L, Onno M, Lamy T, Dauriac C, Le Prise PY, Fauchet R, Drenou B: Loss of HLA molecules in B lymphomas is associated with an aggressive clinical course. Br J Haematol 1998, 100:655-663 [DOI] [PubMed] [Google Scholar]
- 43.Townsend A, Bodmer H: Antigen recognition by class I-restricted T lymphocytes. Annu Rev Immunol 1989, 7:601-624 [DOI] [PubMed] [Google Scholar]
- 44.Garrido F, Ruiz-Cabello F, Cabrera T, Perez-Villar JJ, Lopez-Botet M, Duggan-Keen M, Stern PL: Implications for immunosurveillance of altered HLA class I phenotypes in human tumours. Immunol Today 1997, 18:89-95 [DOI] [PubMed] [Google Scholar]
- 45.Hanahan D, Weinberg RA: The hallmarks of cancer. Cell 2000, 100:57-70 [DOI] [PubMed] [Google Scholar]
- 46.Lengauer C, Kinzler KW, Vogelstein B: Genetic instabilities in human cancers. Nature 1998, 396:643-649 [DOI] [PubMed] [Google Scholar]
- 47.Dams E, Van de Kelft EJ, Martin JJ, Verlooy J, Willems PJ: Instability of microsatellites in human gliomas. Cancer Res 1995, 55:1547-1549 [PubMed] [Google Scholar]
- 48.Levine GD, Rosai J: Thymic hyperplasia and neoplasia: a review of current concepts. Hum Pathol 1978, 9:495-515 [DOI] [PubMed] [Google Scholar]
- 49.Kirchner T, Müller-Hermelink HK: New approaches to the diagnosis of thymic epithelial tumors. Prog Surg Pathol 1989, 10:167-189 [Google Scholar]
- 50.Masaoka A, Monden Y, Nakahara K, Tanioka T: Follow-up study of thymomas with special reference to their clinical stages. Cancer 1981, 48:2485-2492 [DOI] [PubMed] [Google Scholar]
- 51.Shimosato Y, Mukai K: Tumors of the mediastinum. Atlas of Tumor Pathology. 1997, DC, Armed Forces Institute of Pathology, Edited by J Rosai. Washington